
Abstract
Stem cells have tremendous potential for treating various human diseases. Protocols have been established to differentiate stem cells into specific lineages through the provision of signals in the form of growth factors, cytokines, or small molecules. Herein we investigate an alternative strategy for directed differentiation of human embryonic stem cells (hESCs)—extracellular-matrix (ECM) mediated differentiation. Decellularized ECM and conditioned media from the appropriate committed cell lines are used to differentiate stem cells to the required phenotype. Applying this strategy to differentiate hESCs to pancreatic beta cells, we have obtained functional cells that secreted insulin in a glucose-responsive manner, and were able to recover normoglycemia in a streptozotocin (STZ)-induced diabetic mouse model. ECM-mediated differentiation was also demonstrated to be effective for the differentiation of hESCs into kidney tubule cells and cardiomyocytes. Gene expression studies suggested the involvement of integrins and catenins in the beta cell differentiation process; in particular, α1, αv, and β1 integrins, and β-catenin showed the highest upregulation. To further elucidate the biochemical and mechanical cues that have led to effective hESC differentiation to beta cells, we have employed an artificial system that allowed for variation of matrix stiffness and combination of individual ECM proteins at various ratios. The differentiation response of hESCs to the native ECM could be approximated by optimizing this system.
Introduction
S
The micro-environment is comprised primarily of two components, insoluble extracellular matrix (ECM) and secreted soluble factors. The ECM is a complex mixture of collagenous and noncollagenous proteins, proteoglycans and polysaccharides, the specific composition and distribution of which impart a distinct mechanical strength and topography to the tissue concerned. This provides an additional level of signaling over that afforded by ECM ligand binding to cell surface receptors. Both ECM and soluble factor signaling are known to exert their influence on stem cell differentiation in an interactive manner; however, the exact molecular mechanism by which lineage specification occurs is still unclear. Especially intriguing is the ability of decellularized ECM and conditioned media to direct lineage specification of pluripotent stem cells, as demonstrated by earlier decellularized organ studies.9,10 In formulating this work, we asked two questions. Firstly, would the ECM and other factors secreted by cells grown on tissue culture plates exhibit the same ability to differentiate stem cells? Secondly, could the composition of proteins or other ECM components and mechanical stiffness of the substrate be tuned to reproduce ECM-mediated differentiation? In this work, we attempted to answer the above questions, with a focus on beta cell differentiation. A beta cell line was grown on tissue culture plates and decellularized, after which the derived ECM and conditioned media were used for the culture of embryonic stem cells. By means of gene expression studies and functional assays, we investigated the change in phenotype of the stem cells following the differentiation regime. The insulin-secreting function of the differentiated cells was further explored in a streptozotocin (STZ)-induced diabetic animal model.
In the next step, we characterized the composition of the beta cell ECM with respect to three of its major protein components, collagen IV, fibronectin, and laminin. We then investigated the effect of the ECM protein ratios and stiffness of the culture matrix in differentiating human embryonic stem cells (hESCs) to beta cells, so as to obtain a defined and optimized system.
Materials and Methods
Cell culture
HUES-7 cell line (obtained from Howard Hughes Medical Institute) was cultured as described earlier.9,11 Upon expansion, the cells were used in the differentiation protocol described below. Rat pancreatic beta cell line (RIN5F) was obtained from American Type Culture Collection (ATCC) and cultured as prescribed by ATCC. The medium collected from RIN5F cells (70% confluence) was filtered with 0.2-μm filter and used as conditioned medium. The filtered media can be stored in aliquots at −20°C for long-term storage.
Differentiation of hESCs on RIN5F ECM
RIN5F was cultured in six-well plates at least 5 days before use, to attain about 90% confluency. The cellular fraction was removed by incubating with 0.02 M NH4OH for 5 min. The ECM on the tissue culture plates was rinsed twice with water, and stored in phosphate-buffered saline (PBS) at 4°C. About 5×105 hESCs were seeded on each ECM-coated well (10 cm2) and cultured with conditioned medium (diluted 1:1 with RIN5F growth medium). Differentiation was allowed to proceed for 14 days.
