
Abstract
Matrix metalloproteinases (MMPs) and a family of tissue inhibitors of metalloproteinases (TIMPs) may contribute to myocardial remodeling in heart failure. TIMPs are the main inhibitors of MMPs and have other MMP-independent functions. Because little is known of the role of TIMPs in the heart, we examined the effects of TIMPs on cardiac fibroblasts (CFs) and cardiomyocytes. In vitro, TIMP-1–4 enhanced smooth muscle actin (SMA) expression in CFs, and TIMP-1 and TIMP-3 enhanced the expression of phosphorylated Smad-3 and phosphorylated transforming growth factor (TGF)-β type 1 receptor in CFs; this effect was inhibited by TGF-β receptor blocker SB-505124. TIMPs-1, -3, and -4 also inhibited the FAK, AKT, and ERK pathways that induce cardiac hypertrophy. TIMP-1 and TIMP-2 suppressed apoptosis in cardiomyocytes; in contrast, TIMP-4 induced apoptosis in CFs. TIMP-2 stimulated collagen synthesis. Collagen gels containing TIMP-1 or TIMP-3, which exhibit cardioprotective effects in vitro, were transplanted to the left ventricular anterior wall of a rat heart model of myocardial infarction. Gel-released TIMP-1 and TIMP-3 significantly improved cardiac function and myocardial remodeling and enhanced SMA expression in the infarcted area in ischemic cardiomyopathy model rats. Further, the transplantation of TIMP-1 or TIMP-3 gels inhibited apoptosis in the ischemic myocardium and reduced MMP-2 activity. TIMPs may be an ideal target of cardiac regeneration therapy.
Introduction
A
The interaction between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) plays an important role in cardiac remodeling. 1 MMPs are central to the destruction of the ECM.2,3 Although a number of studies have reported the physiological role of MMPs in the heart, the role of TIMPs is unclear. MMP-2 and MMP-9 are induced in a variety of experimental heart failure models as well as in the failing heart; they degrade collagen, thereby mediating ECM remodeling.4,5
The TIMP family comprises TIMPs-1, -2, -3, and -4. They are secreted proteins that inhibit MMPs. Although the canonical role of TIMPs-1–4 in the heart is to neutralize active MMPs, there is increasing evidence that TIMPs may have other MMP-independent activities. TIMPs-1–4 are expressed in cardiac fibroblasts (CFs) and cardiomyocytes 6 and their expression is reduced in the failing heart. 7
To understand the role of TIMPs in the heart, we focused on their effects on CFs and cardiomyocytes. We also examined the function of the TIMPs with the greatest cardioprotective activity in a rat model of ischemic cardiomyopathy.
Materials and Methods
Animal ethics
Experimental animals were housed in accordance with the Guide for Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996). The Ethics Review Committee for Animal Experimentation of Osaka University Graduate School of Medicine approved the experimental protocols.
Construction of recombinant rat TIMPs
pcDNA3.1 containing the cDNA encoding C-terminally Flag-tagged rat TIMPs-1, 2, 3, and 4 (pcDNA3.1-rTIMP-1–4) was constructed (Fig. 1A). Recombinant TIMP was produced by calcium phosphate transfection of HEK 293T cells with pcDNA3.1-TIMPs. The recombinant protein was expressed and secreted into the culture medium, which was harvested. Recombinant TIMPs were purified by immunoprecipitation with anti-flag antibody (Sigma). The purity of the recombinant TIMPs was established by Coomassie Brilliant Blue staining.
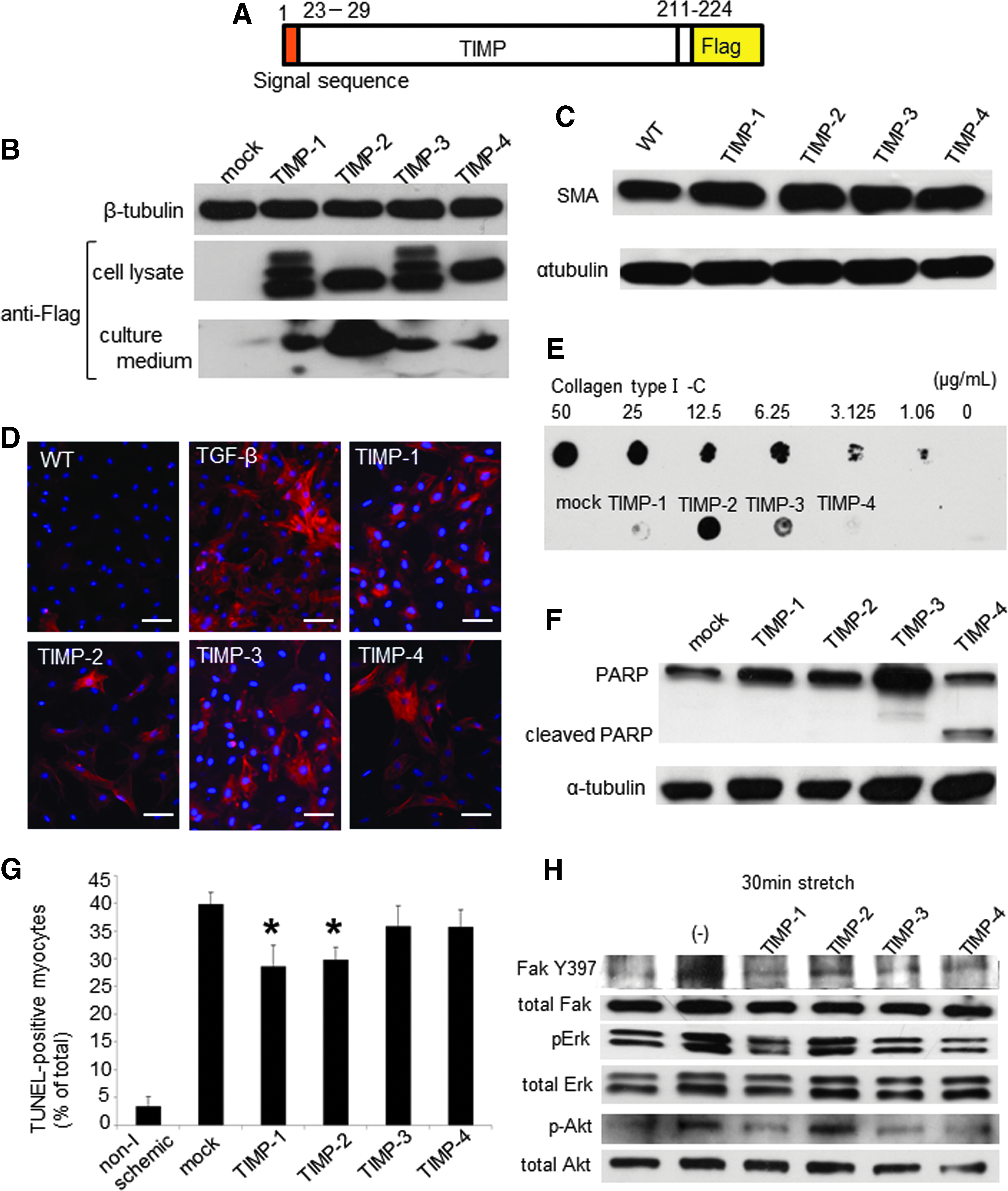
Characterization of CFs transfected with plasmid vector containing rrTIMP-1–4.
