
Abstract
Human bone marrow stem cells (hBMSCs) represent a promising regenerative material because of their mutipotency, including their ability to regenerate collagenous soft tissues. We previously found that water-soluble chitin (WSC) enhances the ability of human periodontal ligament stem cells (hPDLSCs) to synthesize collagen tissue. The aim of this study was to determine the effects of WSC on hBMSCs and hPDLSCs for the collagen synthesis both in vitro and in vivo. hBMSCs and hPDLSCs were isolated and expanded with or without 0.3 mg/mL WSC. A series of in vitro and in vivo analyses were performed to evaluate their characteristics as stem cell populations. Then, collagen and hydroxyproline assays were conducted using both in vitro and in vivo assay models, and the real-time polymerase chain reaction was performed to analyze the expression of collagen-related markers. WSC-treated and nontreated hBMSCs and hPDLSCs were transplanted into immunocompromised mice, and histology and immunohistochemistry analyses were conducted after 8 weeks. The in vitro results showed that those cells possessed the characteristics of mesenchymal stem cells. The amount of soluble collagen synthesized was significantly greater in WSC-treated hBMSCs than in the nontreated group; conversely, treatment of hPDLSCs with WSC decreased the formation of soluble collagen. The amount of insoluble collagen synthesized was greater in the WSC-treated groups than in the nontreated groups for both hBMSCs and hPDLSCs. The hydroxyproline contents of the regenerated soluble and insoluble collagens were similar. The expressions of mRNA for collagen types I–V, hyaluronic acid synthase 1 (HAS1), HAS2, and HAS3, and the LOX family were higher in WSC-treated hPDLSCs than in the nontreated group, whereas WSC increased the expression of collagen type III and decreased that of collagen type I in hBMSCs. The histology and immunohistochemistry results revealed that WSC significantly increased the amount of collagen formed in vivo by both types of stem cells. Collectively, treatment with WSC significantly enhanced the collagen-forming potentials of hBMSCs and hPDLSCs, but the collagen they produced exhibited distinctively different characteristics. These findings suggest that the appropriate stem-cell source should be chosen based on the purpose of the required regenerated tissue.
Introduction
V
Various sources of adult MSCs have been identified, including muscle, skin, adipose tissue, synovial membrane, dermis, trabecular bone, periosteum, pericyte, teeth, and blood.8–13 Among these various types of MSC, human bone marrow stem cells (hBMSCs) were the initial source of adult MSCs to be utilized, and are the most frequently investigated cell type, often being described as the gold standard. 13 Previous studies have shown that hBMSCs are applicable to various forms of tissue regeneration and wound healing,14,15 but recent studies have explored a potentially new application for soft-tissue healing purposes.12,13 In collagen regeneration, transplanted MSCs seem to attenuate the inflammatory phase and induce controlled collagen deposition through the indirect repression of myofibroblast differentiation.9–11 Previous studies have revealed that the characteristics of collagen tissue regeneration differ according to the source of MSCs utilized.12,13,16 These considerations are necessary to select a valid and purpose-driven source of MSCs 17 ; however, a few, if any, studies have investigated this.
One interesting feature of stem cells is that their biologic activities are influenced by the surrounding microenvironment, such as the type of scaffold, growth factors, and biomaterials. 18 Among various biomaterials, chitin and its derivatives have been extensively investigated due to their excellent biocompatibility and various effects on cells and immune responses.19–21 Furthermore, studies of wound-healing models have shown that chitin derivatives induce collagen-fiber formation by stimulating the cells to produce extracellular matrix (ECM)22,23 and increasing the proliferation of fibroblasts.24–26 In addition, a recent study revealed that chitin-based materials were able to maintain the stemness and induce the differentiation of various stem cells. 27 Collectively, these findings suggest that chitin and its derivatives could modulate the functions of cells and enhance tissue regeneration.
The newly developed water-soluble chitin (WSC) appeared to be effective in wound healing, especially in terms of collagen formation,20,28 and it was shown that WSC enhanced the ability of human periodontal ligament stem cells (hPDLSCs), which have been widely investigated for their clinical application because they can be easily obtained without causing too much morbidity to the patients and without causing ethical issues, to regenerate collagen tissue. 28 Previous studies indicated that MSCs from different tissue origins produce different tissue products, 17 and so the authors were curious as to how the hBMSCs would respond to WSC and how different the resulting tissue would be in comparison to hPDLSCs. These considerations prompted us to postulate that a combination of hBMSCs and WSC can also regenerate collagen tissue both in vitro and in vivo, but in a different way from hPDLSCs.