Characterization of ECM
The ECM isolated was characterized by enzyme-linked immunosorbent assay (ELISA) and atomic force microscopy (AFM). Sandwich ELISA was used to identify the ECM proteins. The amount of ECM proteins was determined with the help of a standard graph for each ECM protein. Antibody against collagen IV (ab6586; Abcam), antifibronectin (AB1954; Millipore) and antilaminin (ab14055; Abcam) were used in the study. The mechanical properties of the ECM were determined by the AFM (BioScope; Veeco Instruments, Inc.) at room temperature in PBS with silicon nitride cantilever (force constant=0.32 N/m, resonance frequency=56 kHz). The spring constant was calculated in advance by the thermal tune method. The force curve was conducted at 1-μm scale, and the elastic modulus was calculated by using Hertz model. 12
STZ animal model
All the animal experiments described in this manuscript were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). Diabetes was induced in SCID mice with a single intraperitoneal injection of STZ (freshly prepared in citrate buffer) at 100 μg/kg body weight. Blood glucose was monitored to confirm the diabetes in the mice. Approximately 1×105 differentiated cells in the form of clusters (∼100 μm) were encapsulated in 25 μL of Matrigel (growth factor reduced) and incubated at 37°C for 10 min. The gel construct was implanted into a 2-cm incision made in the skin of the diabetic mice (n=8). A subcutaneous site was chosen for implantation as it is easily accessible and well vascularized. Many researchers have used kidney capsule transplantation; however, we chose subcutaneous transplantation as a less invasive surgical protocol for implantation and eventual excision of the cells. Previous work showed that implantation of pancreas using either protocol did not lead to differences in the regulation of blood glucose level. 13 Blood glucose level was monitored on specified days. The implant was surgically excised on day 91. The study was terminated a day after excision of the implant. The control diabetic animals received either undifferentiated cells (n=5) or no cells (n=5). The controls were sacrificed at day 42 due to sickness.
Blood glucose measurement
Ten microliter of blood was drawn from tail vein of the mice. During the glucose tolerance test (GTT) experiments, blood was drawn every 30 min over a period of 2 h. The blood was analyzed using a domestic glucometer (Accu-Chek Compact Plus; Roche).
Intraperitoneal GTT
Animals were fasted for 16–18 h (overnight). Blood glucose level was measured as described above. Freshly prepared and filtered glucose solution was used at 2 g of glucose per kg of body weight. Glucose solution was injected intraperitoneally, and blood glucose level was measured after 30, 60 and 90 min. In a separate set of experiments, animals with similar blood glucose levels were used for the measurement of c-peptide in the serum during GTT. Animals (n=5) were sacrificed after overnight fasting and after 30 min of glucose injection. Blood from the sacrificed animals was collected, and serum c-peptide level was measured as described below. Nondiabetic animals (n=5) served as the controls.
c-Peptide measurement in serum
c-Peptide measurement was conducted using human c-peptide ELISA kit (Millipore). Briefly, 10-μL aliquots of the collected serum were assayed in triplicates. The assay was performed as described by the manufacturer. The plates were read at 450 nm in a plate reader. A standard curve was established for the conversion of absorbance units to relative c-peptide level.
ECM protein extraction
ECM proteins were collected by scraping, followed by sonication. Presence of various ECM proteins was quantified by ELISA. ELISA kits (Abcam) were used for the quantification of fibronectin (ab 108849) and laminin (ab 119599). Collagen IV was measured by the sandwich ELISA method using collagen IV antibody (ab 6586; Abcam). Standard curves were used to determine the concentration of the ECM proteins.
Hyaluronic acid hydrogel
A commercially available hyaluronic acid (HA) hydrogel system was used in the study. Glycosil™ (thiol-modified HA) and Extralink™ (polyethylene glycol diacrylate) were purchased from Glycosan Biosystems. The stiffness of the hydrogel was manipulated as described by Vanderhooft et al. 14
RNA isolation and two-step reverse transcription polymerase chain reaction
The total RNA isolation and cDNA synthesis were carried out as described earlier. 9 The specific gene of interest was amplified by PCR using Advantage 2 polymerase (BD Biosciences). The PCR products were resolved in a 2% agarose gel containing ethidium bromide. The primers used in the study and their expected product size were given in the Supplementary Table S1 (Supplementary Data; Supplementary Data are available online at www.liebertpub.com/tea). For real-time PCR, total RNA was reverse transcribed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was conducted using gene-specific Taqman assays listed in the Supplementary Table S1.
