
Abstract
Autologous mesenchymal stromal cell (MSC)-based therapies offer one of the most promising and safe methods for regeneration or reconstruction of tissues and organs. Routine procedures to obtain adequate amount of autologous stem cells need their expansion through culture, with risks of contamination and cell differentiation, leading to the loss of cell ability for therapies. We suggest the use of human bone marrow (BM) as a physiological bioreactor to produce autologous MSC by injection of autologous platelet-rich plasma activated by recombinant human soluble tissue factor (rhsTF) in iliac crest. A trial on 13 healthy volunteers showed the feasibility and harmlessness of the procedure. The phenotype and cellularity of BM cells were not modified, on day 3 after injection. Endothelial progenitor cells (EPC) were mobilized to the bloodstream, without stimulation of hematopoietic stem cells (HSC). MSC level in BM increased with a specific commitment to preosteoblastic cell population both in vivo and in vitro. This self-stimulation system of BM seems thus to be a promising feasible process 3 days before clinical cell therapy applications.
Introduction
T
As most relevant, the need for a critical mass of autologous stem cells is fundamental to achieve success in tissue engineering. This is particularly important for MSC, present at low levels in BM and requiring in vitro expansion for clinical application. 7 The general approach to MSC culture involves isolation of BM MNC and in vitro expansion in fetal bovine serum (FBS)—a supplemented medium or in human-sourced alternatives such as platelet lysate. 8 However, the freshness of BM samples, culture media used, cell density, and number of passages have a significant impact on the heterogeneity of MSC preparations and the selection of subpopulations with divergent proliferation and differentiation potentials. 9 Long-term culture was associated with chromosome variability after four passages. 10 Furthermore, in vitro expansion of MSC led to replicative senescence, including alterations in phenotype, 11 differentiation potential, 12 telomere length, 13 proliferation rates, 14 immunosuppressive activity, 15 global gene expression patterns, 16 and DNA methylation. 17 The production of cytokines and chemokines is also significantly modified during expansion. 18
Platelet-rich plasma (PRP) purified from whole blood comprised many growth factors (GF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF), transforming growth factor-β (TGF-β),…and was defined as GF cocktail. 19 Indeed, platelets store GFs in their α-granules and their aggregation and activation result in the release of GFs. These factors are involved in wound healing and the repair of mineralized tissue.20,21 PRP may be prepared from the patient's own blood, eliminating thus concern about immunogenic reactions and disease transmission. PRP provides an autologous source of essential GFs and has been widely used from more than 10 years in various applications with controversial clinical results. 22 The heterogeneity of these studies is probably due to PRP preparation techniques, cell types, biomaterials, or implantation sites. One major variable in PRP therapy is the platelet activation before or during administration. The platelet activation is not a spontaneous event, but a regulated process leading to sequential changes in platelet form and function (adhesion, shape change, aggregation, and secretion). Platelets can be activated by a number of physical or chemical influences such as thrombin, CaCl2, or both, 23 type I collagen 24 or a freeze–thaw cycle that physically disrupts platelet membranes. 25 Platelets represent thus a source of bioactive substances for in vivo wound healing and tissue repair, but allow also the expansion of human stem and progenitor cells in vitro. Indeed, PRP promotes the in vitro proliferation of MSC and affects their differentiation.26,27 More recently, it has been demonstrated that PRP can be a safe substitute for FBS in clinical settings using MSC. 28
The aim of our study is to evaluate the level of MSC after injection of autologous PRP activated by recombinant human soluble tissue factor (rhsTF) in the iliac crest. Tissue factor (TF), a 47-kDa-transmembrane glycoprotein with structural homology with class-II cytokine receptor family, is a cellular receptor for clotting factor VIIa. TF is the major initiator of the coagulation cascade and has been used in maxillofacial surgery as replacement for thrombin to induce gel formation of PRP in bone preparation.29,30
Materials and Methods
Subjects
With the agreement of the Regional Ethics Research Committee and according to the principles expressed in the Declaration of Helsinki, we performed studies on 13 healthy volunteer adult donors (8 women and 5 men), mean age of 38 years (range 20–62), from 2007 to 2009. Each subject was informed about the procedures, which the participant approved and provided written consent. The timeframe of 3 years between the last procedure and the issuance of this publication was dedicated to the follow up of clinical parameters of the subjects.
Collection and preparation of PRP
PRP was obtained from the venous blood of each donor. Briefly 50 mL of blood was anticoagulated with anticoagulant citrate dextrose-A at a ratio of 10:1 volume. The Magellan® Autologous Platelet Separator System (Medtronic) was used to produce PRP (6 mL). The PRP was activated by the rhsTF produced by Henogen under good manufacturing practice (GMP). rhsTF is a recombinant soluble version of the human TF purified from GRAS microorganisms (yeast). To show its full activity, rhsTF (334 μg/2 mL) has to be mixed with phospholipids (2 mg) and CaCl2 (50 mM, 2 mL). After mixing, the product (4 mL) is ready for use and is stable at 20°C for 24 h.
Procedure
At day 0, BM (10 mL) was collected from the left iliac crest by puncture. Autologous PRP (6 mL) was activated by the adjunction of rhsTF. The activated PRP was rapidly injected in the iliac crest before clotting. Ten milliliters of peripheral blood (PB) was also obtained. At day 3, two BM samples were obtained: one puncture in the same area of activated PRP injection and one in the opposite site (right crest) to evaluate the specific effect of this activation procedure (Fig. 1). Ten milliliters of blood was obtained to evaluate the different parameters. The in vivo level of HSC, endothelial progenitor cells (EPC), and MSCs after injection of autologous activated PRP solution in the BM was evaluated by various assays described thereafter. The presence of these cells in the PB has been also investigated.