Primary culture of adult ventricular fibroblasts and neonatal cardiomyocytes
CFs were isolated from 8-week-old adult male Sprague-Dawley rats 4 weeks after induction of MI by left anterior descending (LAD) coronary artery ligation. Rats were anesthetized with intraperitoneal injection of pentobarbital (300 mg/kg) and heparin (150 U); the heart was removed and transferred to Hank's buffered salt solution (HBSS). The minced tissues were digested in 100 U/mL type II collagenase (Worthington Biochemical Corp.) and 0.1% trypsin at 37°C. The cells were centrifuged and suspended in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS).
Primary rat neonatal cardiomyocytes were isolated from the heart of 0–1-day-old Sprague-Dawley rats (n=40) by digestion with 0.2% type II collagenase at 37°C. The neonatal rats were anesthetized by intraperitoneal injection of pentobarbital (300 mg/kg). The isolated cells were plated on gelatin-coated culture dishes in Medium-199 (Sigma) containing 6% FBS. After 24 h, the medium was replaced with serum-free Medium-199.
Simulated ischemic cardiomyocyte model
To produce ischemia, cells were sealed in a small flask-type culture vessel. The flasks were filled with normal medium (2.5 mL) and phosphate-buffered saline (PBS; 47.5 mL) containing 137 mM NaCl, 8.1 mM Na2HPO4/12H2O, 2.7 mM KCl, 1.5 mM KH2PO4, 0.9 mM CaCl2/2H2O, and 0.3 mM MgCl2/12H2O containing each rrTIMP-1–4 (0.1 μg/mL), and bubbled with 5% CO2–95% N2 for 2 min to fix the initial pH at 7.4 and eliminate oxygen from the remaining air space. In the control group, cells were incubated in 2.5 mL medium equilibrated with a 5% CO2–95% air atmosphere. 8
Cyclic stretch
Isolated cardiomyocytes were placed on gelatin-coated six-well BioFlex culture plates (Flexcell International Corp.) in Medium-199 containing 6% fetal serum. After 24 h, the medium was replaced with serum-free Medium-199 containing rrTIMP-1–4 (0.1 μg/mL) and incubated at 37°C in 5% CO2. After 48 h, the cardiomyocytes were stretched in a Flexcell FX-3000 strain unit to 120% of resting length at a frequency of 1 Hz for 30 min.
Immunofluorescence staining
Isolated CFs were incubated with rrTIMP-1–4 (0.1 μg/mL) or transforming growth factor (TGF)-β1 (25 ng/mL) for 48 h. The cells were fixed with 4% paraformaldehyde and incubated with anti-smooth muscle actin (SMA) antibody (Dako), incubated with cyanine-3-conjugated secondary antibody (GE Healthcare), and observed by fluorescence microscopy (ECLIPSE E600; Nikon).
Dot blotting
The culture media from rrTIMP-1–4-treated CFs were used for dot blot assays. CF cultures were treated for 72 h with rrTIMP-1–4. Each sample was spotted onto the nitrocellulose membrane. To evaluate collagen production, a serial dilution of collagen type I-C was also spotted onto the membrane as a control. The membrane was probed with primary antibody against collagen type I. Secondary-antibody-linked horseradish peroxidase (GE Healthcare) was added and the blots were imaged with an ECL kit (GE Healthcare).
TUNEL staining
The ApopTag Fluorescein In Situ Apoptosis Detection Kit (Chemicon International, Inc.) was used to determine the levels of apoptosis by TdT-mediated dUTP nick-end labeling (TUNEL). The culture medium from isolated cardiomyocytes was replaced with serum-free Medium-199 containing rrTIMP-1–4 (0.1 μg/mL) and incubated for 48 h under ischemic conditions. Detection of digoxigenin covalently attached to the nucleotide can be accomplished by using anti-digoxigenin antibody conjugated to fluorescein, which fluoresces green.
Western blotting
Stretch-stimulated cells were lysed in lysis buffer (50 mM Tris at pH 8.0, 120 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40). The samples were separated by SDS-PAGE, transferred to a polyvinylidene fluoride transfer membrane, and probed with anti-SMA, anti-FAK (Santa Cruz Biotechnology), anti-FAK-pY397 (Millipore), anti-AKT (Cell Signaling Technology), anti-pAKT (Cell Signaling Technology), anti-ERK1/2 (Cell Signaling Technology), and anti-pERK1/2 (Cell Signaling Technology) antibodies.
To examine the activity of TGF-β receptor-Smad signaling induced by TIMP-1 or TIMP-3, the phosphorylation of Smad3 and TGF-beta receptor I (TβRI) was studied by western blotting. The CFs were incubated with TβRI inhibitor SB-505124. After 1 h, TIMP-1 (0.1 μg/mL), TIMP-3 (0.1 μg/mL), or TGF-β1 (25 ng/mL) was added and incubated for another 24 h. Primary antibodies against phospho-Smad3, Smad3 (Cell Signaling), TβRI (phospho S165; Abcam), and SMA were used.
Real-time quantitative PCR
Total RNA was isolated from CFs incubated with rrTIMP-1 or -3 (0.1 μg/mL) for 48 h. Quantitative RT-PCR was performed with Power SYBR Green master mix (Toyobo). The Ct values for each sample were obtained by subtracting the values for glyceraldehyde-phosphate dehydrogenase (GAPDH) from the target gene (fibroblast growth factor-1 [FGF-1], hepatocellular growth factor [HGF], insulin-like growth factor-1 [IGF-1], stromal-cell-derived factor-1 [SDF-1], and vascular endothelial cell growth factor [VEGF]).
