
Abstract
Functional activation of stem cells after transplantation is a main concern in stem cell therapy. For local transplantation, mesenchymal stem cells (MSCs) are usually administered via scaffolds, either by direct implantation or after preculturing of cells, and it is unclear which is better for the activation of transplanted cells. In this study, we investigated the in vivo gene expression activity of human MSCs (hMSCs) transplanted into calvarial defects either directly post-seeding on collagen sponges (Group 1) or after overnight in vitro culturing post-seeding (Group 2). Real-time reverse transcription–polymerase chain reaction at days 7 and 14 after transplantation identified a time-dependent, rapid decrease in gene expression by the hMSCs, which in Group 1 was slightly more attenuated than in Group 2. Both groups exhibited a limited range of human-specific gene expression, which involved type I collagen (ColI), fibronectin, stromal cell-derived factor (SDF-1), and osteoprotegerin. Among these, ColI expression was the most efficient, with higher levels in Group 1 than Group 2. There was a lack of evidence for the expression of osteoblast differentiation-related markers or trophic factors, while resident cells showed clear expression of those genes. Rat-specific β-actin expression in Group 2 was least among the scaffold control, Group 1, and Group 2, and this pattern was repeated in the expression of other rat osteogenic genes. Group 1 transplants positively influenced the osteogenic process of the defect tissue in part, and rat IGF-1 expression was significantly increased in Group 1. This tendency of gene expression by hMSCs in a rat model was very similar to what was observed in transplantations using immunodeficient mice. The current study showed that a main gene expressed by transplanted hMSCs during the initial weeks following transplantation is ColI, with a lack of differentiation-related markers or growth factor expression by hMSCs. Our data suggest that direct transplantation of hMSCs loaded on a collagen sponge is more efficient for gene activation in transplanted hMSCs, and more favorable to the local host tissue than transplantation after preculturing of cells.
Introduction
H
Delivery of MSCs to treat generalized skeletal disease is accomplished by systematic administration or with the aid of scaffolds. 17 For regeneration of bone defects, tissue engineering studies recommend combining cells with the appropriate scaffolds and osteogenic signals to stimulate bone repair. 4 Scaffold or osteoconductive bone substitutes are critical for increasing survival rates and the differentiation potential of the cells, leading to effective acceleration of the osseous regeneration of bone defects.5,18 It is possible for scaffolds to be designed to encourage the ingrowth of marrow stromal elements and to repopulate the entire construct with osteoprogenitor cells or stem cells derived from surrounding tissues. Because bone regeneration requires a long time period, in cases of extremely large (critical size) defects, additional biocomponents that increase regeneration or improve structure are preferable, such as MSCs, growth factors, or a combination of both using suitable biomaterials. MSCs can be extensively expanded in vitro to obtain sufficient numbers, making them very attractive to researchers. 19 While each scaffold has unique advantages for bone tissue engineering, three-dimensional scaffolds that contain ceramics (usually hydroxyapatite/tricalcium phosphate) as part of their formulation appear to be the most reliable with respect to the formation of bone and support of hematopoiesis when seeded with MSCs.4,20 Incorporation of growth factors with MSCs is used to stimulate transplanted cell activity and differentiation, as well as to recruit undifferentiated osteoprogenitor cells into the carrier. Numerous studies have shown that codelivery of growth factors and MSCs both in vitro and in vivo enables regenerative potential more efficiently than MSCs alone.6,21,22 When cotransplanted with MSCs and growth factors, a collagen sponge is preferred. This is especially the case when BMP-2 is used as a growth factor; collagen sponges have characteristics that allow for sustained release of BMP-2 in addition to their biocompatible, osteoconductive properties. 23
In stem-cell-based tissue engineering, animal studies that investigate hMSCs in xenogeneic settings suggest that ex-vivo-expanded hMSCs improve tissue regeneration and wound healing after in vivo transplantation into animals without notable immunological rejection.6,7,24 These studies, which target local bone tissue, utilized a variety of nonstandardized strategies, including a post-treatment process where hMSCs were seeded on biomaterials followed by either direct implantation or preculturing in vitro until transplantation. It is expected that preculturing of MSCs on a scaffold before transplantation might be beneficial for increasing MSC potential, as well as enhancing viability, after transplantation in vivo. However, it is uncertain whether inclusion of this step results in a better or worse outcome than direct implantation, as the use of both methods has resulted in successful outcomes.5,20
The major purposes of this study were threefold: (a) to compare in vivo gene expression of transplanted hMSCs that were seeded on scaffold and were implanted directly (Group 1) with that of transplanted hMSCs that were seeded on scaffold and cultured in vitro overnight prior to implantation (Group 2), (b) to compare gene activation in the defect tissue of host rat cells in either of Groups 1 or 2 with that of the nontransplanted vehicle control group, (c) to demonstrate whether there is an effect of immune rejection on the differentiation of hMSCs into the osteoblast lineage in xenogeneic animals by comparing gene expression between immunodeficient Balb/c nude mice after direct implantation of hMSCs, as done in Group 1, and the immune-competent host in Group 1. Gene activation of transplanted hMSCs or host cells in vivo was determined by the expression of osteogenesis-related genes using real-time reverse transcription–polymerase chain reaction (RT-PCR) with human- or rat/mouse-specific primers, respectively, at 7 or 14 days after surgery.
Materials and Methods
Isolation and culture of hMSCs
Primary hMSCs from two different sources were prepared. hMSCs#1 were isolated from the bone marrow of a healthy donor (man, aged 22 years) after receiving consent, and the study was approved by the local ethics committee (IRB of Seoul National University Dental Hospital, Nr:CRI05008) according to the legal regulations for human tissue and organs in Korea. The marrow suspension was collected in a syringe containing 6000 U/mL heparin and was mixed with phosphate-buffered saline (PBS) solution in a 1:1 volume ratio and centrifuged at 2500 rpm for 10 min. After aspiration of the upper PBS layer, the marrow suspension was layered on Ficoll-paque (Amersham Biosciences, Uppsala, Sweden) in a 1:5 ratio and centrifuged at 1200 g for 30 min. 25 Nucleated cells concentrated at the interface were collected and washed with PBS. Adherent cells were plated at a density of 2×106 cells/100-mm plate and cultured in expansion medium containing low-glucose Dulbecco's modified Eagle's medium, 100 U/mL of penicillin, 100 mg/mL of streptomycin, and 10% heat-inactivated fetal bovine serum (FBS) under a humidified atmosphere of 5% CO2 at 37°C. The medium was changed every 3 or 4 days. Cells were passaged when they reached 70% confluence. Third-to-sixth-passage hMSCs#1 were used for all experiments described in this study and were verified to express MSC surface markers; over 90% of cells were positive for CD105 and CD90, and <10% were positive for CD45. hMSCs#1 were also confirmed to be able to differentiate into adipogenic, chondrogenic, and osteogenic lineages (shown in Supplementary Data; Supplementary Data are available online at www.liebertpub.com/tea). hMSCs#2, a commercially available hMSC line (PT-2501; Lonza, Walkersville, MD) from the bone marrow of a man aged 36 years, were cultured in the specific medium provided according to the manufacturer's instructions.
hMSC transplantation into a calvarial defect model
A total of fifty-four 8-week-old Sprague–Dawley rats were used in this study. hMSCs#1 or #2 (1×106 cells/defect) were seeded on collagen sponges 2 mm in thickness and 8 mm in diameter prepared from the cross-reaction of chondroitin-6-sulfate (Sigma-Aldrich, St. Louis, MO) and type I collagen (ColI; Bioland Co., Osong, Korea) as previously described. 26 Acellular vehicle controls were implanted with the collagen sponge alone and were sacrificed on days 7 or 14 (each n=7) after surgery for RNA extraction. Experimental groups transplanted with hMSCs#1 or #2 (n=20, each) were divided into two groups according to whether or not cells were precultured overnight prior to transplantation after seeding on the collagen sponge. Cells were either administered into the defect immediately after seeding onto the collagen sponge (Group 1) or after post-seeding overnight incubation in culture medium (Group 2). After disinfection of the calvarial skin with 10% betadine (Potadines; Sam-Il Pharm, Seoul, Korea) and subcutaneous injection of 2% lidocaine containing 1:100,000 epinephrine (Lidocaine HCL Injs.; Yuhan, Seoul, Korea), an incision was made along the sagittal suture. The periosteum was elevated, and an 8-mm diameter, critical-sized calvarial bone defect was created with a trephine burr without dural perforation. After implantation of the collagen sponge with or without cells, suturing was done using 4-0 absorbable nylon (Ailee CO., Ltd., Busan, Korea). Post-operatively, all animals received antibiotics with intramuscular injection of Cefazolin (55 mg/kg; ChongKunDang Pharm., Seoul, Korea) for 3 days. The same strategy as used for Group 1 with hMSCs#1 was performed in immunodeficient Balb/c nude mice (n=3) to investigate gene expression 7 days after surgery.