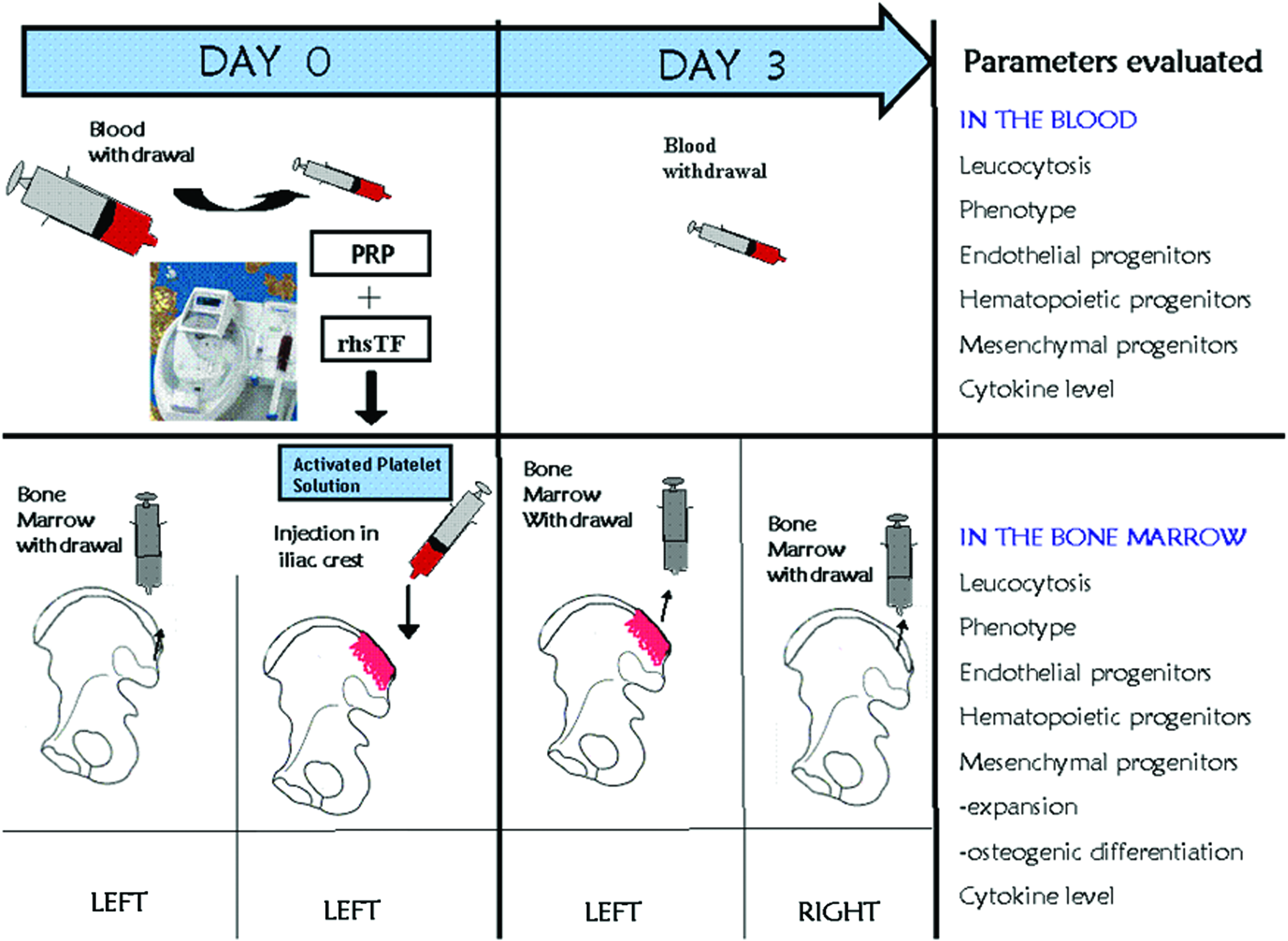
Procedures to activate bone marrow (BM) by platelet-rich plasma (PRP) and recombinant human soluble tissue factor (rhsTF). Various parameters are then evaluated in the peripheral blood (PB) and BM at day 0 and 3. Color images available online at www.liebertpub.com/tea
Flow cytometry analysis
BM and PB cells were evaluated for the expression of CD3, CD14, CD19, CD31, CD45, CD146, and HLA-DR (Immunotech), CD34 and CD133 (Miltenyi Biotec), CD73 and CD166 (BD Biosciences), and CD105 (Ancell) and KDR (VEGFR2; R&D Systems). Cells were incubated for 30 min at room temperature with primary phycoerythrin (PE)- or fluorescein isothiocyanate (FITC)- or PerCP-conjugated antibodies. Uti-Lyse reagent A (Dako) was mixed with labeled cells and incubated for 10 min. Reagent B (1 mL) was added and the cells incubated for a further 10 min. All labeled leukocyte samples were kept at 4°C and analyzed as soon as possible by flow cytometry. Flow cytometry was performed using MACSQUANT flow cytometer (Miltenyi Biotec) and 10,000 events for each sample were acquired.
Hematopoietic colony assay
The number of multipotential granulocyte-erythroid-macrophage-megakaryocyte CFU (CFU-GEMM), myeloid granulocyte-macrophage CFU (CFU-GM), erythroid burst-forming units (BFU-E), and clonogenic progenitors was evaluated at day 0 and 3 in the blood and BM. Briefly 40,000 MNC were plated in 1 mL of semisolid medium (Methocult SFbit H4536 from StemCell Technologies). After 12/14 days, the number of colonies was counted using an inverted microscope.
EPC culture assay
BM and PB MNC were plated on fibronectin-coated 24-well dishes in the EndoCult Liquid medium (StemCell Technologies) for 2 days to remove adherent cells, including mature endothelial cells. The nonadherent cells (containing EPC) were then plated on fibronectin-coated 24-well dishes in the EndoCult medium. After 5 days, colonies consisting of round cells and elongated sprouting cells were enumerated.
Matrigel angiogenesis assay
PB MNC were incubated in the endothelial cell growth medium (Promocell) for 10–15 days to allow endothelial cell proliferation. Endothelial cells were then harvested with TrypLE Select (Gibco; Life Technologies) and cultured at 104 cells until 24 h in μ-slide angiogenesis (Ibidi) precoated with 10 μL Matrigel (BD Biosciences) in the presence of vascular endothelial growth factor (VEGF) at 100 ng/mL. The culture was monitored and photographed at 2, 4, 6, and 24 h with an inverted microscope.