Therefore, the aim of this study was to determine the effect of treating hBMSCs with WSC on collagen synthesis both in vitro and in vivo, and to compare the regenerated tissue with that synthesized by WSC-treated and nontreated hPDLSCs.
Materials and Methods
Isolation and culture of hBMSCs and hPDLSCs
Fresh human bone marrow (BM) was harvested from the vertebrae of five volunteers (one man and four women; median age, 46 years; age range, 41–58 years) during orthopedic surgery (Naeun Hospital, Anyang, Korea). The patients provided informed consent for their tissue being used in accordance with guidelines approved by the Institutional Review Board, College of Dentistry, Yonsei University (IRB No. 2-2012-0050). The mononuclear cell fraction was separated from the BM aspirates for the isolation of MSCs by conventional density-gradient centrifugation with a density gradient solution (Ficoll-Paque Plus; GE Healthcare Bio-Sciences) using a slightly modified version of a previously described protocol. 29
The hPDLSC culture protocol which was used is described by Seo et al., 30 and it is a slightly modified version of that used in our previous study. 31 The experimental protocol was approved by the Ethics Committee of the College of Dentistry, Yonsei University (IRB No. 2-2012-0049), and written informed consent was obtained from all subjects before they were enrolled in the study. hPDLSCs were isolated from healthy teeth that had been extracted from three adults aged 11–19 years receiving orthodontic treatment.
hBMSCs (10–100×106) and hPDLSCs (5×105) were seeded into T75 cell culture flasks with a growth medium and incubated at 37°C in 5% CO2. Basically, the basic methods to isolate hBMSCs and hPDLSCs are different, and, therefore, it is difficult to regulate the initial seeding numbers. In addition, initial fresh BM cells are fully heterogeneous, including a large number of red blood cells or other hematopoietic cells as well as MSCs. Therefore, there was a greater initial cell seeding number of hBMSCs. However, after passage of 2–4, both hBMSCs and hPDLSCs used for this study were seeded under the same cell number. Colony formation in the culture was observed using a light microscope (CK40; Olympus Optical). Adherent MSC-like cells were obtained after 2 weeks of culture.
For the purposes of this study, the cells were further expanded in the presence of 0.3 mg/mL WSC for WSC-treated hBMSCs and hPDLSCs; for control (i.e., nontreated) groups, hBMSCs and hPDLSCs were expanded using a conventional method that did not involve WSC. WSC was manufactured using the method described by Kurita et al., 32 as described in our previous study. 28 The expansion medium was exchanged every 3–4 days, and the cells were harvested when they reached confluence. Cells from passage 2 (P2) to P4 were used for this study. We have previously provided full details of the methods used to characterize these cultured MSCs.31,33
Evaluation of surface-marker expression: fluorescence-activated cell sorting
Cell-surface-marker characterization of the isolated cells in the four experimental groups (i.e., nontreated hBMSCs, WSC-treated hBMSCs, nontreated hPDLSCs, and WSC-treated hPDLSCs) was performed using flow cytometry analysis (fluorescence-activated cell sorting [FACS]), the procedure that is described elsewhere.29,31,33 The cells were then incubated with specific fluorescein-isothiocyanate- or phycoerythrin-conjugated mouse monoclonal antibodies raised against human CD105, CD73, CD90, CD44, CD29, CD14, CD34, CD45, CD146, and STRO-1 for 20 min at room temperature. The cells were washed twice and subjected to flow cytometry analysis using a flow cytometer (Beckman Coulter), and analyzed using FACScan software (FACS Calibur; BD Biosciences).
Morphology of WSC-treated hBMSCs and hPDLSCs
WSC-treated hBMSCs and hPDLSCs were continuously expanded in the presence of 0.3 mg/mL WSC, while those in the nontreated groups were expanded in the usual expansion medium. Cell morphologies were observed at P2 using a light microscope (CK40; Olympus Optical).
Assessment of cell proliferation and doubling time
The proliferation rates were assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at P4. Cells (1×103) were plated onto 48-well plates, cultured, and then subjected to MTT assay after 1, 3, and 5 days. After incubation with 10% MTT solution (5 mg/mL; Amresco) at 37°C at 5% CO2 for 2 h, the supernatant was discarded and dimethylsulfoxide (Amresco) was added to each well. The absorbance was measured at 540 nm using a microplate spectrophotometer (Benchmark Plus enzyme-linked immunosorbent assay [ELISA] reader; Bio-Rad).