Verification of hMSC amounts before transplantation
hMSCs#1 were inoculated on a collagen sponge at the same density as transplanted cells, with 4×105 cells in 20 μL of HIFBS on a 5-mm-diameter sponge. Specimens for Group 1 (n=3) were prepared by transferring collagen disks into a fresh plate within 2 h after cell loading (2 h is the mean time needed for surgery), and those for Group 2 (n=3) were prepared by transferring collagen disks into a fresh plate after overnight cell culture. The cell amounts were assessed using a Cell Counting Kit-8 (Dojindo Laboratories, Tokyo, Japan), which employs the tetrazolium salt WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] according to the manufacturer's protocol. The amount of yellow-colored product was measured at a wavelength of 450 nm using a microplate reader and is directly proportional to the number of viable cells in a culture media. The optical density (OD) values of samples represent an index of cell amounts.
DNA content assay
The sponge constructs that were used for verification of cell amounts were reused for DNA content assay after determination of OD values. For the DNA content assay, the scaffolds with cells were washed with PBS solution and chopped into small pieces. The chopped sponge was homogenized in lysis buffer, and the lysate was assayed for DNA content using a QIAamp DNA mini kit (QIAGEN GmbH, Hilden, Germany), and subsequently treated as prescribed in the manufacturer's instructions. DNA quantity and quality were measured by UV/VIS spectrophotometry (NanoDrop™ 1000; Thermo Fisher Scientific, Wilmington, DE). DNA quality was evaluated by the ratios of A260/A280 and A260/A230 (absorption values at the different wavelengths).
RT-PCR/real-time RT-PCR
For RNA extraction, implanted collagen sponges loaded with hMSCs#1 or #2 were removed and washed with PBS solution and chopped into small pieces. After adding 0.5 mL of TRIzol reagent (Invitrogen, Life Technologies, Carlsbad, CA) directly to the chopped sponge, total RNA was extracted and subsequently treated as prescribed in the manufacturer's instructions. One microgram of RNA from each sample was subjected to cDNA synthesis using SuperScript™ Reverse Transcriptase II (Invitrogen) and oligo (dT)12–18 primer (Invitrogen) in a 20-μL reaction volume according to the manufacturer's instructions, and RNA complementary to cDNA was removed using Escherichia coli RNase H (Invitrogen). A total of 1 μL of cDNA was subjected to PCR using the following amplification profile: predenaturation at 94°C for 40 s, amplification (denaturation at 94°C for 40 s; annealing at 60°C for 40 s; extension at 72°C for 1 min) for 30 cycles, followed by a final extension step at 72°C for 10 min. PCR was performed in a DNA thermal cycler (model PTC-200; MJ Research, Inc., Waltham, MA). Electrophoresis was done using 10 μL of each PCR mixture on a 1.5% agarose gel in the presence of ethidium bromide, and bands were visualized using a Gel Documentation System.
Real-time RT-PCRs were performed using SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. Briefly, real-time RT-PCR was carried out in triplicate in three independent experiments (n=3). The human- or rat-specific primers used in our study were designed using Real-Time PCR System Sequence Detection Software v1.3 (Applied Biosystems), and their sequences are provided in Table 1. For positive controls, cDNA was prepared from in-vitro-cultured hMSCs#1 or #2. Fold differences in the levels of each gene were calculated for each treatment group using normalized CT values of the housekeeping gene 18S rRNA according to the Applied Biosystem instructions. The human- and rat-specific primers used for real-time RT-PCR are shown in Table 1 (Supplementary Table S1 lists human-, rat-, and mouse-specific primers for RT-PCR.).
Primers are designed according to human (h) or rat (r) sequence of each gene.
ALP, alkaline phosphatase; ColI, type I collagen; FN, fibronectin; IGF-1, insulin-like growth factor-1; OPG, osteoprotegerin; SDF, stromal cell-derived factor; VEGF, vascular endothelial growth factor; 18S rRNA, 18S ribosomal RNA.
Tracking transplanted hMSCs in vivo
hMSCs#1 were labeled with the fluorescent dye diakylcarbocyanines (Dil; Molecular Probes, Inc., Eugene, OR) by incubating cells in a Dil-dye-containing medium at a final concentration of 10 ng/mL for 24 h. Dil-labeled hMSCs#1 were seeded on collagen sponges and implanted into the calvarial defect. Specimens were prepared from collagen sponges implanted into the calvarial defect after sacrifice at 7 or 14 days post-surgery. The half-cut specimens were decalcified by incubation in a solution of EDTA (7%, pH 7.0) for 10 days, and the solution was changed every 2 days. Specimens were dehydrated in 70% ethanol, sectioned longitudinally along the axial plane, and embedded in paraffin by positioning the center portion of bone chip up. After the decalcified paraffin sections were cleaned for 10 min with xylene, the deparaffinized sections were washed three times with PBS, fixed in 4% paraformaldehyde for 30 min, and blocked in an undiluted serum solution for 30 min. Slides were incubated with proliferating cell nuclear antigen (PCNA) antibody (anti-rabbit, mouse polyclonal, 1:100; Dako Cytomation, Carpinteria, CA) for 1 h at room temperature, washed twice with PBS, and then incubated with secondary antibody labeled with fluorescein isothiocyanate (FITC; anti-rabbit immunoglobulin G [IgG], 1:400; Biomeda Corp., Foster City, CA) for 40 min. Primary antibody controls were processed in parallel using only the secondary antibody. Cover slips were mounted in crystal mounting solution (Biomeda Corp.) and observed under a confocal laser-scanning microscope (Carl Zeiss LSM7000, Oberkohen, Germany).
Fluorescence-activated cell sorting for immune typing
A total of 2×105 hMSCs#1 were cultured with or without interferon (IFN)-γ (100 IU/mL; PeproTech, Rocky Hill, NJ) for 3 days. IFN-γ-treated or nontreated hMSCs were incubated with FITC-conjugated mouse anti-human major histocompatibility (MHC) class I (HLA-ABC; BD Biosciences, San Jose, CA), anti-human MHC class II (HLA-DR; BD Biosciences), or anti-mouse IgG isotype control for 30 or 90 min in the dark after being washed with a washing buffer (PBS buffer containing 1% bovine serum albumin). Cells were washed twice with the washing buffer and suspended in washing buffer containing 0.1% paraformaldehyde (Sigma-Aldrich) for fluorescence-activated cell sorting (FACS) analysis. Subsequently, the cells were fixed in 70% alcohol and sorted using the BD FACS Aria Cell Sorting System (BD Biosciences).