Human MSC culture and expansion
BM MNC were seeded at 2×104 cells/cm2 in the Dulbecco's modified Eagle's medium (DMEM) low glucose (Lonza) supplemented with 15% FBS (Sigma-Aldrich), 2 mM
CFU-F assay
CFU-F assay was used to evaluate the number of mesenchymal progenitors in BM and PB. About 105 MNC or 5000 cells obtained after each passage were incubated in Petri dishes in the complete DMEM. Ten days later, fibroblastic colonies with more than 50 cells were counted under light microscopy after May-Grunwald-Giemsa coloration. Formation of osteoblast progenitors was detected using an alkaline phosphatase (ALP) assay (Sigma-Aldrich). ALP-positive (ALP+) colonies were scored on an inverted microscope.
Induction of osteogenic differentiation
For osteogenic differentiation, MSC were seeded at 1000 cells/cm2 in 48-well dishes (for calcium and ALP assays) or in Petri culture plates (for real-time polymerase chain reaction [RT-PCR]) containing the α-MEM culture medium (Lonza) supplemented with 10−7 M dexamethasone (Aacidexam®; Aaciphar), 6×10−5 M ascorbic acid (Sigma-Aldrich), and 10−2 M β-glycerophosphate (DAG; Sigma-Aldrich), and cultured for up to 3 weeks. The osteogenic medium was changed weekly and experiments were stopped after 7, 14, or 21 days to assess the osteoblastic phenotype of MSC. Osteogenic differentiation was monitored by mineralization assay, calcium determination, ALP activity measurement, and osteoblast-related gene expression evaluation.
Mineralization assay
Mineralization of the extracellular matrix was assessed by Alizarin Red S (AR-S) staining. Cells were washed in phosphate-buffered saline (PBS) and fixed in 70% ethanol at room temperature for 5 min followed by several washes in H2O. Cells were stained in 40 mM AR-S (Sigma-Aldrich) pH=4.2 for 15 min at room temperature, rinsed in H2O, and then air-dried. Red staining was examined by light microscopy
Measurement of calcium accumulation (quantitative determination)
The matrix was demineralized by addition of 500 μL of 0.6 M HCl and overnight incubation at 37°C. Solutions were then collected and centrifuged at 2000 rpm for 5 min. The calcium concentration in the supernatant was determined by colorimetric assay (QuantiChrom Calcium Assays Kit; BioAssay Systems) as described by the manufacturer.
ALP activity determination (quantitative analysis)
The ALP activity was determined using the LabAssayTM ALP (Wako Chemicals Gmbh), according to the manufacturer's recommendations. This measurement consists of the determination of the p-nitrophenol amount released from the substrate. The cell layers were lysed with 100 μL of ice-cold 0.1% Triton X-100 in PBS. Samples were then frozen and thawed twice, and the cell lysates were collected.
Cytokine level
BM plasma and PB serum samples were analyzed for PDGF-bb (sensitivity<15 pg/mL), FGF-2 (<3 pg/mL), IGF-1 (26 pg/mL), VEGF (9 pg/mL), SDF-1α (18 pg/mL), MIP-1α (<10 pg/mL), Rantes (2 pg/mL), and BDNF (<20 pg/mL) by ELISA (Quantikine; R&D Systems).
Bone-associated marker expression of mesenchymal cells by semiquantitative RT-PCR
By multiplex semiquantitative RT-PCR using β-actin as a housekeeping gene, the expression of genes implicated in osteogenesis (osteocalcin, osteopontin) was evaluated. Total RNA from each cell culture was extracted by the Tripure method (Roche Diagnostics). The samples (1 μg RNA) were treated by DNAse (Invitrogen Life Technologies) at 37°C for 30 min in a final volume of 10 μL containing 1 μL DNAse, 10×buffer, and 1 U DNAse RQI. The reverse transcription was performed with 1 μg DNAse-treated RNA using the M-MLV reverse transcriptase (Invitrogen Life Technologies) in a final volume of 20 μL containing a 4 μL first-strand buffer, 10−2 M DTT, 1 μM each of dNTP, 50 URNase inhibitor, and 100 U M-MLV reverse transcriptase. The reverse transcription led to the production of 5 ng cDNA, which were used for PCR reaction in a final volume of 50 μL containing 25 μL multiplex PCR mix and 200 nmol upstream sense and downstream sense primers (Sigma-Genosys):
Osteopontin (395) (forward, ctaggcatcacctgtgccatacc; reverse, cagtgaccagttcatcagattcatc). Osteocalcin (119) (forward, gactgtgacgagttggctga; reverse, ctggagaggagcagaactgg). β-actin (610) (forward, tgacggggtcaccacactgtgcccatcta; reverse, ctagaagcatttgcggtggacgatggaggg).
Each cycle consisted of activation at 95°C for 15 min, denaturation at 94°C for 30 s, annealing at 60°C for 90 s, extension at 72°C for 90 s, and elongation at 72°C for 10 min. After 35 cycles for each primer sets, PCR products were separated by electrophoresis on 2% (w/v) agarose gel and were visualized by ethidium bromide staining. Gels were scanned with a FLA-5000 imaging system (Fujifilm; Raytest®) and a software Image Reader (Raytest). Band intensities were quantified using the software AIDA® Image Analyzer 3.45 (Raytest) and normalized with β-actin used as endogen loading control.
RT quantitative PCR
mRNA expression of relevant osteoblast-related markers (Runx2, OPG, BSP, OPN) was also quantified by quantitative PCR (qPCR) using SYBR Green PCR Master Mix (Applied Biosystem) and sequence-specific primers as previously described by our group. 32 qPCR was performed on an ABI PrismR 7900HT Sequence Detection System with 40 cycles of a two-step PCR (95°C for 15 s and 60°C for 60 s) after an initial activation step (95°C for 10 min). Melting curves from 60°C to 99°C were assessed to evaluate specificity. Relative quantification was calculated by normalizing the test crossing thresholds (Ct) with the β-actin amplified control Ct.