Enzyme-linked immunosorbent assay
Culture medium from the isolated CFs incubated with rrTIMP-1 or -3 (0.1 μg/mL) for 72 h was analyzed by FGF-1, HGF, IGF-1, VEGF, and SDF-1 enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's protocol (HGF, IGF-1, and VEGF; R&D Systems, and FGF-1 and SDF-1; Uscn Life Science, Inc.). The captured growth factor was bound to a second specific monoclonal antibody. The amount of specifically bound monoclonal antibody was detected using an enzyme-labeled antibody. Following incubation with chromogenic substrates, the intensity of the color precipitate was measured.
Animal model and collagen gel sheet treatment
The MI models were generated by ligation of the LAD coronary artery in 8-week-old Sprague-Dawley rats. The rats were anesthetized by isoflurane inhalation (2%, 0.2 mL/min), intubated, and then placed on a respirator during surgery to maintain ventilation. The adequacy of anesthesia was monitored by electrocardiography and pulse rate. Two weeks after ligation of the LAD coronary artery, MedGels® (MedGEL Corporation), which are sustained-release collagen gels, were transplanted on the LV anterior wall of rat heart model of MI. The gels were affixed to the epicardium with a fine suture. The gel was impregnated overnight with a solution containing 50 μg recombinant TIMPs (150 μL) at 4°C. The rrTIMP-containing collagen sheets were retained for about 2 weeks in vivo.9,10 The impregnated rrTIMPs were released for two weeks as the gel dissolved. The rats were divided into (1) a PBS group (implanted with 1/10 PBS-perfused collagen gel, n=20); (2) a TIMP-1 (implanted with rrTIMP-1-sustained collagen gel [50 μg/gel], n=20); (3) a TIMP-3 (implanted with rrTIMP-3-sustained collagen gel [50 μg/gel], n=20); and (4) a control group (reopened chest at 2 weeks when the other groups were implanted with collagen gel, n=20). Sustained release of TIMP-1 and TIMP-3 from the gel is 0.3–0.4 μg/h in vitro (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/tea).
Measurement of cardiac function
Cardiac function was evaluated by echocardiography 1, 2, 4, 6, and 8 weeks after treatment (baseline, 1, and 2 weeks, n=20; 4 weeks, n=15; 6 weeks, n=10; 8 weeks after gel treatment, n=5). Baseline represents pretreatment results. Measurements were made with a SONOS 5500 sonograph (Philips Electronics) with a 12-MHz transducer under isoflurane inhalational anesthesia (2%, 0.2 mL/min). The LV end-systolic area, the LV end-diastolic area, and the LV dimensions at end-diastole and end-systole (LVIDd and LVIDs, respectively) were determined. The ejection fraction (EF) and fractional shortening (FS) were calculated as follows:
(1) LVEF (%)=(LVDd3 − LVDs3)/LVDd3×100 (%) (2) LV%FS=[(LVDd − LVDs)/LVDd]×100 (%)
Heart weight/body weight ratio
Body weight (g) was measured before the rats were anesthetized with intraperitoneal pentobarbital (300 mg/kg) and heparin (150 U) by intraperitoneal injection and their hearts were removed. The weights of the removed hearts (mg) were also measured and the heart weight/body weight (HW/BW) ratio was calculated. The HW/BW was calculated using five rats in each group at 2, 4, 6, and 8 weeks after treatment.
Histological analysis
Five rats in each group were used for histology at 2, 4, 6, and 8 weeks after treatment. LV chamber diameter and anterior wall thickness were measured for sections stained with hematoxylin–eosin or Masson Trichrome. Infarcted wall thickness, posterior wall thickness, and LV chamber diameter were measured with the scale loupe. Dilation of the LV chamber was evaluated as the diameter of the LV chamber divided by the LV posterior wall thickness. Sirius red staining was used to detect fibrosis. The percentage of fibrosis was calculated from the fibrotic ratio in the infarcted border area. Periodic acid–Schiff staining for cardiomyocyte hypertrophy was performed. We selected 100 cardiomyocytes and measured the two-point shortest axes at the level of the nucleus. The distribution of elastin fibers was evaluated by Elastica van Gieson staining. We estimated the percentage of elastin fiber (purple-stained area) for collagen fiber (red-stained area) in the infarcted area. The distribution of myofibroblast-like cells was evaluated by immunohistochemical staining with anti-SMA antibody (Dako). The sections were incubated with biotinylated secondary antibody (Dako) and peroxidase-conjugated streptavidin (GE Healthcare). Visualization was performed with biphenyl-3,3′,4,4′-tetramine solution (Sigma). SMA-positive cell density was calculated as SMA-positive area/infarcted area×100 (%).
Western blotting of gel-treated myocardium
Samples of myocardial tissue removed at 2, 4, 6, and 8 weeks after gel treatment were divided into infarction, border, and remote areas. Each sample was homogenized in SDS buffer, and the supernatants were used for western blotting with anti-PARP (Cell Signaling Technology) antibody. The samples were obtained from five hearts in each group at each timepoint.
Zymography
The left ventricle samples were homogenized in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% [wt/vol] Nonidet P-40, and 0.1% [wt/vol] SDS). The samples were obtained from five hearts in each group at 2, 4, 6, and 8 weeks after treatment. Each sample was loaded per lane of a polyacrylamide-gel-containing gelatin. Gels were incubated in 2.5% Triton X-100 for 45 min, and then exchanged with developing buffer (20 mM Tris [pH 7.4], 5 mM CaCl2, and 1 μM ZnCl2) and incubated for 18–24 h at 37°C. The gels were stained with Coomassie Brilliant Blue R-250.
Statistical analyses
Data are presented as the mean±standard deviation. Cardiac function was analyzed by repeated-measures analysis of variance (ANOVA) for differences across the entire time course, as well as one-way ANOVA, whereas the Tukey–Kramer post hoc test was used to examine significant differences at each time point. To assess the significance of the differences between individual groups for other data, statistical comparisons were performed using an unpaired Student's t-test. p<0.05 was considered statistically significant.
Results
Construction of recombinant rat TIMPs
Recombinant rat TIMPs were detected in the cell lysate and culture medium of HEK293T transfected with pcDNA3.1-rTIMP-1–4 (Fig. 1B). Each recombinant TIMP was purified with anti-flag antibody and used for the study.
Effect of rrTIMPs on CF-SMA expression
SMA expression increased with rrTIMP-1–4 addition to isolated CFs (Fig. 1C). SMA expression varied little between the four rrTIMPs and immunofluorescence staining showed that expression of SMA increased in CFs treated with rrTIMP-1–4 (Fig. 1D).
Effect of rrTIMPs on CF collagen synthesis
We performed dot blots and found that rrTIMP-1 and -4 had no significant effect on collagen synthesis (Fig. 1E). In contrast, TIMP-2 stimulated collagen synthesis in comparison to untreated CFs. Although TIMP-3 also induced collagen synthesis, the degree of induction was less than that of TIMP-2.