Histological and immunohistochemical staining
The host immune response was observed at 7 days after implantation. Immunohistochemical (IHC) staining for CD8 enabled visualization of T cells because of cell-specific expression of these surface markers. The presence of macrophages was determined by evaluation of conventional hematoxylin-eosin (H&E) stains and macrophage IHC staining. hMSCs were detected with anti-human-specific Nucleolin antibody. The collagen disks were removed and decalcified through incubation in ethylenediaminetetraacetic acid solution (7%, pH 7.0) for 3–4 days (with a solution change on day 2). The specimens were then dehydrated in 70% ethanol and embedded in paraffin. Decalcified paraffin sections were cleaned for 10 min with xylene and stained with H&E, or, for IHC staining; the xylene-cleaned sections were treated with an undiluted serum solution for 30 min. Specimens were then incubated with anti-macrophage (1:100; Serotec, Oxford, United Kingdom), anti-CD8 (1:100; Serotec), or anti-human-specific Nucleolin (1:100; R&D Systems, Minneapolis, MN) antibodies at 4°C overnight. After incubation, the sections were incubated with R.T.U biotinylated universal antibody using a Vectastatin kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Staining was detected with diaminobenzidine (DAB; Sigma-Aldrich) substrate, and slides were mounted with Crystal/Mount (Biomeda Corp.). Images of stained cells were captured by bright-field microscopy.
Enzyme-linked immunosorbent assay of TGF-β and IL-10
hMSCs#1 were treated with IFN-γ (100 IU/mL) for 3 days and inactivated with 25 mg/mL of mitomycin C. Approximately 104 cells were cultured in a 96-well plate and IL-10 and TGF-β detection was performed after 3 and 6 days. TGF-β/IL-10 levels in the culture supernatants were determined using enzyme-linked immunosorbent assay (ELISA) kits (Quantikine®; R&D Systems). Standards for cytokines (0–2000 pg/mL) were run in each series. After incubation, aspiration, and washing, either human TGF-β conjugate (50 mL/well) or human IL-10 conjugate (100 mL/well) was added based on the manufacturer's instructions. The OD of each well was determined within 30 min using a microplate reader set to a 450-nm wavelength with correction for optical imperfections in the plate. ELISAs were repeated in triplicate for three or four independent samples (n=3–4).
Statistical analysis
All data are presented as means±standard errors of the mean or standard deviation. Statistical analyses were performed using IBM SPSS statistics 20.0 software (IBM, Armonk, NY). Data between two groups were evaluated using a two-tailed Student's t-test, and the comparison of data in more than two groups was done using one way ANOVA with post hoc using Tukey method. Differences with p<0.05 were considered significant.
Results
In vivo viability of hMSCs after transplantation into rat
The initial cell densities in the two groups were verified in vitro by cell proliferation assays and by quantifying DNA amounts (Fig. 1). Specimens were prepared as described in the “Materials and Methods” section. Cell proliferation assays showed that there was no difference between two groups, even though the hMSCs in Group 2 were implanted following in vitro overnight preculturing on the collagen sponge. The same tendency was obtained from the measurements of DNA using the same disks used for the cell proliferation assay.
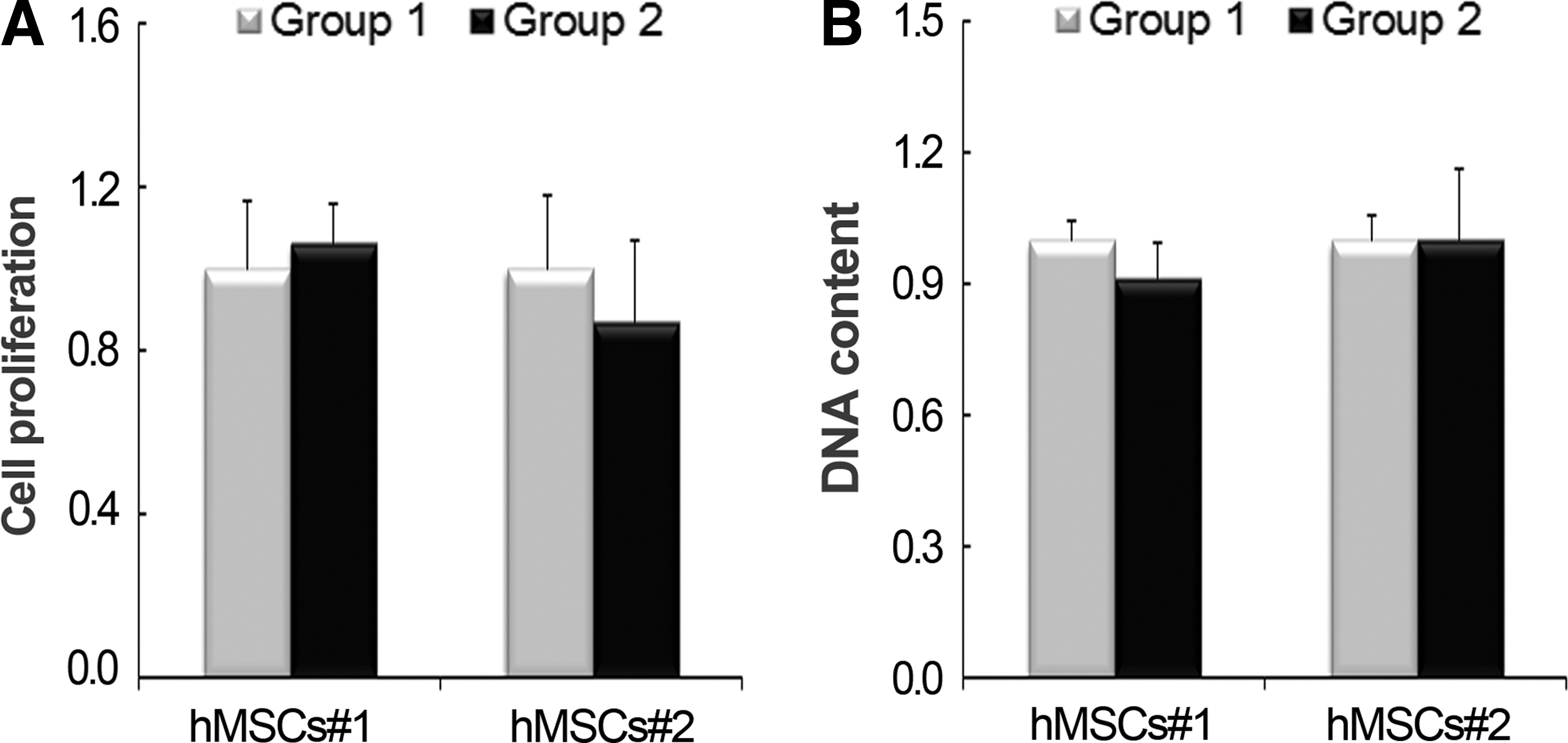
Verification of cell amounts in Group 1 and Group 2 before transplantation. Experimental groups transplanted with human mesenchymal stem cells (hMSCs)#1 or #2 were divided into two groups according to whether cells were directly administered (Group 1) or precultured overnight prior to transplantation after seeding on the collagen sponge (Group 2). To verify initial cell amounts before transplantation, hMSCs#1 and #2 each were inoculated on a collagen sponge at the same density as transplanted cells. Specimens for Group 1 (n=3) were prepared by transferring collagen disks into a fresh plate within 2 h after cell loading, and those for Group 2 (n=3) were prepared by transferring collagen disks into a fresh plate after overnight cell culture.
We determined the in vivo viability of primary hMSCs#1 at 7 and 14 days after transplantation by evaluating the expression of the housekeeping gene GAPDH (Fig. 2). The primer for human GAPDH was designed to be specific to the human gene, but there was minimal expression in the vehicle control implanted with collagen sponge alone due to partial similarity of nucleotide sequence between the rat and human genes. A positive control for human gene expression was used to confirm the expected size corresponding to the transcripts for most of the analyzed genes using cultured hMSCs#1 in vitro. Gene expression was investigated at the same two time points, 7 and 14 days after transplantation of hMSCs#1 in Group 1, with normalization to expression of the housekeeping gene 18S rRNA using primers common to both rat and human sequences. After transplantation, the GAPDH expression level of hMSCs#1 in Group 1 animals at 7 days was decreased to approximately one-sixth of in-vitro-cultured hMSCs#1, which corresponded to the same number of transplanted cells measured by GAPDH expression (Fig. 2A). In Group 1 animals at 14 days after transplantation, human GAPDH expression drastically declined to 1.66% of that at seen 7 days. Notably, GAPDH expression in the same cell line declined even more rapidly in transplants pretreated with overnight culturing (Group 2) (Fig. 2B). The hMSCs#1 in Group 2 showed GAPDH expression of 24.68% of the expression seen in Group 1 cells at the same time point of 7 days without preculturing, and declined further to 0.12% of the 7-day Group 1 level at 14 days after transplantation.