Statistical analysis
Data are expressed as the mean±standard error of the mean. Statistical comparisons were performed using the paired Wilcoxon's test. All tests were two-sided. p-Values lower than 0.05 were considered statistically significant. All analyzes were performed using GraphPad Prism Version 5.00 for Windows (GraphPad Software, www.graphpad.com).
Results
BM and PB parameters before and after activation
Before TF-PRP injection and 3 days after, BM and PB samples were obtained for cell counts and cell phenotype evaluation by flow cytometry. CFU-GM, CFU-GEMM, and BFU-E were enumerated by standardized assays. As observed in Table 1, the BM activation did not modify the BM leukocytosis and no statistically significant effect on the hematopoietic progenitor level was observed. Moreover, no modification of the cell phenotype in BM samples was seen in these conditions (Table 2). In contrast, we observed an increase of cells expressing CD34 and CD133, but a decrease of CD45-positive cells in PB at day 3 postactivation. These observations suggest the mobilization of EPC in circulation (Table 2).
CFU-GM, granulocyte-macrophage CFU; CFU-GEMM, granulocyte-erythroid-macrophage-megakaryocyte CFU; BFU-E, erythroid burst-forming units.
The results were expressed as the percentage of positive cells of the total number of leukocytes.
p<0.01.
p<0.04.
BM, bone marrow; PB, peripheral blood.
Increased frequency of CFU-F and CFU-osteoblast after BM activation
CFU assays were performed at days 0 and 3 on MNC obtained from BM and PB before and after activation to evaluate the frequency of mesenchymal progenitors. After 10 days of culture, the CFU-F frequency was significantly higher for activated BM in comparison to inactivated BM (left and right sites) (Fig. 2A). Moreover, the CFU-F frequency determined in primoculture (PM) differs also before and after activation. The total number of CFU-F was 8.7±3.8×103 (left BM) and 15±5×103 (right BM) versus 20.2±8.7×103 after activation by rhsTF-PRP (p=0.042) (Fig. 2B). Interestingly, we have observed a greater proportion of CFU-osteoblast (CFU-O) among CFU-F after BM activation. About 25%±10% of CFU-F obtained from MNC were ALP+ before activation and 32%±9% after activation. In PM, the level of CFU-O reached 49%±12% for activated BM versus 21%±8% without activation (Fig. 2C) (p=0.0156).
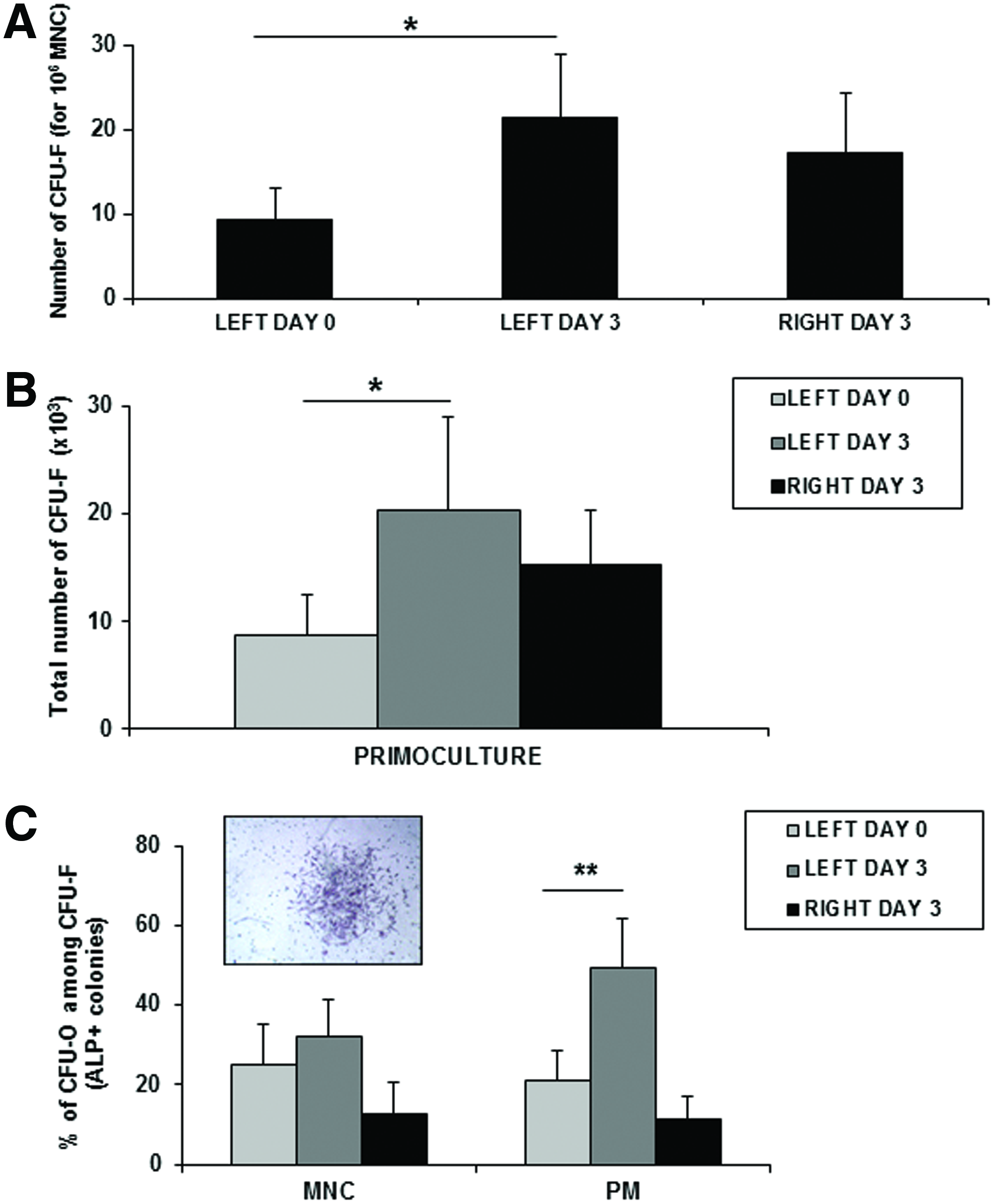
Frequency of colony-forming unit-fibroblast (CFU-F) and CFU-osteoblast (CFU-O) after BM activation.