After confirming the MTT assay, the expressions of mRNAs related to proliferation were observed by real-time polymerase chain reaction (PCR) for the gene encoding Ki-67. 34 Information regarding specific primers are given in Table 1. The real-time PCR protocol is described in our previous study. 28
ACAN, aggrecan; HAS, hyaluronic acid synthase; PPARγ2, peroxisome proliferator-activated receptor γ2; Runx-2, runt-related transcription factor 2.
The doubling time was calculated using the following equation:
where Td is the doubling time, q1 is the quantity grown at time t1, and q2 is the quantity grown at time t2.
Clonogenic assays
The number of colony-forming units (CFUs) was evaluated for all four experimental groups at P2 using a light microscope (CK40; Olympus Optical). The cells were plated at a density of 1×102 cells/mL onto 100-mm culture dishes with growth medium and allowed to grow for 14 days. The dishes were then fixed with 10% neutral-buffered formalin and stained with crystal violet (Sigma-Aldrich). The number of CFUs at P2 was used to determine the colony-forming efficiency (CFE). The colony with the diameter of more than 2 mm wide was counted, and the lightly stained one was included if it was wider than 2 mm.
Cell adhesion activity
Double-label immunofluorescence was used to observe differences in cellular morphology related to the hBMSCs and hPDLSCs adhesion activity for each subpopulation of cells.35–37 The cells were seeded in two-well chamber slides at a density of 1×104 cells per well and expanded for 5 days. The cell, which was in a one-well chamber, was permeabilized with 100% methanol and blocked with 1% bovine serum albumin and 4% goat serum before incubation with the primary antibody raised against integrin alpha 5 (diluted 1:100, ab6131; Abcam) for 30 min at 37°C. After washing with washing buffer, an Alexa-fluor 488-conjugated got anti-mouse lgG (FITC, diluted 1:750, ab6785; Abcam) was used as a secondary antibody for 1 h at room temperature.
The cell was fixed with 10% neutral-buffered formalin and blocked using 10% serum for 30 min at 25°C in another chamber. The primary antibody raised against E-cadherin (diluted 1:25, ab15148; Abcam) was added and incubated for 1 h at 25°C. After incubation, an Alexa-fluor 555-conjugated anti-rabbit lgG (TRITC, diluted 1:300, ab6718; Abcam) was used as a secondary antibody for 1 h at room temperature. The chamber slides were washed thrice and then mounted with mounting medium.
The samples were observed using an Olympus BX41 microscope fitted with an Olympus DP71 camera and a U-RFL-T burner connected to a personal computer running DP-BSW software, and the mean fluorescence intensity of integrin alpha 5 and E-cadherin was analyzed using Adobe Photoshop CS4 software (Adobe Systems).
Induction of osteogenic, adipogenic, and chondrogenic differentiation
Osteogenic and adipogenic differentiation was induced as in the previous study. 31 The cells (P4) were seeded into six-well plates at a density of 1×105 cells/well, and cultured until they reached subconfluence. The culture medium used for osteogenic differentiation was refreshed at 3-day intervals. After 4 weeks of induction, the cells were stained with alizarin red. For adipogenic differentiation, the cells were subjected to three cycles of adipogenic induction/maintenance once confluence had occurred. The adipogenic differentiation media were purchased from Lonza. Mature adipocytes were detected by Oil Red O staining at the end of 21 days of culture. The total areas of mineralized nodule formation and lipid vacuoles were measured using an automated image-analysis system (Image-Pro Plus; Media Cybernetics).
Chondrogenic differentiation was assessed by conducting a pellet culture using a slightly modified version of a previously described method. 38 The cultured cell pellets were left free floating for 21 days and then fixed in 10% neutral-buffered formalin. Paraffin-embedded, 5-μm-thick section were cut and immunostained with Alcian blue to enable the visual assessment of chondrogenic differentiation.
After confirming the differentiation potential of each group of cells, samples were collected for quantitative osteogenic, adipogenic, and chondrogenic marker expressions (Table 1). The expressions of each of the differentiation markers were analyzed using the real-time PCR according to the manufacturer's instructions for each product. The real-time PCR protocol is described in our previous study. 31 Each PCR was performed in triplicate with the same total RNA.
In vitro collagen and insoluble synthesis assay
hBMSCs and hPDLSCs were cultured in culture medium with or without 0.3 mg/mL WSC for 7 days, and the total levels of soluble and insoluble collagen in the culture were measured using the Sircol collagen assay kit (Biocolor). The cell supernatants in the four experimental groups were collected and measured using a previously described soluble collagen assay method, 28 and the insoluble collagen assay was performed using the scraped contents from the culture dishes as also described in the previous study. 28 Then, the content of measured soluble and insoluble collagen was normalized to the final cell number.