Human GAPDH expression by transplanted hMSCs. Collagen sponges implanted into rat calvaria defects were removed at 7 or 14 days after surgery. hMSCs were delivered either immediately post-seeding on collagen sponges (Group 1) or after preculturing in culture medium in vitro post-seeding (Group 2).
We repeated the same strategy used for hMSCs#1 with a commercially obtained cell line, termed hMSCs#2 for this study. As with hMSCs#1, in vivo GAPDH expression from transplanted hMSCs#2 declined over time, with better efficiency seen in Group 1. The GAPDH expression level of transplanted hMSCs#2 on day 7 was about one-sixth that of in-vitro-cultured cells (Fig. 2C). For Group 1 animals treated with hMSCs#2 at 14 days, human GAPDH expression drastically declined to 22.80% of that at 7 days, but this decrease was attenuated compared with that seen in hMSCs#1 (Fig. 2D). However, expression of human GAPDH in Group 2 animals treated with hMSCs#2 declined remarkably after pretreatment using the overnight culturing process; only 6.83% of the Group 1 value was seen at 7 days, and at 14 days only 0.65% of the 7-day Group 1 value was detected.
To identify hMSCs after in vivo transplantation, Dil-labeled hMSCs#1 were traced after 7- and 14-day healing periods in Group 1 (Fig. 3). A considerable portion of Dil-labeled hMSCs (red) were identified at 7 and even at 14 days after transplantation. The number of positively stained cells did not decrease over time, although only a small number of sections within the defect were evaluated. In Group 1, Dil-labeled hMSCs had an indistinct shape and were distributed mostly in the interspaces of the fibers. A small portion of cells were seen along the collagen fiber. However, hMSCs cultivated in vitro on collagen sponges showed sharp cell outlines and were found mostly along the collagen fiber. The PCNA counterstain allowed transplanted donor cells to be distinguished from host tissue cells. PCNA-stained cells (green) indicate actively proliferating cells, including both rat cells infiltrated into the defect region and the Dil-labeled hMSCs. Under visual inspection, the space without hMSCs was filled with rat cells, which seemed to be distributed more abundantly than hMSCs in the defect area. Merged images of Dil-labeled hMSCs with PCNA showed that a portion of hMSCs were still proliferating. It was unclear whether the proliferating hMSCs were present as undifferentiated stromal cells, osteoblasts, or some other kind of cell.
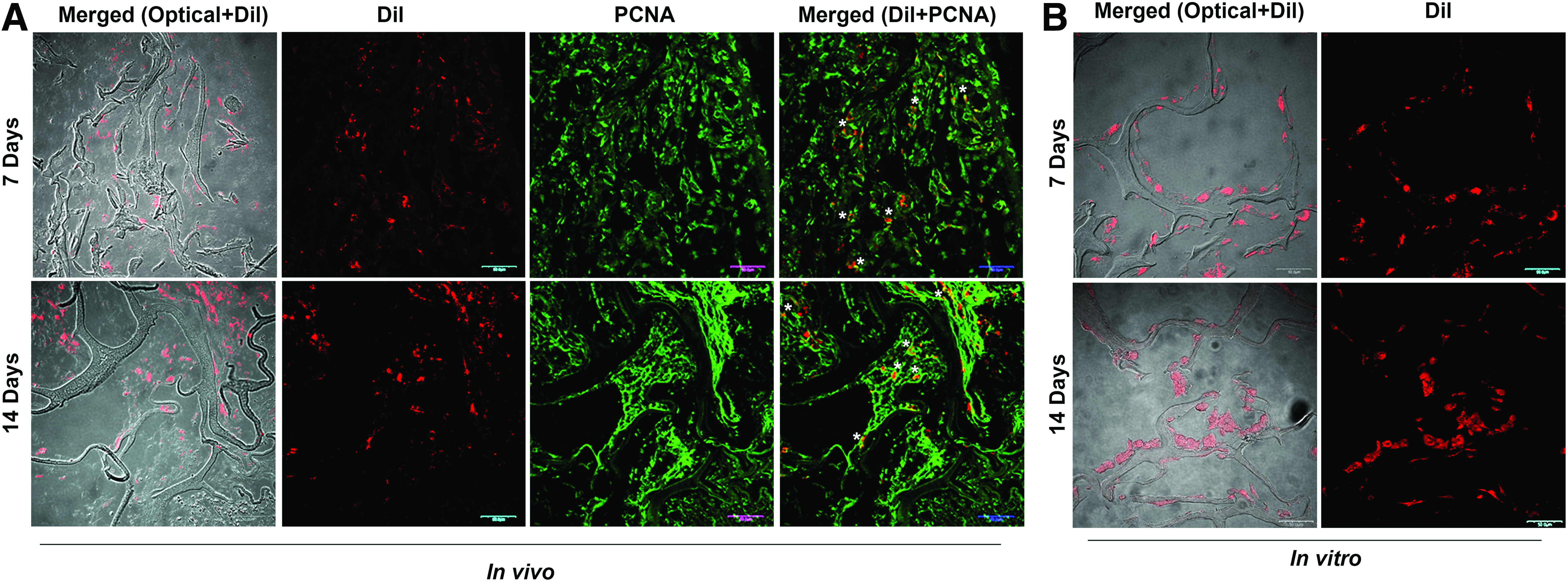
Tracking transplanted hMSCs in vivo in comparison with cells on collagen sponge in vitro.