EPC in PB and BM
EPC are a heterogeneous population in terms of lineage, phenotype, and growth pattern. EPC can be identified as CD34+KDR+ cells by flow cytometry and identified as cells adopting an endothelial phenotype by in vitro culture of MNC. In this study, we evaluated EPC numbers in blood and BM, before and after activation, by colony-forming assay. A colony of EPC consists of multiple thin, flat cells emanating from a central cluster of rounded cells. PB and BM MNC formed distinct colonies on fibronectin-coated dishes (Fig. 3A). EPC isolated in this fashion exhibit many endothelial characteristics, including CD31 and KDR. The number of EPC was significantly increased in PB after BM activation suggesting the mobilization of EPC from BM. About 154±37 colonies were enumerated at day 0 for 2×106 blood MNC versus 369±87 at day 3 post-BM activation (p=0.0034) (Fig. 3D). The levels of EPC in BM before and after TF-PRP injection were very similar. To confirm the mobilization of EPC in the blood, PB MNC were also plated in the endothelial growth medium. After 7–10 days, a confluent layer of adherent cells was observed only in the case of PB obtained after BM activation (Fig. 3B, C). The endothelial nature of these cells was confirmed by flow cytometry (CD34+, KDR+, and CD144+). To examine whether these mobilized EPC can participate in angiogenesis-like endothelial network formation in vitro, endothelial cells expanded in culture were plated on a gelled basement matrix. The formation of capillary-like structures with a lumen was already observed after a few hours (Fig. 3E).
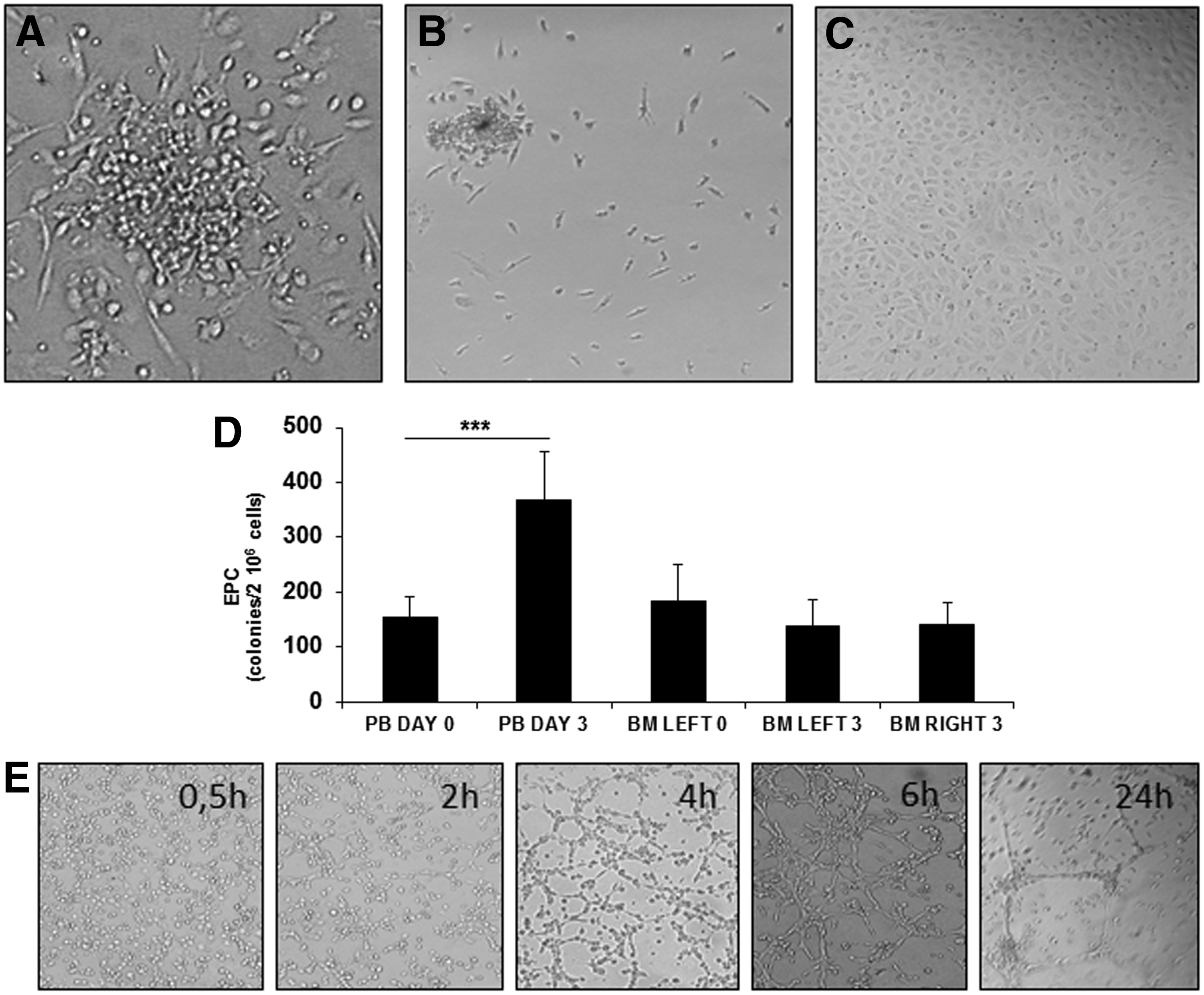
Endothelial progenitor cells (EPC) in PB and BM after rhsTF activation.
Cytokine levels in BM and PB before and after activation
BM plasma and PB serum at day 0 and 3 were first analyzed using RayBio® Human Cytokine Antibody Array C Series 1000 (Gentaur) to detect 120 cytokine expression simultaneously. Any variation was observed between day 0 and 3 in BM plasma samples (Fig. 4A). In contrast, the levels of PDGF-bb, VEGF, Rantes, and BDNF were increased in blood serum samples after 3 days (Fig. 4B). These observations were further confirmed by specific ELISA for each cytokine. Three days post-BM activation, the levels of PDGF-bb, VEGF, Rantes, and BDNF were, respectively, 5±0.76, 0.16±0.03, 70.2±17, and 25.2±4.2 ng/mL versus 3.3±0.7 (p=0.0273), 0.087±0.019 (p=0.0029), 39.4±10.4 (p=0.0117), and 17.2±2.6 (p=0.0488) before activation.