Immunostaining and flow cytometry
The differentiated cells were cultured to 50% confluency, and processed for immunostaining as described earlier. 9 The mounted cover slips were observed under a laser confocal microscope.
About one million cells were suspended in 1 mL of ice-cold PBS containing 10% fetal bovine serum (FBS). The cells were incubated with fluorescent tagged primary antibodies for 30 min at 4°C. The cells were washed thrice at 400 g for 5 min with ice-cold PBS containing 10% FBS. The pellet was resuspended in 1 mL of ice-cold PBS containing 10% FBS. The sample was analyzed with a LSR II 3-Laser FACS Analyzer (BD Biosciences).
c-Peptide assay for insulin measurement
Glucose-dependent insulin assay was performed using the c-peptide ELISA kit (Alpha Diagnostics, Inc.). The cells were exposed to high glucose concentration (20 mM) for 1 h, and the c-peptide secreted in the medium was analyzed. A c-peptide standard graph was used in the calculation to determine the amount of c-peptide released.
Statistical analysis
All data collected were presented as mean±standard deviation of three samples. The unpaired t-test was used to compare the data sets.
Results
Differentiation of hESCs into insulin-producing cells
We tested the potential of the ECM to induce the differentiation of hESCs. hESCs were seeded onto the acellular ECM, and allowed to differentiate in the presence of conditioned media for 15 days. Genes specifically expressed by the pancreatic beta cells were analyzed by reverse transcription–polymerase chain reaction. The results show that the ECM alone did not have any effect on the gene expression levels at 15 days of culture. Interestingly, however, conditioned media alone triggered the expression of Nkx6.1 (a transcription factor required for beta cell development). More interestingly, cells exposed to both ECM and conditioned media were positive for the expression of key beta cell markers, including PDX1, insulin, and Glut2 (Fig. 1A). Gene expression analysis was done to monitor the progress of differentiation. We observed that the stem cell pluripotent genes, NANOG, and Oct3/4, were downregulated (Fig. 1B), while the expression of PDX1 and insulin increased progressively with differentiation (Fig. 1C). Insulin and PDX1 expression levels were detected as early as Day 3 for cells cultured on RIN5F ECM under conditioned media (Fig. 1C). The gene expression results were further confirmed by immunostaining with specific antibodies. Immunostaining with insulin antibody (Fig. 1D) and Glut2 antibody (Fig. 1E) revealed the presence of these proteins in the differentiated cells. We also observed other features that were characteristic of typical beta cells, such as the classical secretory pattern of insulin, in the differentiated cells (Fig. 1G). When the cells were plated on low attachment plates, we observed the formation of clusters varying from 100 to 200 μm in diameter (Fig. 1H). Functionally, these cells were able to produce insulin in a glucose-responsive manner. We compared the amount of insulin secretion between two-dimensional cultured cells and clusters, and observed that the differentiated cells were more responsive to glucose when they were maintained as clusters (Fig. 1I). By ECM-mediated differentiation, we were able to achieve 38% c-peptide positive cells (Supplementary Fig. S1A). Next, we examined the responsiveness of the differentiated cells toward insulin secretion upon glucose challenge. We observed that the differentiated cells responded to cyclic exposure of low (5 mM) and high (20 mM) glucose concentrations for at least three cycles (Supplementary Fig. S1B). We also observed a saturation of c-peptide secretion after 30 min of exposure to high glucose concentration (20 mM), which remained saturated at later time points (Supplementary Fig. S1C).

In vitro characterization of the differentiated cells.
The approach of using decellularized matrix for hESC differentiation was tested on various other cell lines. We have differentiated hESCs into kidney tubule cells (Supplementary Figs. S2 and S3) and cardiomyocytes (Supplementary Fig. S4), using their respective ECM and conditioned media.