Human-specific gene expression by transplanted hMSCs
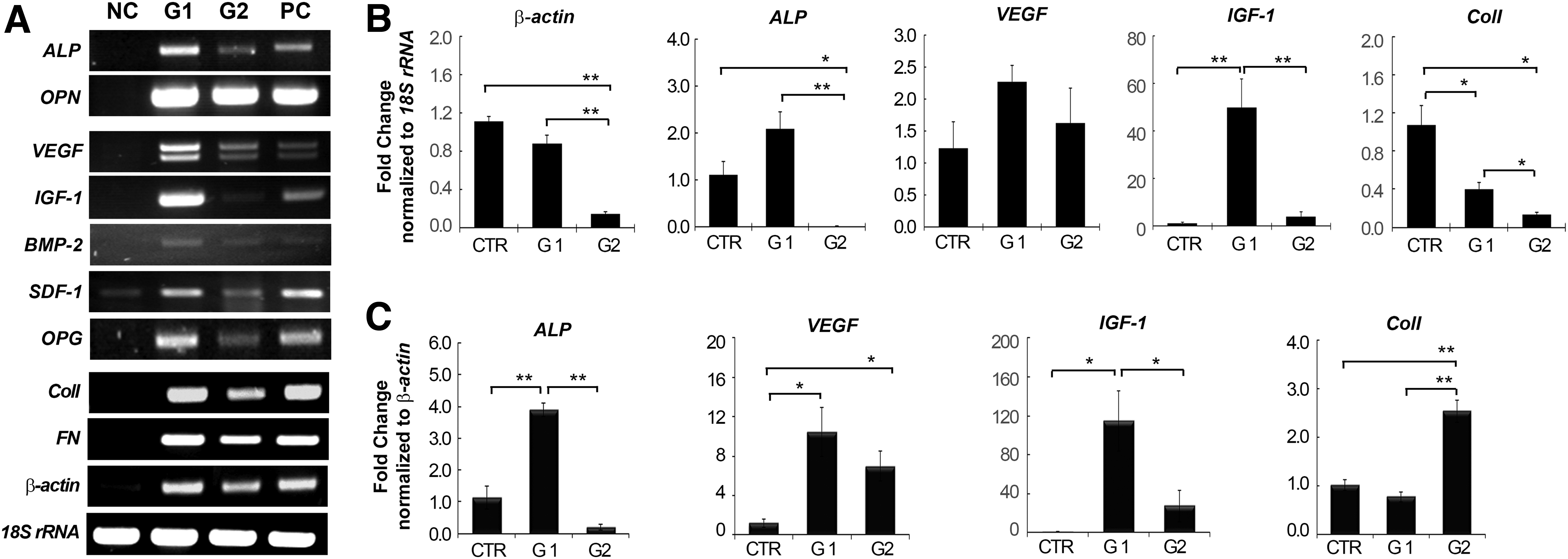
We further investigated the expression of genes related to osteogenesis in transplanted hMSCs#1 (Fig. 4). Because human-specific GAPDH expression rapidly decreased as time passed, with its lowest level at 14 days after transplantation, the expression of osteogenic genes in hMSCs#1 was compared between Group 1 and Group 2 at 7 days using RT-PCR and real-time RT-PCR. The negative control was PCR performed without the cDNA mixture. Representative RT-PCR images of hMSCs#1 showed that the pattern of human GAPDH expression was consistent with the amplification level of fragment containing the human-specific ALU sequence, which was used to confirm transplantation of hMSCs (Fig. 4A). RT-PCR (30 cycles) showed that in both groups, fragments of ColI, fibronectin (FN), stromal cell-derived factor (SDF)-1, or osteoprotegerin (OPG) were successfully amplified by hMSCs#1, while fragments of other genes, such as osteogenesis-related trophic factors (BMP-2, IGF-1, and VEGF) and osteoblast differentiation markers (alkaline phosphatase [ALP] and osteopontin [OPN]), were not detected (Fig. 4A).

The expression of osteoblast-differentiation-related genes by hMSCs after transplantation into bone defects. cDNA was prepared from total RNA extracted from the in-vivo-transplanted collagen scaffold of Groups 1 and 2 as described in Figure 1. Real-time reverse transcription–polymerase chain reaction (RT-PCR) showed fold changes of each gene: type I collagen (ColI)
The expression levels of genes whose fragments were amplified by RT-PCR were analyzed by real-time RT-PCR. Among these genes, ColI expression from Group 1 was remarkably higher at 46.3-fold over that of Group 2 (p<0.05), while GAPDH expression in Group 1 exhibited a 5.5-fold higher value (p<0.01) compared with that of Group 2. FN and OPG expression under Group 1 treatment was higher by 3.6- (p<0.05) and 4.6-fold (p<0.05), respectively, than those of Group 2, which seemed to reflect the difference in GAPDH expression level between the two groups. There was no significant increase in SDF-1 expression, which had a great variance among animal subjects. A comparison of relative levels of ColI gene expression to that of hMSCs#1 cultured in vitro showed that the expression level of ColI was the most active among amplified genes after transplantation into recipients, corresponding to 20% of the expression level of in-vitro-cultured hMSCs#1 (Fig. 4F). The expression levels of other genes were very low as compared with in-vitro-cultured cells; FN expression was 1.65% that of in-vitro-cultured cells, and OPG and SDF-1 expression was under 1% of cultured cells. Real-time RT-PCR showed that osteogenesis-related growth factors (BMP-2, IGF-1, and VEGF) and osteoblast-differentiation-related markers (ALP and OPN) were not amplified by hMSCs#1-specific primers in either group (data not shown).
The pattern of gene expression seen in hMSCs#1 was also seen in hMSCs#2. Genes amplified by RT-PCR included ColI, FN, OPG, and SDF-1, and the expression of all of these genes was significantly higher in Group 1 than Group 2 (Fig. 4G). Consistent with transplantation using hMSCs#1, ColI expression was notable in the level of gene expression by hMSCs, reaching 41.65% of the ColI expression level of hMSCs cultured in vitro on collagen sponges, while FN, OPG, and SDF-1 expression was much lower at 5.2%, 0.83%, and 0.02%, respectively, of the level in hMSCs cultured in vitro. Moreover, the expression of osteoblast-differentiation-related markers was also lacking in hMSCs#2 (data not shown). Although there were minor differences in expression levels of the genes tested, the two hMSC lines showed common characteristics in gene expression after in vivo transplantation.
Influence of transplanted hMSCs on the local tissue of host rats
We expected that hMSC transplantation would guide the host cells into facilitating the osteogenic process. To determine the effect of hMSCs on local tissue cells, gene expression in host rat cells was investigated using RT-PCR and real-time RT-PCR with rat-specific primers (Fig. 5). Vehicle control represents the transplant not treated with hMSCs#1 and implanted with collagen sponge alone, and negative control represents in-vitro-cultured hMSCs#1. The RT-PCR image shows that the designed primers used in the present study were specific for rat genes, with the exception of the SDF-1 gene, which was weakly amplified in the negative control. Contrary to the gene expression pattern by hMSCs, RT-PCR images showed that rat cells in the defect area expressed osteoblast-differentiation-related markers (ALP and OPN), trophic factors (BMP-2, IGF-1, VEGF, OPG, and SDF-1), and matrix genes (ColI and FN) (Fig. 5A). Real-time RT-PCR was analyzed by normalization to the 18S rRNA level, which is expressed both from hMSCs and rat cells (Fig. 5B). The expression of β-actin, which is known as a housekeeping gene, was significantly downregulated in Group 2, compared with vehicle control or Group 1, meaning that the numbers of rat cells infiltrated into the defect regions might be not equivalent among groups, appearing the lowest in Group 2. Real-time RT-PCR showed that many genes followed the same tendency as β-actin expression, and a significant downregulation of rat genes in Group 2 might be the result of low densities of rat cells in the defect area. Among the rat endogenous genes influenced by direct transplantation of hMSCs#1 in Group 1, compared with that of vehicle control group, there was a slight increase in rat ALP and VEGF expression, which was but not significant. Noticeably, rat IGF-1 expression in Group 1 was increased by 40-fold (p<0.01) over that of the vehicle control group. A similar tendency of rat gene expression was also observed when using hMSCs#2 (data not shown).
Gene activation in host rat cells in the bone defect region after hMSC transplantation. Real-time RT-PCR was performed using RNA extracted from cells in the collagen sponge implanted into the calvarial defect in Groups 1 (G1) and 2 (G2) and the vehicle control (CTR).
Rat gene expression was further analyzed by normalization to rat β-actin expression, by which comparison of gene expression is limited to only rat cells infiltrated into the defect region (Fig. 5C). Using this method, we found that rat ALP, IGF-1, and VEGF gene expression under Group 1 treatment exhibited a higher value (p<0.05), compared with that of vehicle control or Group 2. As in Group 1, Group 2 showed a higher expression of VEGF (p<0.05) and ColI (p<0.01) than the control group. Especially, ColI expression of Group 2 was 3.26-fold greater (p<0.01) than that of Group 1.
In vivo gene activity after hMSC transplantation into Balb/c nude mice
The same strategy used for Group 1 was repeated in immunodeficient Balb/c nude mice to determine whether there was immune rejection by the host rat when the hMSCs differentiated into the osteoblast lineage. We expected that hMSCs#1 would be committed to osteoblast differentiation when transplanted into the calvarial defects in nude mice where no immune rejection exists. Immediate transplantation, as in Group 1, was used because the GAPDH expression of hMSCs#1 was very poor in Group 2 using cells precultured after seeding on collagen sponges. RT-PCR using human-specific primers revealed that GAPDH was expressed in three nude mice at different levels. The expression of GAPDH correlated with the expression levels of the other human-specific genes FN, ColI, SDF-1, or OPG (Fig. 6). As in the rat calvarial defect model, fragments of differentiation-related markers, including ALP, osteocalcin (OC), and OPN, were not amplified from hMSCs#1 by RT-PCR. However, host mouse genes, such as ALP, OPN, IGF-1, or OC, were clearly expressed, suggesting that the mouse cells that infiltrated into the defect region differentiated into osteoblasts (Fig. 6B). We confirmed human-specific expression of transplanted hMSCs#1 using a human-specific ALU sequence primer that was consistent with human GAPDH expression and was not detected in the mouse-originated C2C12 cell line (Fig. 6C).