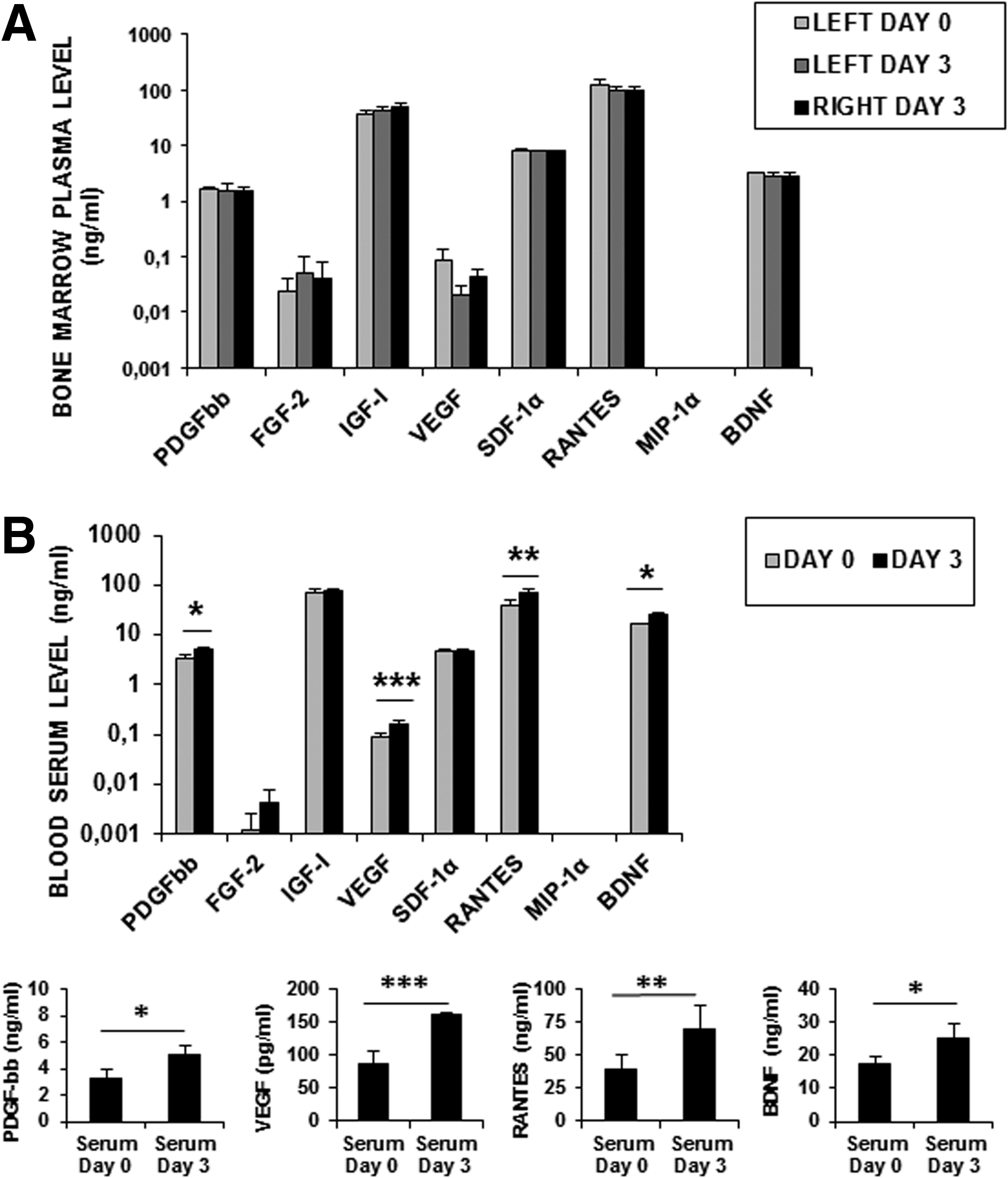
Cytokine levels in BM
Isolation and expansion potential of MSC
The isolation of MSC from BM before and after activation was successful for all donors (n=13). We showed that the cumulative expansion rate was higher when MSC were obtained from activated BM (left day 3). We did not observe a difference between MSC obtained at day 0 versus BM obtained at the right site, but without activation. After two passages, we observed a 176-fold expansion for activated BM compared with a 100-fold expansion for unstimulated BM (Fig. 5A). Indeed, for 5×106 MNC plated in culture, we obtained after two passages, 32.6±16×106 and 89.8±31×106, respectively, before and after activation (p<0.03). The expression of cell surface markers was analyzed by flow cytometry after one and two passages and for all conditions. MSC were highly positive for CD73, CD105, CD166, and CD146, but negative for CD45, CD34, and HLA-DR.

Expansion and characterization of MSC.
Osteogenic differentiation potential of MSC
The influence of rhsTF-PRP activation on the osteogenic differentiation potential of MSC was investigated after appropriate induction. In vitro mineralization, the last step of osteoblastic differentiation is usually obtained after 21 days of confluent culture in the presence of osteogenic inducers (dexamethasone, ascorbic acid, and β-glycerophosphate). Interestingly, MSC obtained from activated BM deposited a more extensive mineralized matrix as demonstrated by strong Alizarin Red staining (Fig. 6A). The mineralization of the MSC extracellular matrix was also determined by colorimetric assay after 7, 14, and 21 days of culture in osteogenic conditions. Calcium deposition was very weak after 7 days, but superior for MSC derived from activated BM (1.9±0.96 vs. 0.32±0.12 mg/dL for unactivated BM-derived MSC). Calcification was induced after 14 days in all conditions with more than 5 mL/dL for activated BM-MSC. After 21 days, matrix calcification was very high and significantly higher for activated BM-MSC (16.11±3.7 vs. 6.38±1.9 mg/dL for unactivated BM, p=0.0156) (Fig. 6B). The increased osteogenic potential of MSC obtained from activated BM was also confirmed by the evaluation of the ALP activity, an early marker of osteoblast differentiation. The ALP activity was maximal after 14 days and higher in the case of MSC obtained from activated BM (0.277±0.109 vs. 0.163±0.075 mM for unactivated BM, p=0.0391) (Fig. 6C).