Effect of rrTIMPs on CF apoptosis and cardiomyocytes
The expression of cleaved PARP was higher in rrTIMP-4-treated CFs than in untreated CFs (Fig. 1F), suggesting that TIMP-4 induced apoptosis. In contrast, TIMPs-1, -2, and -3 had no influence on apoptosis in CFs. In the ischemic cardiomyocytes, TUNEL staining showed that isolated neonatal cardiomyocytes were resistant to ischemia-induced apoptosis when exposed to medium containing TIMPs-1 and -2 (Fig. 1G). TIMP-1- and -2-treated cardiomyocytes reduced the frequency of ischemia-induced apoptotic nuclei.
Inhibition of stretch-induced Fak, Erk, and Akt activation in rrTIMP-treated cardiomyocytes
When cardiomyocytes cultured on a deformable silicone dish were stretched for 30 min, the amount of phosphorylated Fak, Erk, and Akt increases.11–13 However, Fak, Erk, and Akt activation was inhibited by rrTIMPs-1, -3, and, -4 in cardiomyocytes (Fig. 1H). In contrast, rrTIMP-2 had no influence on Fak, Erk, and Akt activation in stretched cardiomyocytes.
Activity of TGF-β/Smad signaling by TIMP-1 or TIMP-3
Phosphorylation of TβRI and Smad3 and the expression of SMA were enhanced in CFs treated with TIMP-1 or TIMP-3 (Fig. 2A). Addition of the TβRI inhibitor SB-5050124 reduced phosphorylation of Smad3 and TβRI and the expression of SMA. TβRI phosphorylation and SMA expression in the negative control were induced by autocrine factors from the CFs.
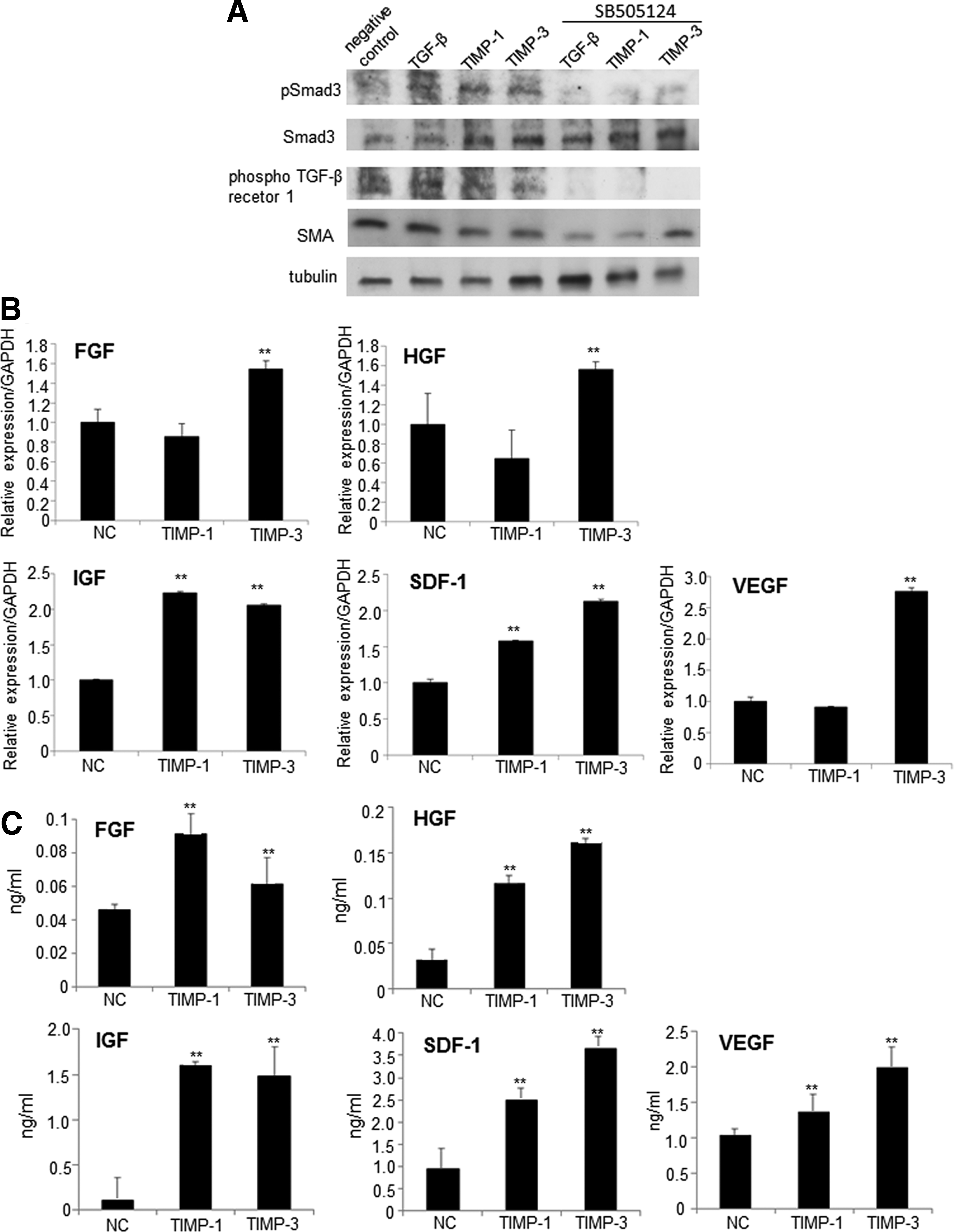
The effects of TIMPs on Smad activation.
Expression of growth factors and chemokine
Effects of TIMP-1 and TIMP-3 on expression of FGF, HGF, IGF, VEGF, and SDF-1 in CFs in vitro were examined by RT-qPCR and ELISA. RT-qPCR revealed significantly higher relative expression of IGF-1 and SDF-1 in TIMP-1- and TIMP-3-treated CFs than in the untreated controls. FGF, HGF, and VEGF expression was significantly higher in TIMP-3-treated CFs versus untreated controls (Fig. 2B). Further, expression of these growth factors and chemokines in the supernatant of CFs incubated with TIMP-1 or TIMP-3 was examined by ELISA. Release of FGF, HGF, IGF, VEGF, and SDF-1 into the supernatants was upregulated in TIMP-1- and TIMP-3-treated CFs (Fig. 2C).