Gene expression from hMSCs and resident cells in an immune-deficient model. hMSCs#1 were transplanted into immune-deficient Balb/c mice using the same strategy as was used for Group 1, where hMSCs were directly implanted without precultivation in vitro post-seeding on collagen sponge. cDNA was prepared from total RNA extracted from collagen scaffolds at 7 days after surgery. RT-PCR images from three animals are presented that show genes expressed using human- and mouse-specific primers.
Host immune response: observation of lymphocytes and macrophages
Lymphocytes and macrophages were observed as a measure of the cellular immune response directed against the cell/scaffold constructs. Anti-CD8 antibody was used for the detection of T cells, and an anti-macrophage antibody was used for macrophage detection (Fig. 7). Additionally, the presence of hMSCs was observed with IHC staining using anti-human Nucleolin antibody that detects human nucleus. Positively stained spots of human Nucleolin were found at surface boundary layer of collagen sponge where hMSCs were loaded in Group 1 and Group 2. CD8-stained cells were relatively rare in both hMSC transplantation groups, and a few positive spots were detected at the boundary with existing calvaria surrounding the defects, rather than near the human-Nucleolin-positive spots or inner side of the collagen sponge. However, macrophages appeared more frequently than T lymphocytes. Notably, positively stained spots of macrophage in the Group 2 (implantation of precultured hMSCs) were more abundant than Group 1 (direct implantation of hMSCs). Macrophage-positive spots of Group 2 were distributed inner side of collagen disks as well as surface boundary layer of collagen sponge while they were relatively rare in the vehicle control group and Group 1. H&E staining showed that a lot of cells were present in the sections of Group 1 and Group 2 while there were relatively a smaller number of cells in the section of vehicle control.
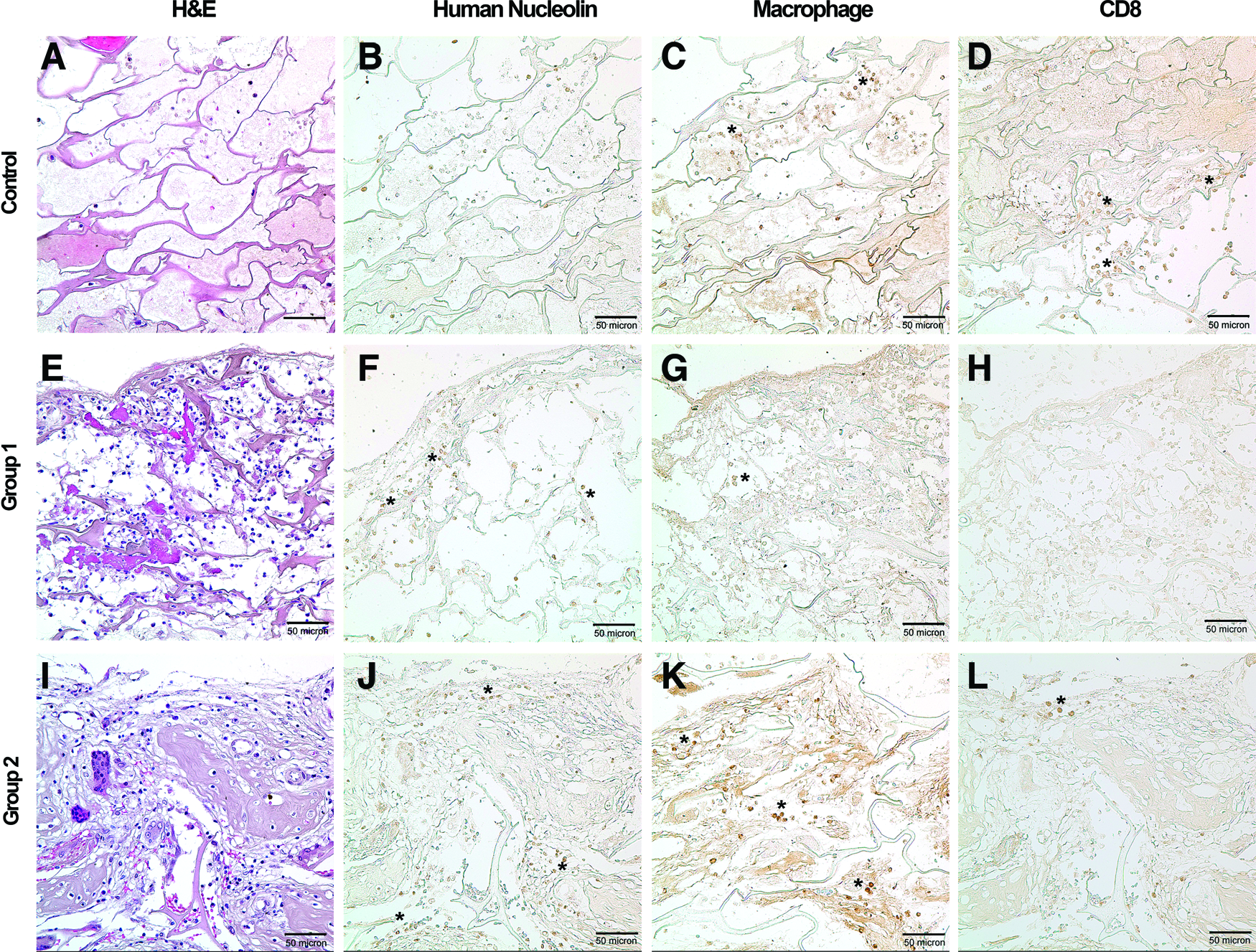
Observation of immune cells (macrophage and T cells) within implanted collagen disks of vehicle control
Phenotype analysis and immunogenicity of hMSCs
The hMSCs#1 used in this study were negative for CD45 but expressed CD90 and CD105, which are known as surface markers of MSCs. hMSCs#1 could differentiate into adipocytes or chondrocytes in the presence of specific differentiation media (data not shown). The immune characteristics of hMSCs#1 were investigated to show that no noticeable immunological rejection was associated with the xenogeneic use of hMSCs. FACS analysis showed that <1.27% of hMSCs#1 expressed HLA-DR (MHC class II), while about 54.8% of cells expressed HLA-ABC (MHC class I) (Fig. 8A). However, over 92% of cells were positive for HLA-ABC after treatment with the pro-inflammatory cytokine IFN-γ (100 IU/mL), while a deficiency in HLA-DR was maintained under these same conditions. In a previous study, commercially available hMSCs (here described as hMSCs#2) showed a similar phenotype with hMSCs#1. 7 Because IL-10 and TGF-β are known to be anti-inflammatory cytokines and to regulate immunity, we assessed their secretion by hMSCs. Neither IL-10 nor TGF-β expression from hMSCs#1 exhibited any significant change at days 3 and 6 in the presence of IFN-γ (Fig. 8B). Similar to our in vitro results, transplanted hMSCs#1 and #2 in our xenogeneic model showed a lack of expression of human-specific HLA-DR, but both had clear expression of human-specific HLA-ABC, which was consistent between Groups 1 and 2 (Fig. 8C). After transplantation of hMSCs#1 into immunodeficient Balb/c mice, human-specific HLA-ABC and -DR expression showed a similar pattern as seen in the rat xenogeneic model (Fig. 8D).