Expression of integrin subunits during differentiation
In the current differentiation methodology, much of the information dictating the differentiation of the stem cells is conveyed from the ECM to the interior of the cell. This outside-in signaling mechanism is mediated by transmembrane proteins, an important class of which is the integrins. 15 We compared the expression level of integrin subunits by cells differentiated using decellularized ECM and conditioned media with those cultured on Matrigel (undifferentiated cells) over a 15-day period (Fig. 2A). The expression of integrin subunit α1 increased progressively for the cells subjected to the differentiation conditions. We noticed a maximum expression (35-folds as compared to undifferentiated cells grown on Matrigel) of α1 subunit at 12–15 days after seeding cells onto the acellular matrix. Similarly, αv expression increased during differentiation, leading to a 35-fold increase (as compared to undifferentiated cells) in the expression level after 12 days. The expression levels of the other alpha subunits, that is, α2, α3, α5, and α6 fluctuated during differentiation, but in general, their expression levels were lowest midway through the differentiation period (Day 3–8), and their final expression levels were higher compared with the undifferentiated cells. We analyzed the expression of three beta subunits (β1, β4 and β5) during the differentiation of hESCs. Expression of β1 increased during the differentiation, and we noticed a seven-fold increase as compared to undifferentiated cells at Day 12. The expression of β4 subunit fluctuated during the differentiation. More interestingly, the expression of β5 subunit decreased during the differentiation period as compared to the undifferentiated cells.
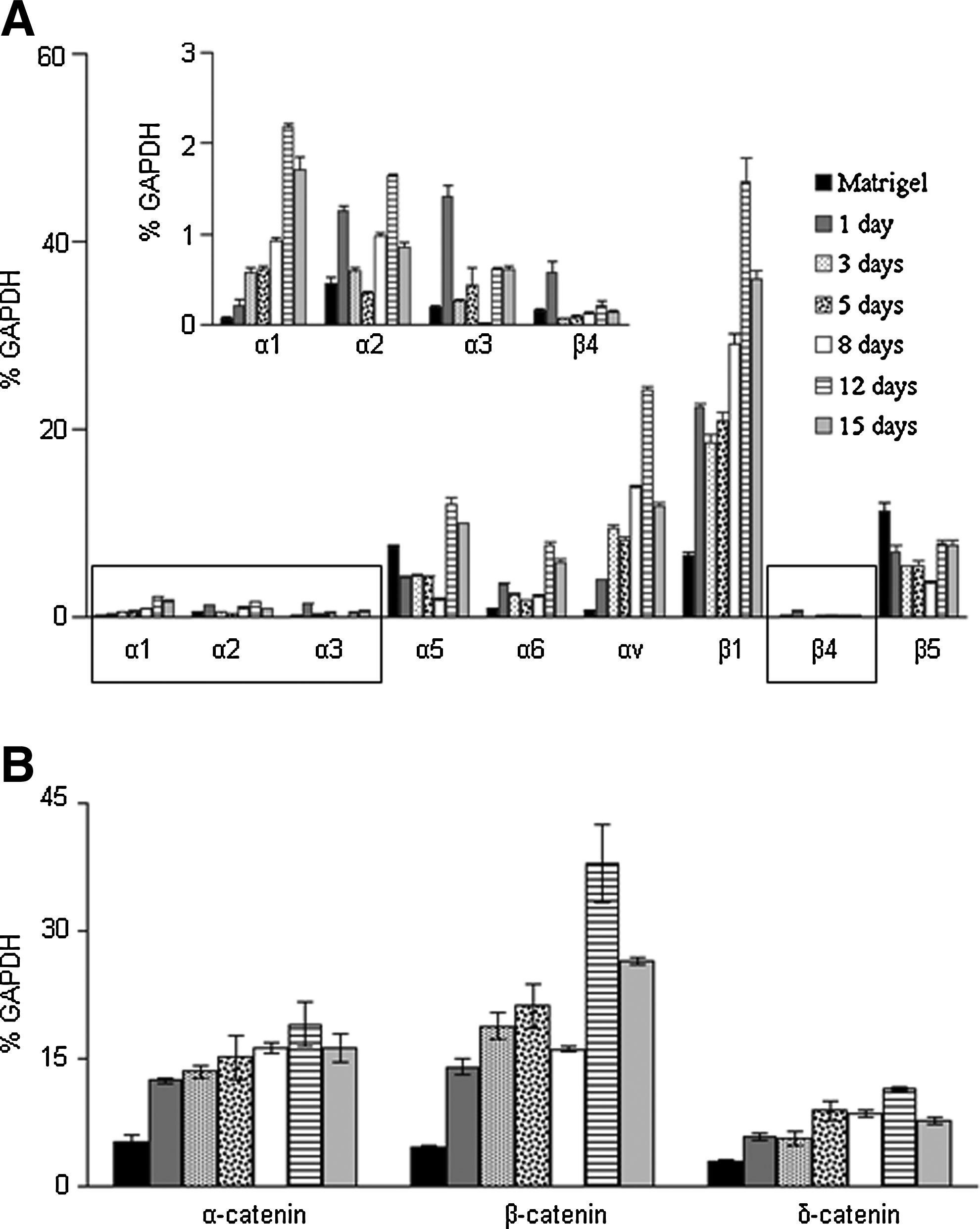
Expression of integrin subunits and beta catenin on the acellular surfaces.