Osteogenic differentiation potential of MSC. MSC were cultured for 21 days in the osteogenic differentiation medium or in the Dulbecco's modified Eagle's medium (DMEM) as control.
Osteoblastic gene expression
Osteoblastic gene expression was analyzed after MSC amplification (P1) obtained from unactivated and activated BM, without adding differentiation agents. MSC from different BM were analyzed using RT-PCR. The expression of osteoblast-related genes, osteocalcin and osteopontin, was upregulated in MSC obtained from activated BM (left day 3) in comparison with unactivated BM (left day 0 and right day 3). These results indicate that rhsTF-PRP injection induces osteoblastic gene expression in MSC (Fig. 7A). By qPCR, we have confirmed the significant overexpression of osteogenic genes (osteopontin, osteocalcin, and bone sialoprotein) in MSC obtained from activated BM. However, no modification of Runx2 expression was observed (Fig. 7B).
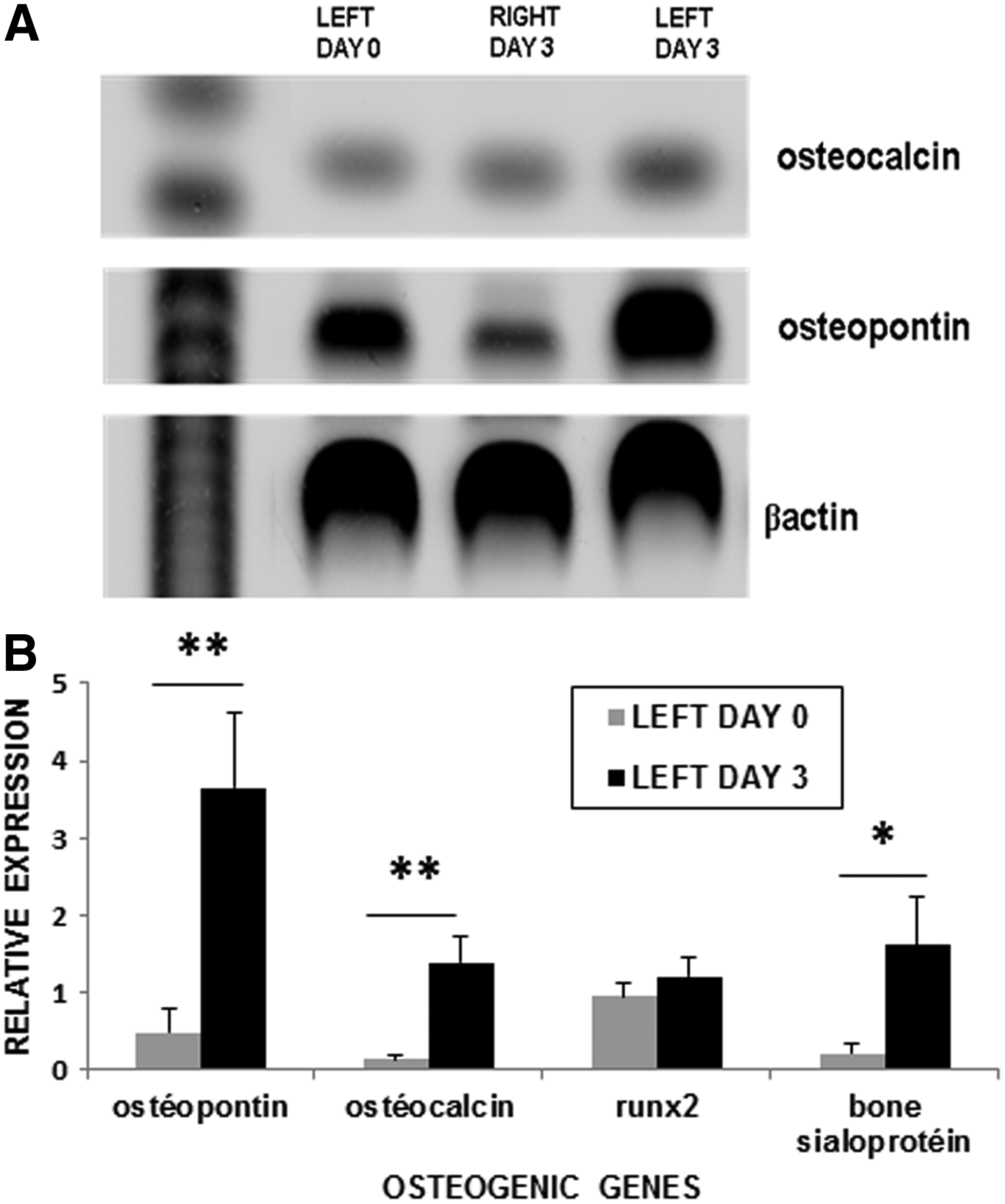
Osteogenic gene expression.
Discussion
The cell-based approach is a promising strategy in the field of regenerative medicine and is widely studied in maxillofacial surgery as bone regeneration and implant surgery. Since the GFs released from platelets are capable of promoting cell proliferation and angiogenesis, the PRP from PB has been widely used in preclinical and clinical applications to enhance bone regeneration. In this article, for the first time, we report a safe in vivo procedure to multiply and activate MSCs in the BM and to mobilize EPC in the blood stream. Contrary to the procedure using G-CSF or Neupogen (Filgrastim; Amgen) aiming to the production of stem cells harvested from PB by apheresis, 30 a time-consuming technique, the lack of HSC phenotype stimulation and multiplication in all clinical cases must be consider as a critical advantage of the described method. The unmodified bloodstream hemostasis status, both hyper- and hypocoagulability is a fundamental prerequisite for any clinical application. Associated with the natural rheological blood feature, circulating EPC should easily reach the targeted specific tissue presenting an acute need for possible requirement of tissue-engineered blood vessels. Furthermore, a long lasting EPC flow should enable better homing in tissue engineering versus damaged tissue, in comparison with time point intravessel injections currently reported. On the other hand, withdrawing of bone-committed MSC is a reliable and easy to use routine clinical practice for injection during bone surgery.