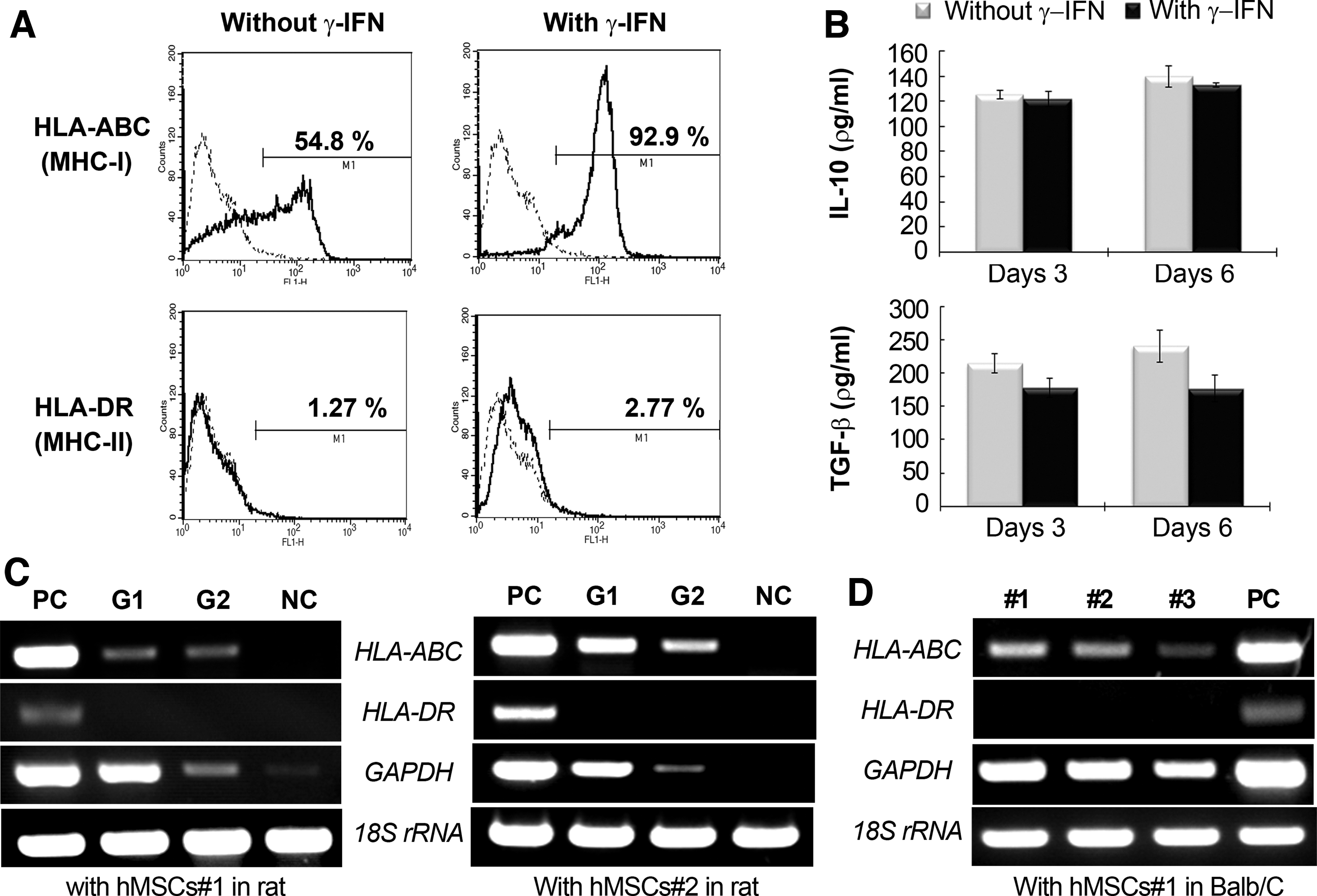
Immunogenicity of hMSCs.
Discussion
To accelerate bone regeneration, particularly for large-size defects, stem cell therapy using sufficient numbers of ex-vivo-expanded hMSCs in association with suitable biocompatible scaffolds is regarded as a practical method.4,17,27 In tissue-engineered construction protocols, preculturing of hMSCs on a scaffold in vitro until transplantation has been suggested to enhance the efficiency of stem cell therapy because the preculturing step allows cells to stabilize by attaching on the matrix surface. However, it is not clear that this is an advantage for enabling transplanted cells to be functionally more active over direct implantation after seeding on the scaffold. In the current study, we focused on comparing the in vivo gene activity of hMSCs after transplantation using two different pretreatment protocols in association with a collagen sponge scaffold at the levels of transplanted cells and native rat cells, respectively. Our results showed that direct loading enhanced the viability of transplanted hMSCs, resulting in more efficient in vivo gene expression in the initial week, compared with the transplantation of cells precultured in vitro on the collagen carrier. This difference was seen despite a lack of evidence of the direct differentiation of hMSCs to osteoblasts. This pattern is consistent with the promoting effect on gene expression of host rat cells.
To accomplish our experimental objective, we chose two conditions that differ in the preculturing period and differentiation state of hMSCs. We set up the preculturing period to be within 1 day of transplantation because preculturing cells on the scaffold for several days makes it impossible to directly compare the results from the direct loading and preculturing protocols due to the different cell numbers present after preculturing. The initial cell densities in these two conditions were confirmed to be equal if cells are cultured overnight. An additional reason for a 1-day culture term in the preparation of precultured hMSCs is the loose deformation of the collagen sponge seen after long culture in vitro. The issue around whether to transplant predifferentiated or undifferentiated MSCs for bone regeneration has been controversial, and related studies show conflicting results, either from the superior potential of bone regeneration using hMSCs in an undifferentiated state, 28 or with the reverse result that differentiated MSCs resulted in greater efficacy in bone formation as compared with undifferentiated cells.29,30 Nevertheless, the undifferentiated state of hMSCs has attractive properties over hMSCs in a differentiated state, including secretion of trophic factors or the great potential for the differentiation into cells of injury tissue type. An additional factor is that the undifferentiated state of hMSCs shows local immune suppression more effectively than differentiated hMSCs in xenogeneic transplantation, which was proven by the finding that differentiated MSC constructs showed increased histologic infiltration of lymphocytes and macrophages compared with undifferentiated MSC constructs. 31 Taken together, undifferentiated hMSCs were chosen for this study because the main purpose of the study was to investigate the in vivo translational capacity of hMSC potential, which had been proven in vitro.
One of the main concerns in stem-cell-based tissue engineering is the low survival rate of the transplanted cells given the rare evidence of the transplanted cells in tissue repair, even after the administration of millions of cells. Consistent with other reports,5,32 our current study revealed that ex-vivo-expanded cells rapidly lose cell bioactivity in terms of GAPDH gene expression, which is regarded as an estimate of cell viability. 33 GAPDH levels from administered hMSCs were detected only at minimum levels 2 weeks after transplantation. This phenomenon appeared independently of whether cells were directly administered or precultured on the scaffolds, and was common to both hMSC lines tested. However, we found that the loss of gene expression was attenuated if cells were administered without the preculturing step, contrary to our initial expectations. It is unclear as to why the preculturing step is counterproductive for in vivo transplantation because it stabilizes cells by adhering them to the scaffold fiber. In a related study on the survival of transplanted cells, a rapid decrease in cell number within 7 days after implantation was also observed. 32 They used precultivated MSCs in an isogenic rat model to avoid immunologic reactions resulting from xenogenic cell transplantation. Zimmermann's group suggested that the reason for the relatively short survival of transplanted MSCs was an inadequate oxygen supply in the seeded constructs due to insufficient vascularity at the subcutaneous recipient site. Although our study has several differences in experimental design, it is possible that our treatment of precultivating hMSCs might expose cells to more drastic changes from the culture medium condition to the hostile injury microenvironment than the cells that were directly administered encountered, leading to an inadequate oxygen or nutrient supply, which might result in lowered gene activation by hMSCs after transplantation. In addition, IHC staining showed that more macrophages were present in the group using precultivated hMSCs (Group 2) than direct-implanted group (Group 1), implying a reasonable factor for lower viability of Group 2 by host immune response.