Activation of catenin pathway
β-catenin plays a major role in cell proliferation and endoderm differentiation. 16 These results prompted us to investigate the expression of catenin during the differentiation of stem cells on the acellular substrate. We observed a steady increase in the expression of α-catenin and β-catenin, and to a lesser extent, for δ-catenin. The expression of β-catenin increased up to seven-folds, while α-catenin expression increased up to three-folds during differentiation, as compared to the undifferentiated cells (Fig. 2B).
Recovery of STZ-induced diabetic mice
The functional properties of hESCs differentiated by exposure to RIN5F ECM and conditioned media were studied by implanting the cells subcutaneously in a STZ-induced diabetic mouse model. The mice were confirmed to be diabetic for at least 1 week before implantation, according to the criterion of blood glucose level above 25 mM. Animals implanted with undifferentiated cells failed to recover from diabetes and remained hyperglycemic. On the other hand, animals implanted with differentiated cells exhibited recovery within a week. At the end of the second week, the animals were close to normoglycemic. Starting from the fourth week after implantation, the animals showed similar blood glucose levels as the untreated control mice (between 10 and 12 mM). We observed long-term diabetic recovery in animals for up to 13 weeks. At the end of the 13th week, the implants were excised. The surviving animals showed a higher blood glucose level, similar to that of the diabetic mice. These results suggested that mice implanted with the differentiated cells were able to recover to normoglycemia in a STZ-induced diabetic model (Fig. 3A).
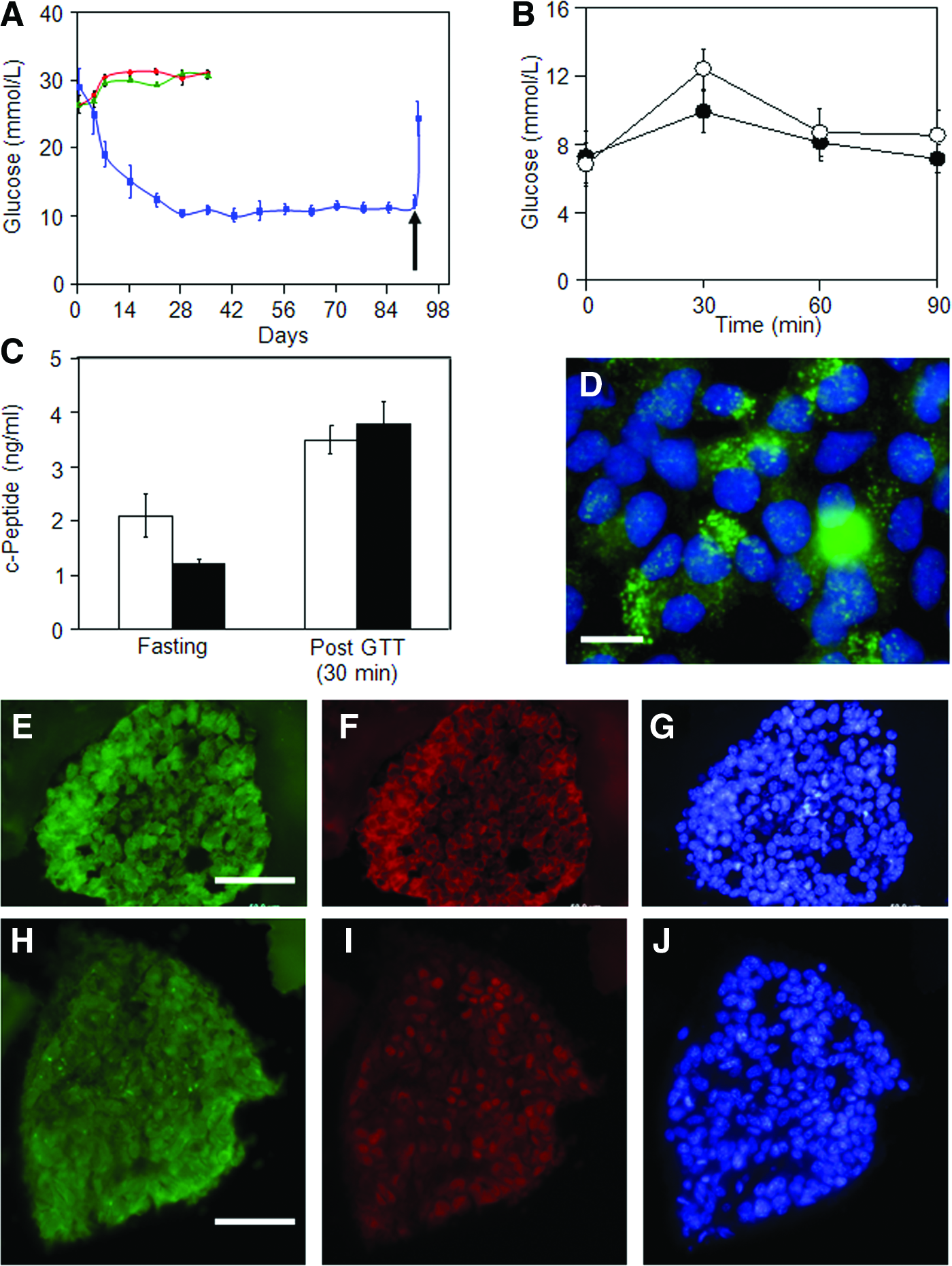
In vivo characterization of the differentiated cells.
We further extended our animal studies to calculate the time required for the animals to respond to high glucose concentrations. The GTT was used to determine the time required to restore normal glycemia in animals. Animals subjected to overnight fasting were injected with a solution of high glucose concentration as described in the Materials and Methods section. Blood glucose levels were measured at a time interval of 30 min. Results indicated that the blood glucose level in implanted diabetic mice reached a maximum at 30 min. Subsequently, the glucose level was brought down to normoglycemia within 60 min (Fig. 3B). This response to high glucose