Analysis of hydroxyproline in soluble and insoluble collagen
Hydroxyproline in the culture supernatant and the collagenous ECM that had accumulated on the culture dishes were quantified using a slightly modified version of the method of Reddy and Enwemeka.28,39 In brief, after 7 days of culture with or without WSC, the cell supernatants in the four experimental groups were collected and a 200-μL aliquot of the supernatant was placed into a 2-mL clear wide-opening crimp-top vial (Agilent Technologies) and closed with an 11-mm crimped cap (Agilent Technologies). The ECM that had accumulated on the cultured dishes was collected into 100 μL of dH2O and 100 μL of concentrated HCl (Duksan Pure Chemicals) in a same vial. The content of hydroxyproline in soluble and insoluble collagen in the culture was measured using the hydroxyproline assay kit (Biovision), and the content of hydroxyproline was normalized at the final cell number to be measured in the same way as the sircol assay.
Expression of collagen, hyaluronic acid synthase, and lysyl oxidase-family-related genes
mRNA was collected from cultured cells and then the expressions of mRNAs related to collagen formation were observed by real-time PCR. The quantitative real-time expressions of the genes encoding collagen types I, II, III, IV, and V, hyaluronic acid synthase 1 (HAS1), HAS2, HAS3, lysyl oxidase (LOX), lysyl oxidase-like 1 (LOXL1), LOXL2, and LOXL3 were detected. Information regarding each specific primer is given in Table 1.
In vivo collagen synthesis assay (transplantation of hBMSCs and hPDLSCs with HA into immunodeficient mice)
The collagen synthesis of the WSC-treated and nontreated hPDLSCs and hBMSCs was evaluated using an in vivo assay model. 31 For a single transplantation, 6×106 cells were mixed with 0.15 mL of HA filler (ReDexis; Prollenium Medical Technologies) and then injected into subcutaneous pockets that had been made on the dorsal surface of 5-week-old immunocompromised mice. The mice were allocated to one of five groups (one nude mouse with three ectopic transplantations per group) with the following specific transplant conditions: (1) HA carrier only (negative control), (2) nontreated hBMSCs with HA carrier, (3) WSC-treated hBMSCs with HA carrier, (4) nontreated hPDLSCs with HA carrier, and (5) WSC-treated hPDLSCs with HA carrier. The mice were killed at 8 weeks post-transplantation, and the implants were harvested. All animal experiments were carried out in accordance with the Guidelines and Regulations for the Use and Care of Animals of the Animal Care Committee of the Medical College, Yonsei University.
In preparation for frozen sectioning, tissue samples were weighed on a balance (Adventurer; Ohaus) and the tissue was then frozen and sectioned serially at 6 μm. The obtained sections were mounted onto glass slides and then stained with Masson's trichrome and immunohistochemically against human-specific mitochondria (hMito), collagen types I and III, and hydroxyproline: human-specific mitochondrial ribosomal protein (diluted 1:50, ab74285; Abcam), collagen type I (diluted 1:200, ab7778; Abcam), collagen type III (diluted 1:1000, ab292; Abcam), and hydroxyproline (diluted 1:500, bs-7512R; Bioss). After washing the sections thrice, immunodetection was performed using a commercially available kit (EnVision Detection System; Dako REAL) according to the manufacturer's instructions. The sections were then counterstained with hematoxylin.
Statistical analysis
All in vitro experiments were performed at least thrice. Means and standard deviations were calculated, and unpaired t-testing was used to analyze the differences between two groups. For multiple analyses, repeated-measures ANOVA was performed followed by Scheffé's comparison. Differences were considered statistically significant at p<0.05.
Results
Comparison of the effects of WSC on the characteristics of hBMSCs and hPDLSCs
The FACS analysis of nontreated hBMSCs and hPDLSCs revealed the expressions of representative MSC markers, testing positive for CD29, CD44, CD73, CD90, and CD105, and negative for CD14, CD34, and CD45. WSC appeared to have little effect on the level of surface-marker expression in either hBMSCs or hPDLSCs (Fig. 1A). Cells that expanded in the presence of WSC appeared to be more vigorous and longer than those grown in normal medium, producing a greater cell yield in the same-size culture dishes (Fig. 1B).