PRP is an autologous material, without toxicity, disease transmission, or immune response. rhsTF, the only external component, is GMP produced and phase I clinical trial approved. Because TF is a transmembrane protein, three types of molecules can be obtained by genetic engineering: a complete 263aa-TF, a truncated 243aa (tTF) without cytoplasmic tail, and a soluble 219aa-TF (sTF), comprising only the extra-cytoplasmic portion, packaged as liquid in vials (Henogen). Recombinant TF was used in first clinical trials for bone reconstruction with no adverse reactions reported from 1997.29–31 Moreover, it was further shown that no residual coagulating disturbance in any clinical case could be seen using tTF. 29 The rhsTF coagulating feature is 4% of full length TF allowing the injection of activated PRP as a fluid in the BM. Up to now, few groups reported the feasibility of an intrabone procedure in the context of stem cell transplantation for malignant hematopoietic diseases 33 or autoimmune diseases. 34 Our study demonstrates clearly the feasibility of coagulating blood injection in iliac crest as a harmless and painless procedure under local anesthesia.
The fundamental basis of our procedure is linked to the pathophysiological events observed during fracture. Indeed, fracture healing is pictured like a playground, where growth and differentiation factors, hormones, cytokines, and the extracellular matrix play with bone primary cells and MSC in a well orchestrated series of biological events. 35 The inflammatory phase is a trauma response, including different stages. The haematoma stage is the stage in which the vascular disruption leads to acute necrosis and activation of thrombotic factors allowing blood clot formation in the fracture site. The inflammatory stage is the stage where macrophages and other immune cells are recruited to the fracture site and essential in secreting proinflammatory cytokines, which have a chemotactic effect on mesenchymal cells. 36 Activated platelets release growth and differentiation factors inducing ossification. The formation of hematoma associated with an inflammatory environment will temporarily transform the BM of the patient into an autologous bioreactor. In our procedure mimicking fracture, activated PRP is the source of signaling molecules initiating the cascades of cellular events. Biological signals are released in the bloodstream and targeted to BM inducing the mobilization of EPC and MSC activation. Moreover, the only clinical adverse event reported by our volunteers is fatigue on day 1, after injection, proving a subject's reaction to BM stimulation. Indeed, in the case of G-CSF-induced mobilization of PB stem cells from healthy donors, the major adverse effects include also general fatigue. 37 In contrast to this mobilization, HSC were not stimulated by our procedure and the total blood cell count and hemostatic parameters were not modified.
During fracture repair, the recruitment, proliferation, and differentiation of MSC and EPC are induced by cytokines, chemokines, and GFs secreted at the fracture site and released in the peripheral circulation. Similar to leukocyte trafficking, EPC are thought to mobilize from the BM into the circulation under the guidance of these factors. EPC mobilization may contribute to new bone formation in fracture healing and begins as early as 24 h after traumatic fracture. 38 In our study, at day 3, an increased EPC level was observed in the bloodstream and the level of four cytokines (VEGF, Rantes, BDNF, and PDGF-bb) is significantly higher. Several studies have shown that VEGF is a potent mobilizer of EPC from the BM to the peripheral circulation 39 and is released into the circulation during vascular trauma and early postfracture. 40 Rantes seems to play a pivotal role for the recruitment of EPC in wound healing. 41 Recently, several proangiogenic effects of BDNF have been observed. It promotes revascularization in ischemic injury and stimulates VEGF production and endothelial proliferation. 42 PDGF-bb has been also described to modulate endothelial proliferation and angiogenesis using PDGFβ-receptors. 43 These feedings suggest a link between EPC mobilization and the increased levels of these cytokines in the bloodstream. After mobilization, EPC have the capacity to home in and to be incorporated into the sites of vascular injury and ischemia, with evidence of improvement in the function and viability of tissue. Since insufficient vascularization remains one of the major problems in tissue engineering, our approach to mobilize EPC is thus of importance.
During bone repair, MSC originating from the BM near the fracture are believed to invade the wound site, proliferate and differentiate into osteoblasts necessary for bone regeneration. It has been also suggested that circulating MSC in PB are mobilized during fracture healing and may contribute to fracture repair. However, we did not observe circulating MSC after BM activation. However, our study was performed in healthy volunteers and we do not know if, in case of tissue injury the MSC will be mobilized to the damaged site. Our prospective trial showed a MSC proliferation in stimulated BM. Three days postinjection, we observed an increased frequency of CFU-F, a better MSC expansion, and the overexpression of some osteogenic genes. Interestingly, these cells showed a marked commitment to bone forming cells, evidenced by the ALP activity, calcium release, and bone mineral matrix deposition under in vitro culture with appropriate stimuli. rhsTF-PRP injection can serve as a feasible approach to manipulate in vivo MSC fate (increasing proliferation and osteogenic differentiation) for bone regeneration. Different strategies for recruiting or directing endogenous MSC into the osteogenic lineage such as BMP-2 delivery or proinflammatory cytokine challenging, has been used but seems not optimal.44,45 As a first clinical perspective, withdrawing of bone-committed MSC is a reliable procedure and easy to use in routine clinical practices for in situ injection during bone surgery. Associated with circulating EPC, preoperative BM stimulation would offer a new gold standard for safer and faster bone tissue regeneration with blood vessel formation, requested for graft survival. We believe that our ongoing work evaluating practical methods for optimizing the use of BM-derived cells harvested by simple needle aspiration will offer a new powerful tool and result in more effective and less invasive advanced bone grafting procedures.
Footnotes
Acknowledgments
This work was supported by the Walloon Region, Alpha Stem Source, and Les Amis de l'Institut Bordet.
Disclosure Statement
No competing financial interests exist.