concentration was observed to be similar to that of the control untreated mice (which are normal and nondiabetic). These results indicate that the differentiated cells acted swiftly in restoring normal blood glucose levels. We also measured the c-peptide in the serum before and after GTT. The basal level of c-peptide was noted to be slightly higher in the implanted diabetic mice compared with the untreated control mice. However, the level of c-peptide released in the serum 30 min after GTT for the implanted diabetic mice was similar to that of the untreated control (Fig. 3C). These results further supported that the differentiated cells responded to glucose challenge in a similar way as the beta cells in untreated, normal mice.
Immunostaining of the explants with insulin antibody (Fig. 3D) further indicated that these cells secreted insulin in a pattern similar to the cultured, differentiated cells (Fig. 1G). Immunostaining with additional antibodies against beta cell-specific markers Glut2 (Fig. 3F) and PDX1 (Fig. 3I) was also positive, and substantiated the presence of these markers in the 13-week explants. The sustained expression of these marker proteins suggested that these cells maintained their beta cell phenotype and supported the observed functionality of these cells in diabetic mice.
Effect of compositional ECM proteins and matrix stiffness
We had shown above that the hESCs differentiated in the presence of beta cell ECM and soluble factors could recover normoglycemia in diabetic mice. Next, we studied the physico-chemical aspect of the ECM to elucidate its effect on cell differentiation. ECM of the beta cell (RIN5F) was used as a model. We extracted the ECM proteins and examined them with ELISA. A total of three different extractions were used to study three different ECM proteins. The proteins of interest were collagen IV, laminin, and fibronectin. The amount of each protein present in the ECM was measured. Results of the three independent studies revealed that these proteins exist in a specific ratio. The ratio of collagen IV:fibronectin:laminin was estimated to be 1:3:3 (Table 1).
Collagen IV, fibronectin, and laminin were measured by enzyme-linked immunosorbent assay using specific antibodies against these proteins. Three different preparations were used to measure the level of proteins (Extractions 1, 2 and 3). The measurements were done in triplicate. Standard curves for each protein were used for the quantification.