Host cell activation by transplanted MSCs is also suggested as one possible mechanism by which MSCs promote tissue regeneration. We interpreted our results with two possibilities, which include an explanation either for rat gene expression from rat cells that have infiltrated into the defect region by normalization of gene expression level to rat-specific β-actin expression, or for that from total cells that involve rat cells and hMSCs in the defect region by normalization to 18S rRNA expression. In the analysis with reference to total cells, the expression levels of rat β-actin in the transplantation with precultivated hMSCs (Group 2) were the lowest among the vehicle control, direct-loaded hMSCs (Group 1), and precultured hMSC group (Group 2), implying that the infiltration of neighboring cells into collagen fibers in the defect region in Group 2 might not be favorable. The expression of osteogenesis-related marker ColI or ALP of Group 2 was decreased, compared with vehicle control, which seems to reflect the lowered β-actin expression. However, the effect of directly transplanted cells on rat gene expression was very minor, except for IGF-1 expression, which was greatly enhanced over the nontransplanted vehicle group. Studies in animal models and in humans have established critical roles for IGFs in skeletal growth and development, including cellular growth, differentiation, bone density, and bone renewal. 34 This result indicates that hMSC transplantation facilitates bone regeneration by host cells, although it did not induce noticeable increases in the expression of rat genes related to osteogenesis. However, in another analysis of gene expression levels, by normalization to β-actin expression (limited to infiltrated rat cells), hMSC transplantation significantly promoted rat ALP, VEGF, or IGF-1 expression, which was higher following direct implantation than after implantation following preculturing treatment. A positive effect of transplanted hMSCs on the host system was also reported in a rat focal cerebral ischemia model. In this model, intravenously transplanted hMSCs induced functional improvement, reduced infarct volume, and provided neuroprotection in ischemic rats, possibly by providing IGF-1 and inducing VEGF, EGF, and bFGF neurotrophic factors in host brain. 35 An additional study showed that treatment with MSCs enhances stroke-induced angiogenesis in the ischemic brain and that the host tissue trophic factor network can be activated by the MSC-mediated JAK-STAT3 signaling or Notch signaling pathway for tissue repair.36,37
We investigated in vivo gene expression at 7 or 14 days after creation of a calvarial defect, when the defect region is presumed to undergo early osteogenesis. The administered hMSCs failed to express the ALP gene, a representative marker of early osteoblast differentiation, while the resident rat cells showed definite ALP expression. This result means that osteogenesis proceeded normally in the bone defect region, while osteoblast differentiation of hMSCs was blocked, despite the cells being in the same local microenvironment. In addition to the ALP gene, hMSCs did not express growth factors, including BMP-2, VEGF, or IGF-1, which are normally expressed in host rat cells as well as in hMSCs in vitro. hMSCs expressed a limited range of genes after in vivo transplantation, including ColI, FN, SDF-1, and OPG. We found the expression of SDF-1 and OPG by hMSCs to be less activated than that of the ECM genes (ColI and FN), which probably play to provide an anchorage site for cytokines expressed by host tissues in the local defect region, 38 finally facilitating tissue regeneration.
Transplanted MSCs rarely differentiate into cell types involved in repair of the injured tissue itself, 39 although the multilineage differentiation potential of MSCs has been demonstrated in vitro. Because there is much debate for the direct role of transplanted hMSCs, many researchers have questioned the efficient in vivo translation of the in vitro capacity of hMSCs to differentiate or to secrete a variety of trophic factors. Rai et al. suggested that the in vivo survival of transplanted hMSCs and the trophic factors they secrete must be examined, since the hypoxic environment of a typical bone defect site has been shown to reduce the secretions of pro-angiogenic and -osteogenic factors from resident stromal cells that help to initiate new bone formation. 40 However, there is rare evidence that confirms that hMSCs still show the expression of trophic factors after in vivo transplantation, even into immune-deficient mice, as efficiently as that seen in in vitro culture despite the definite positive effect of the presence of these cells on bone regeneration. Moreover, data from autogeneous or allogenic transplantation of MSCs derived from animals is unavailable to track gene expression of transplanted cells at the transcriptional level. Another limiting factor is the variable experimental conditions in previous studies, including diverse implant materials, injection methods, or in vitro precultivation on scaffolds with or without osteogenic differentiation steps, which make a comparison with other data complicated. In a previous study, we approached a xenogeneic setting using hMSCs coupled with mechanical loading in a rabbit distraction osteogenesis (DO) model 7 and found that hMSCs showed a similar pattern, with no definite signs of differentiation into tissue-specific cells and with a rapid decrease of human gene expression. Interestingly, we found that in the DO model, transplanted hMSCs express IGF-1 when the transplanted local region microenvironment is subjected to mechanical stretching, suggesting that mechanical stimuli may be involved in the activation of transplanted cells.
The International Society for Cellular Therapy proposed a set of minimal criteria to label a cell as an MSC; the lack of HLA-DR surface molecule expression is included as an immune-response-related factor. 41 Immunophenotypic analysis of hMSCs used in this study showed that undifferentiated hMSCs#1 express HLA-ABC, but not HLA-DR. It has been reported that MSCs expressing HLA-ABC are not recognized by the immune system, while MSCs expressing HLA-DR respond to pro-inflammatory cytokines and display phagocytic properties.42–44 We observed a similar pattern of HLA-ABC/DR expression in vivo from administered hMSCs as seen in the in vitro results in both direct and precultured transplantation in rats and nude mice. Additionally, T lymphocytes in the hMSC-transplanted groups (Groups 1 and 2) appeared very rarely around hMSCs in the defect area, with a similar pattern seen in the vehicle control group. However, transplantation using precultured hMSCs is likely to be more vulnerable to attack by macrophage, which might be linked to faster decrease of cell viability at initial weeks post-transplantation, compared with direct implantation of hMSCs. This might be caused by remained constituents from cell culture such as FBS. In the current study, we replicated the xenogeneic setting created in the rat calvarial defect model using an immune-deficient mouse. As with the rat, transplanted hMSCs expressed a limited range of genes, including ColI, FN, SDF-1, and OPG, but failed to express osteogenic genes even in an immune-deficient host lacking the capacity for immune rejection of the hMSCs. Even if we assume that the reduced gene expression was due to cellular disintegration and early cell death of the transplanted MSCs, 32 why there is only limited gene expression in the living transplants is still not understood. Taken together, these data indicate that the suppression of hMSC differentiation into tissue-specific cells may be caused by a reason from separate from immune rejection, which will require further studies to elucidate the mechanisms for the loss of differentiation potentials of transplanted cells.
Our current findings demonstrate that, for local bone tissue engineering, hMSCs transplanted in vivo display more active gene expression if immediately administered after seeding on collagen sponges rather than preculturing overnight in vitro. Genes expressed by transplanted hMSCs include ColI, FN, SDF-1, and OPG, among which ColI expression was most active, while other osteoblast-differentiation-related markers or growth factors were not expressed by hMSCs in our xenogeneic model or in immune-deficient animals. The direct implantation protocol was more favorable to the osteogenic process of the host system, with enhanced host IGF-1 expression seen with direct implantation rather than preculturing in culture medium until transplantation. Our data suggest that hMSCs in vivo do not have the same potential as in vitro to exert their osteogenic stimulus on the local host tissues. Transplanted hMSCs were shown to survive only for a short time interval and to express few genes. Further study is required to determine what stimuli or conditions lead hMSCs to be more functional in vivo after transplantation, or why ex-vivo-expanded cells have almost silent gene expression after in vivo transplantation.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF); funded by the Ministry of Education, Science and Technology (2011-0026313); and a grant of the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (A120313). The authors thank J.H. Oh, and B. Lee for assistance in the preparation of the article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.