Heart weight/body weight
We used the HW/BW ratio as an indicator of the degree of cardiac hypertrophy. The HW/BW ratio was significantly reduced in the TIMP-1 and TIMP-3 groups at 2, 4, 6, and 8 weeks after treatment in comparison to the control and PBS groups (p<0.05; Fig. 3A).
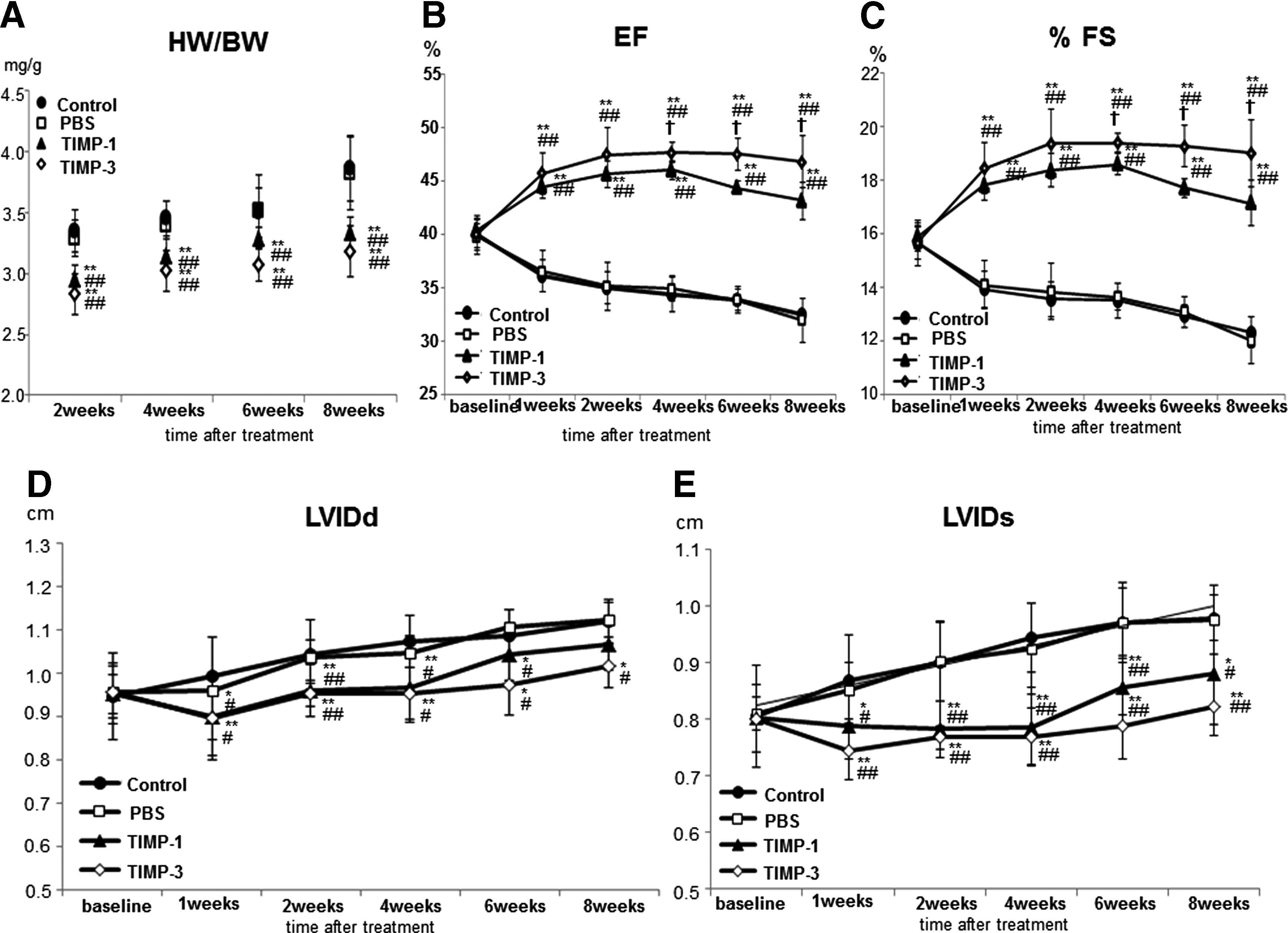
Cardiac function
Echocardiography showed a significant improvement in LVEF and %FS in the TIMP-1 and TIMP-3 groups versus the control and PBS groups at all time points after gel treatment (p<0.01) (Fig. 3B, C). In addition, LVEF and %FS in the TIMP-3 group was significantly improved at 6 and 8 weeks after gel treatment in comparison to the TIMP-1 group (p<0.05) (Fig. 3B, C). LVIDd and LVIDs in the control and PBS groups gradually expanded; however, this expansion was inhibited in the TIMP-1 and TIMP-3 groups from 1 to 8 weeks after gel transplantation (Fig. 3D, E). Inhibition was strongest in the TIMP-3 group.
Histology
Masson-Trichrome-stained LV sections demonstrated thinning of the infarcted wall and dilation of the LV chamber in the control and PBS groups, whereas the thickness of the infarcted wall was maintained and dilation was inhibited in the TIMP-1 and TIMP-3 groups 2, 4, 6, and 8 weeks after gel treatment (Fig. 4). Statistical analysis demonstrated that the infarcted wall in the TIMP-1 and TIMP-3 groups was significantly thicker than that in the control and PBS groups (p<0.01; Fig. 5A), and LV chambers of these groups were also significantly less dilated than those of the control and PBS groups (Fig. 5B). There were no significant differences between the control and PBS groups in these indices.

Masson-Trichrome-stained section of the left ventricle.