Next, we performed experiments to show that combination of the ECM proteins could induce the differentiation of hESCs into beta cells. We chose the same three proteins of interest (collagen IV, laminin and fibronectin) and used them in different ratios. We examined their differentiation by real-time PCR for beta cell specific genes, such as insulin, Glut2, and PDX1. We observed the highest expression at a collagen IV:fibronectin:laminin ratio of 1:3:3 (Fig. 4A). Next, we investigated the optimal mechanical strength to induce the differentiation of hESCs into beta cells. A tunable HA hydrogel system was used in the study, the stiffness of which was adjusted by varying the crosslinker. Gene expression analysis clearly indicated that the HA hydrogel with a mechanical stiffness of 2100 Pa was optimal for the differentiation of hESCs into the beta cell phenotype (Fig. 4B). We incorporated collagen IV, fibronectin, and laminin into the hydrogel with a mechanical stiffness of 2100 Pa. Gene expression analysis of these samples revealed that addition of the individual ECM proteins (collagen IV, fibronectin, or laminin) did not improve the differentiation (Fig. 4C). On the other hand, when we incorporated the combination of collagen IV, fibronectin, and laminin proteins at the ratio observed in the RIN5F ECM (collagen:fibronectin:laminin ratio=1:3:3), upregulation of the beta cell gene expression was noted (Fig. 4D). The expression level was comparable to that achieved by differentiating hESCs on RIN5F ECM. These results suggested that differentiation of the hESCs was dictated by the compositional ECM proteins and the mechanical stiffness of the matrix.

Gene expression analysis on different substrates. Pancreatic beta cell specific marker genes, insulin (black bars), Glut2 (white bars) and PDX1 (grey bars) were analyzed.
Discussion
Existing protocols of stem cell differentiation rely on mimicking the sequence of signaling events that underlies the differentiation of tissues in embryological development. 17 Apart from specific soluble growth factors, the role of ECM on the differentiation of stem cells is largely ignored. In this paper, we presented an alternative method of differentiating stem cells, ECM-mediated differentiation, which was based on the in vivo tissue repair mechanism. Under in vivo conditions, stem cells are recruited to the damaged site for repair, whereby they undergo differentiation into the cell or tissue concerned. In contrast to conventional methods, ECM-mediated differentiation makes use of both the chemical composition of the ECM itself, as well as the resulting mechanical properties of such a composition. Supplementary Figure S5 shows the protein microarray of ECM obtained by decellularizing the beta cell line, RIN5F, giving an idea of the chemical complexity inherent in the ECM secreted by committed cell types. Table 2 compares the elastic modulus of ECM from a variety of cell types to that of tissue culture plate. The tissue culture plate surface exhibited an elastic modulus 1.5 to 2 times higher than the cell-secreted ECM, and amongst ECM from the different cell types, a clear variation was observed. In addition, different topographies are also exhibited by the different surfaces (Supplementary Fig. S6). Thus, mechanical signaling acts as an important component of ECM-mediated differentiation.
An area of 1×1 μm was scanned to deduce the force curve using the Hertz model. The spring constant was measured in advance in phosphate-buffered saline to be 0.32 N/m.
HPTCs, human proximal tubule cells; MEFs, mouse embryonic fibroblasts; TCP, tissue culture polystyrene.
In the present paper, we have established ECM-mediated differentiation as a method for differentiating cells, to obtain a high population of functional cells. By culturing embryonic stem cells on surfaces obtained by decellularizing confluent monolayers of different committed cell types in the presence of conditioned media from the same cells, the stem cells were shown to gradually acquire the phenotypic markers and function of the committed cell types concerned (Fig. 1, Supplementary Figs. S2 and S4). This was illustrated by performing a detailed study on insulin-secreting pancreatic beta cells. Our method of differentiation afforded up to 38% of insulin-positive cells, comparable to the recently reported directed differentiation methodology. 17 Gene expression studies and immunostaining of the differentiated cells indicated the presence of beta cell specific markers, such as PDX1, Glut2, and insulin. Furthermore, insulin antibody staining revealed a typical granular secretory pattern of packaged insulin particles, as reported earlier. 18 Subcutaneous implantation of these cells in a diabetic mouse model restored normoglycemia in a long-term study. Intraperitoneal GTT results were similar to the previously reported results.19,20 It was noted that teratomas formed after week 10 in the diabetic animals implanted with differentiated cells, indicating the presence of residual pluripotent cells. We are currently exploring methodologies to isolate the insulin-secreting beta cells from the undifferentiated pluripotent cells.