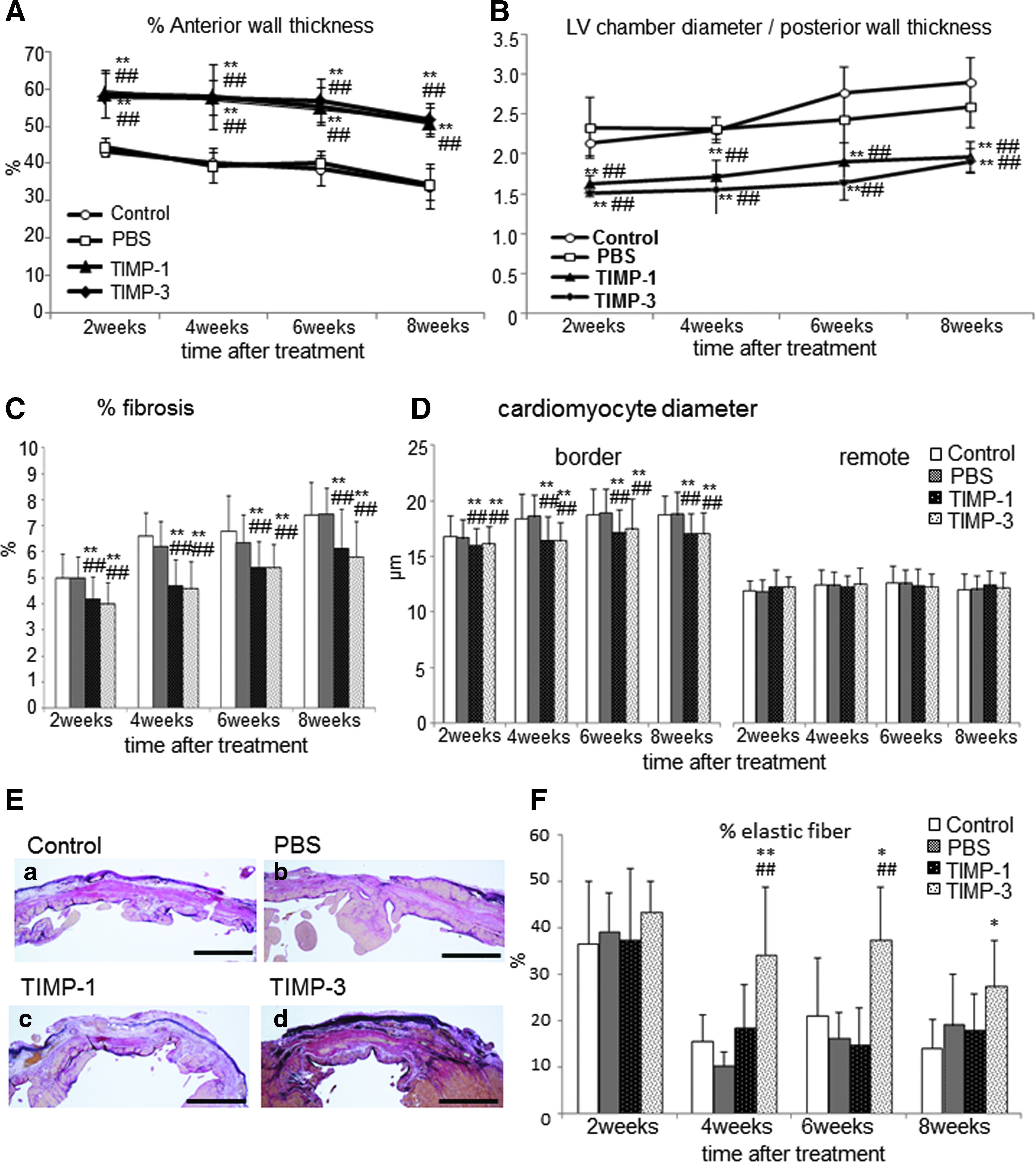
Histological evaluations of % anterior wall thickness and LV chamber diameter. Quantitative estimation of
In the infarcted border area, staining with Sirius red showed significantly reduced fibrosis in the TIMP-1 and TIMP-3 groups in comparison to the control and PBS groups at 2, 4, 6, and 8 weeks after gel treatment (2 weeks, p<0.05; 4 weeks, p<0.01; 6 weeks, p<0.05; 8 weeks: TIMP-1 p<0.05, TIMP-3 p<0.01) (Fig. 5C). Cardiomyocyte diameters in the border area of the TIMP-1 and TIMP-3 groups were significantly smaller than that in the control or PBS group (p<0.01) (Fig. 5D). However, there were no significant differences between groups at 2 and 4 weeks in remote areas. We investigated the percentages of elastin and collagen fiber by Elastica van Gieson (Fig. 5E, F). There was no significant difference between groups at 2 weeks; however, the TIMP-3 group had a significantly higher percentage of elastin fiber to collagen fiber in comparison to the control and PBS groups at 4, 6, and 8 weeks after treatment, and the elastin fibers were maintained in the infarcted area of the TIMP-3 group (Fig. 5F). Elastin fibers were observed in the epicardial area of the infarcted wall in the TIMP-3 group (Fig. 5E). There was no significant difference between the control, PBS, and TIMP-1 groups at all time points.
The distribution of SMA-positive cells
Immunohistochemical staining with an anti-SMA antibody showed that the distribution of SMA-positive cells increased in the TIMP-1 and TIMP-3 groups at 2, 4, 6, and 8 weeks after gel treatment, and many clusters of SMA-positive cells were distributed in the infarcted areas of the TIMP-1 and TIMP-3 groups at 4, 6, and 8 weeks after treatment (Fig. 6A). The rate of SMA positivity in the infarcted area in TIMP-1 and TIMP-3 groups was significantly higher than in the control and PBS groups 4, 6, and 8 weeks after treatment (Fig. 6B).
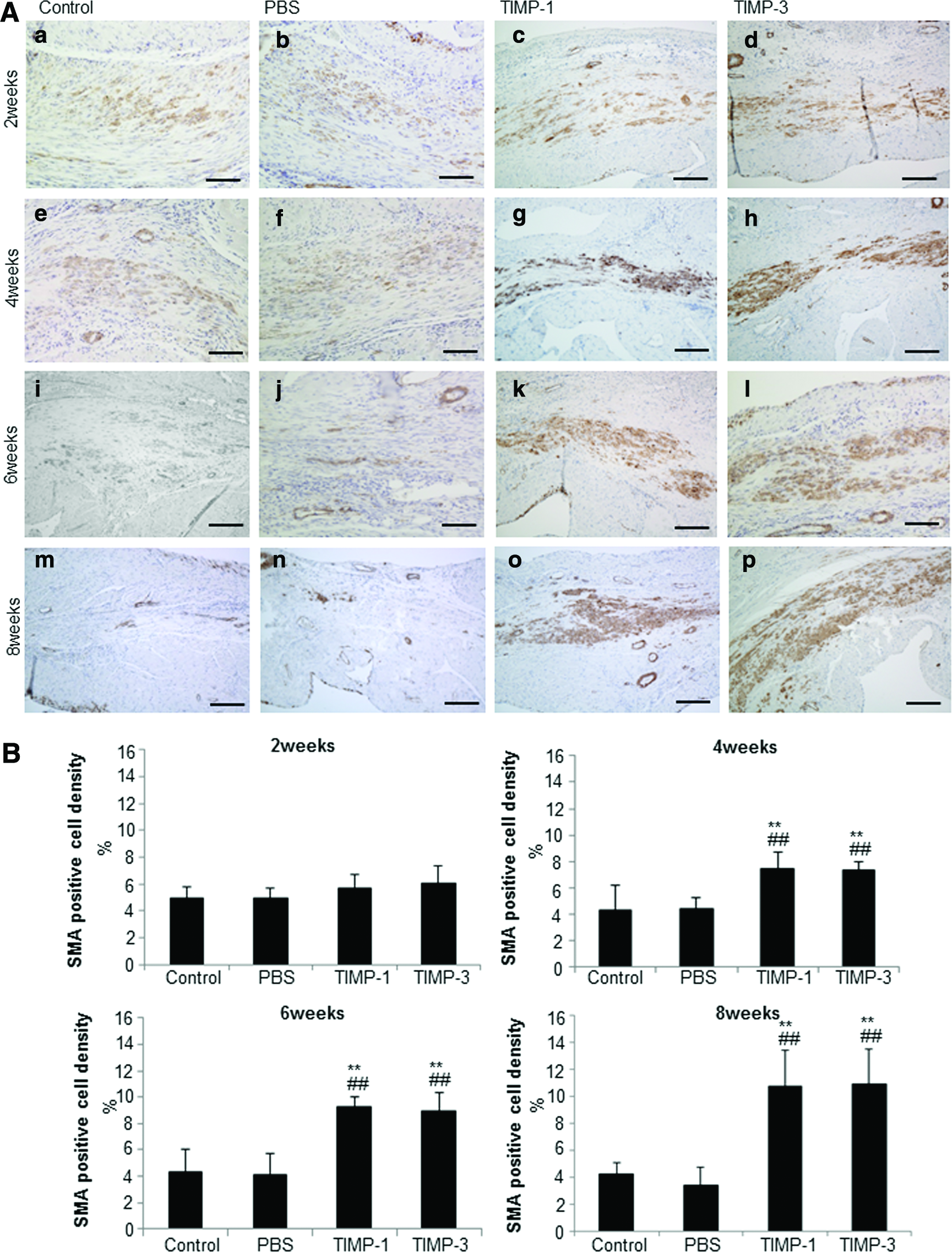
The distribution of SMA-positive cells in infarcted area.
Inhibition of apoptosis of ischemic myocardium by TIMP-1 and TIMP-3
TIMP-1 and TIMP-3 inhibited expression of cleaved PARP in the infarcted border and remote area (Fig. 7A). Particularly in the border and remote area of the TIMP-1 group, the expression of cleaved PARP was lower than that in the control and PBS groups at all time points.
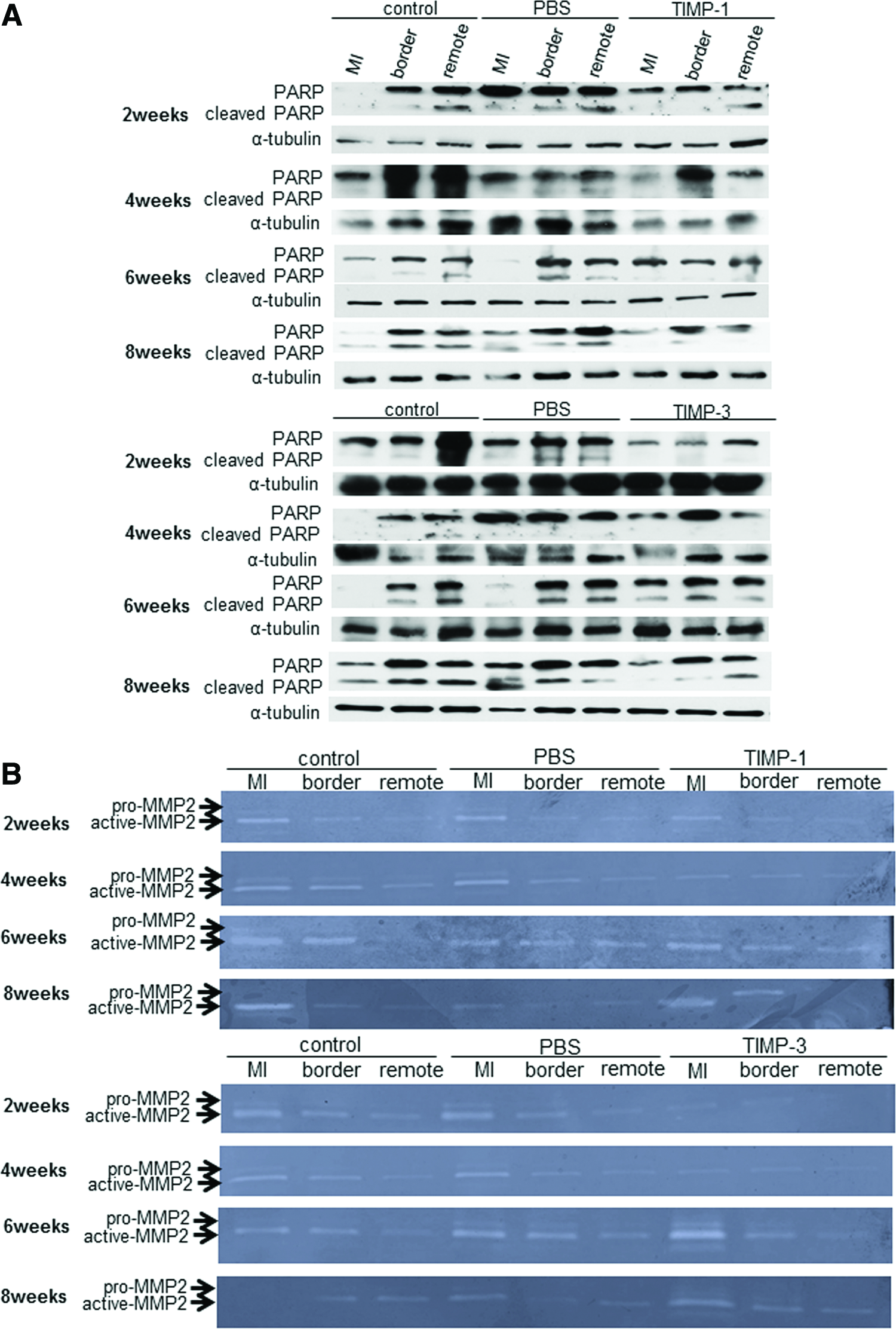
Inhibition of MMP-2 activity in the TIMP-1 and TIMP-3 groups
The gel-released TIMP-1 and TIMP-3 inhibited MMP-2 activity in all areas at 2 and 4 weeks after gel treatment (Fig. 7B). At 6 and 8 weeks after treatment, MMP-2 activity increased in the infarcted area of TIMP-1 and TIMP-3 to the same degree as the control and PBS groups.