The current results allow us to postulate that insoluble components of the ECM plus secreted soluble factors present in the conditioned media provide the combined signaling that directs differentiation of stem cells into the cell lineage and phenotype concerned. We speculate that the ECM plays a role in determining the final effect of the soluble factors, via spatially and temporally controlled engagement of integrin receptors and other cell adhesion molecules.1,21–24 The increased expressions of α1, αv, and β1 subunits during differentiation correlated with earlier observations. For example, in human fetal beta cells, integrins αvβ1, and α1β1 enabled attachment, motility and insulin secretion on vitronectin and collagen IV matrices, respectively.25,26 These previous results signify that αvβ1 and α1β1 are important in the development of pancreatic beta cells, and it is likely that the same receptors are involved in ECM-mediated differentiation of stem cells to beta cells in the present work. Similarly, Saleem et al. have demonstrated that the β1 subunit is required for the complete differentiation and survival of human fetal islet cells. These authors have shown that signaling via β1 subunit to focal adhesion kinase is required for the activation of ERK pathway leading to PDX1 activation. 27 In our work, collagen IV, fibronectin, and laminin were identified to be major components of the acellular ECM. All three ECM proteins are ligands for the integrin heterodimer receptor α3β1 28 ; fibronectin is a well-known ligand for α5β1, and laminin is preferred by α6β1.29,30 The present results strongly suggest an important role for integrin-binding (in particular, binding that involves the β1 subunit) in the ECM-mediated differentiation of embryonic stem cells to insulin-secreting beta cells.
Besides integrins, β-catenin plays a major role in cell proliferation, determination of cell fate and endoderm differentiation. 31 Deletion of the β-catenin gene results in a morphological change in the endodermal cells, and biases the cell fate toward heart mesoderm. 32 It is also noted that the WNT signaling pathway, which controls β-catenin, plays a critical role in the differentiation of embryonic stem cells into foregut endoderm. 33 The expression of β-catenin was reported to be inversely related to the Oct4 expression. 34 In our current study, we observed an increase in β-catenin expression during the differentiation of hESCs to beta cells.
Apart from macromolecular signaling, ECM also provides unique mechanical stiffness and topography that act as additional signals for cellular differentiation. Many reports have emerged in the past few years on the role of ECM mechanical properties in stem cell differentiation.35,36 Tuning the mechanical stiffness of a variety of materials has led to the differentiation of stem cells into various phenotypes.37–40 In the second part of our work, we investigated if some of the above-mentioned components of ECM-mediated differentiation could be “resynthesized” to approximate the physico-chemical signaling of decellularized ECM. We endeavored to combine substrate matrix stiffness with ECM composition (by varying the ratios of collagen IV, fibronectin, and laminin) in an artificial system to stimulate hESC differentiation. We analyzed the expression of three beta cell-specific genes, namely insulin, Glut2, and PDX1 in the artificial system, and compared these to expression levels for cells obtained by RIN5F ECM-mediated differentiation of hESCs. The expression of insulin in the artificial system was almost similar to that in the native ECM-mediated differentiation, while the other genes were expressed at lower levels. These results emphasized the role of the ECM in differentiation–we believe that soluble factors in the conditioned media prime the cell response and trigger the initial signaling cascade, while ECM signaling is critical in directing the exact response. The combinatorial effect of these three components directs differentiation of the stem cells specifically towards the beta cell phenotype, rather than other pancreatic cell phenotypes. Our future research would focus on creating a defined system for differentiation, by closely mimicking the physicochemical properties of native ECM and the biochemical composition of the conditioned media.
In conclusion, this paper introduces ECM-mediated differentiation as a simple and effective method for directed differentiation of stem cells into committed cells with functional properties similar to the ECM-providing cells. How exactly the stem cell processes the signals from the ECM, and whether the various stages of embryonic development are recapitulated in the process of ECM-mediated differentiation would be the subject of future investigations. The methodology and understanding gained from this work would impact further efforts in developing therapies for tissue regeneration, in particular, for augmenting the source of differentiated cells in cell-based therapy.
Footnotes
Acknowledgments
This work is funded by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.