Discussion
TIMPs are the major inhibitors of MMPs, but they also have MMP-independent functions. In this study, we examined the effects of TIMPs on isolated CFs and cardiomyocytes in vitro. TIMP-1 and TIMP-3 produced especially effective cardioprotective functions in vitro. We also examined the effects of rrTIMP-1 and -3 on TGF-β/Smad activation and the production of growth factors and cytokines in vitro, and investigated their functions in the development and progression of heart disease in ischemic cardiomyopathy model rats.
In vitro, the increased expression of SMA on CFs was the same for all members of the TIMP family; however, their effects on cardiac hypertrophy, apoptosis, and production of collagen fiber in isolated CFs and cardiomyocytes differed between TIMPs. TIMPs-1, -3, and -4 also inhibited the signaling pathways that cause cardiac hypertrophy. TIMP-1 and TIMP-2 suppressed apoptosis of cardiomyocytes; in contrast, TIMP-4 induced apoptosis of CFs. TIMP-2 stimulated collagen synthesis, which may promote myocardial fibrosis. Therefore, we examined the effects of TIMP-1 and TIMP-3, which have a greater cardioprotective effect than TIMP-2 and TIMP-4 in vitro on ischemic cardiomyopathy model rats.
When the infarcted hearts were treated with TIMP-1 or TIMP-3 sustained-release gel, we observed continuous improvements in cardiac function and attenuation of cardiac remodeling. Myofibroblasts have a hybrid phenotype with fibroblast and smooth muscle characteristics and increased expression of particular contractile proteins, such as α-SMA, SM-MHC type 2, and vimentin.14,15 They also exhibit greater contractility and synthetic properties than fibroblasts, enabling repair and remodeling of the cardiac interstitium to manage local devastation caused by MI.
In this study, many clusters of SMA-positive cells were observed in infarcted areas of the TIMP-1 and TIMP-3 groups 4, 6, and 8 weeks after treatment. Accumulation of SMA-positive muscle-like cells triggered by the release of TIMP-1 and TIMP-3 from the gel adds contractility to the infarcted wall. SMA-positive cells are myofibroblast like; thus, TIMP-1 and TIMP-3 facilitate long-term improvement of cardiac function in the failing heart. The accumulation of SMA-positive muscle-like cells in the infarcted area may reduce the adverse impact on the surviving and uninjured myocardium and may attenuate cardiac remodeling.
Further in isolated CFs, TIMP-1 and TIMP-3 increased the expression of phosphorylated Smad-3 and TGF-β type 1 receptor, and activated TGF-β/Smad signaling. This increase was inhibited by TGF-β receptor blocker SB-505124. TGF-β plays an important role in the activation of fibroblasts in wound repair and induces SMA expression via Smad signaling. 16 In the ischemic cardiomyopathy model, gel-released TIMP-1 and TIMP-3 may enhance expression through this signaling pathway.
Cardiac function dramatically improved in the TIMP-1 and TIMP-3 groups by 1 week after treatment. In vitro, TIMP-1 or TIMP-3 significantly enhanced secretion of growth factors and cytokines, such as HGF, VEGF, FGF-1, IGF-1, and SDF-1, from CFs. Although the mechanism of secretion is unknown, the gel-released TIMP-1 or TIMP-3 stimulates the production of these factors in CFs within the infarcted area, thereby improving early cardiac function after treatment.
At 4, 6, and 8 weeks after gel treatment, cardiac function in the TIMP-3 group improved in comparison to the TIMP-1 group. TIMP-3 is ECM bound and a potent endogenous inhibitor of a disintegrin and metalloproteinase (ADAM) that mediates cardiac disease. 17 Although TIMP-3 is strongly expressed in the heart, its expression is reduced in heart failure and its loss is associated with the progression of myocardial remodeling. 18
Elastin fibers were maintained in the infarcted area of the TIMP-3 group 8 weeks after gel treatment. TIMP-3 inhibits tumor necrosis factor-α (TNF-α)–converting enzyme (TACE; ADAM-17). 19 TACE primarily activates TNF-α, which is a key inflammatory cytokine during cardiac remodeling and the development of heart disease. 20 Inhibition of TACE suppresses the invasion of inflammatory cells and the degradation of elastin fibers. 21 In the TIMP-3 group, the elastin fibers were maintained in the epicardial area for 8 weeks after gel administration. These fibers provide elasticity to the infarcted wall22,23 and may therefore enhance motility of the infarcted wall in the TIMP-3 group. The long-term improvements of cardiac function in the TIMP-3 group might be attributable to the elastin fibers in the infarcted region.
TIMP-3 modulates neonatal cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling. 24 The effect of TIMP-3 on epidermal growth factor receptor activation is probably established by the inhibition of ADAM12-mediated shedding of epidermal growth factor from the cell surface.24,25 Human angiotensin-II-type-2 receptor also interacts with TIMP-3, thus linking the enzyme with the renin–angiotensin system.26,27 Although the pathological roles and signaling mechanisms of angiotensin-II-type-2 receptor remain unknown, it suppresses cardiac hypertrophy during ischemic heart disease.28,29 In this study, TIMP-3 likely suppressed cardiac hypertrophy by inhibiting ADAM-12 and angiotensin-II-type-2 receptor signaling in vitro and in vivo.
TIMP-1 and TIMP-3 reduced expression of cleaved PARP in the infarcted border and remote area, and inhibited apoptosis of ischemic myocardium. TIMP-1 strongly inhibited apoptosis. Some studies have shown that TIMP-1 prevents apoptosis through the activation of cell-survival pathways, including Akt.30,31 In this study, TIMP-1 promoted survival in the ischemic myocardium. Further, MMP-2 activity was inhibited in TIMP-1 and TIMP-3 groups 2 and 4 weeks after gel treatment. MMP-2 decomposes cardiac ECM compartments, advances LV remodeling, and leads to LV rupture. Therefore, suppression of MMP-2 activity by TIMP-1 or TIMP-3 attenuates LV remodeling. However, the detailed functional mechanism of TIMPs in myocardium was not revealed by this study. To explain the effect of TIMPs in improving cardiac function and their clinical utility, the mechanistic details must be defined.
In summary, TIMP-1 and TIMP-3 extended-release gels provided continuous improvement of cardiac function and LV remodeling in ischemic cardiomyopathy model rats. Further, TIMP-1 or TIMP-3 was accumulated muscle-like cells of SMA positive in the myocardium—without the need for cell transplantation. These results suggest that TIMP-1 and TIMP-3 could become the target of a novel cardiac regeneration therapy.
Footnotes
Disclosure Statement
The authors have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.