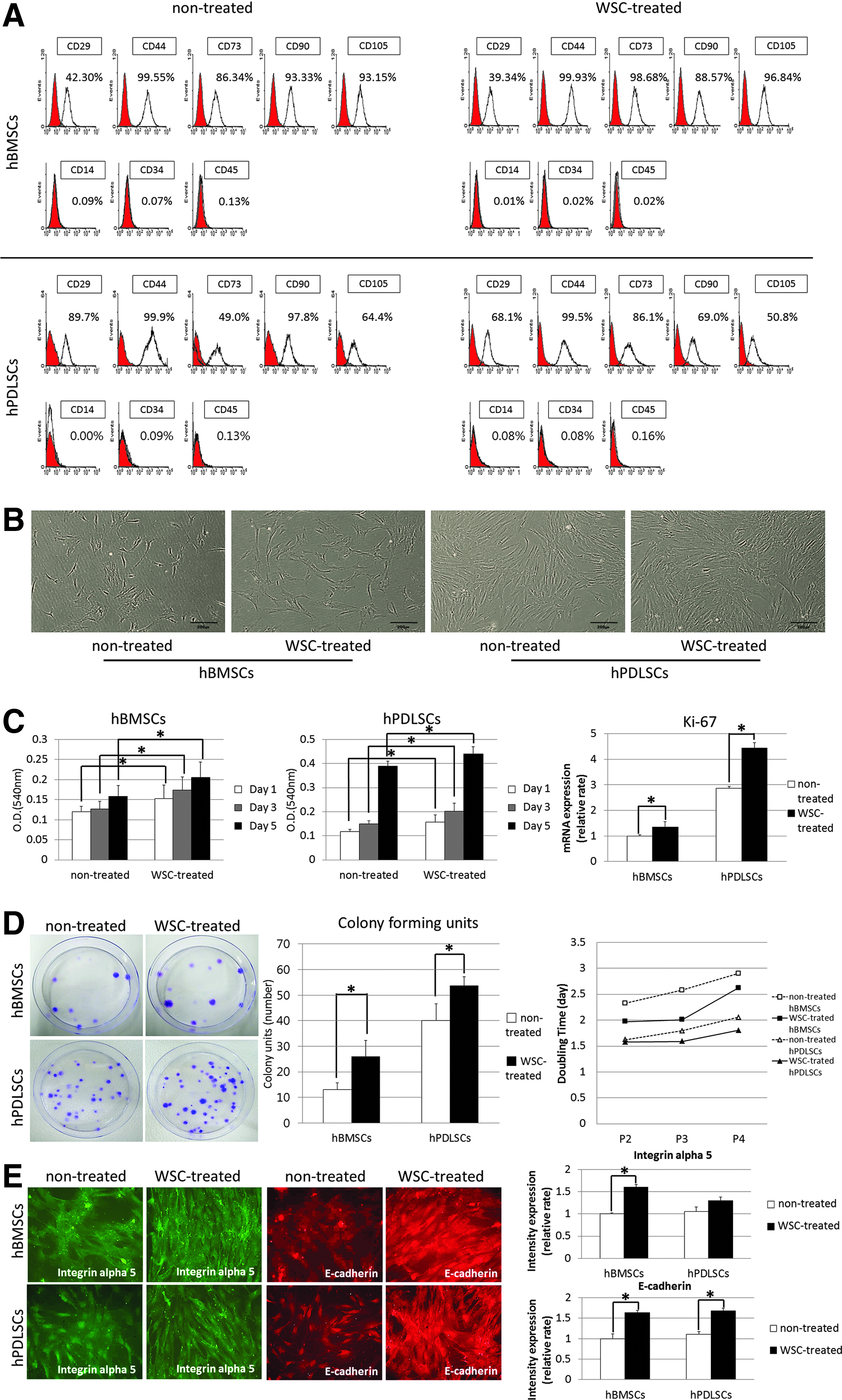
Influence of water-soluble chitin (WSC) on the characteristics of human bone marrow stem cells (hBMSCs) and human periodontal ligament stem cells (hPDLSCs).
The proliferative potential of the WSC-treated cells was assessed relative to that of nontreated cells using the MTT assay and statistically significant differences were found among the four cell groups investigated, particularly at day 5. WSC significantly increased the proliferative potentials of both hBMSCs and hPDLSCs. The real-time PCR performed to determine the changes in proliferation expressed at the mRNA level revealed that the expression of mRNA for Ki-67 was significantly increased in WSC-treated hMSCs compared with nontreated hMSCs (p<0.05; Fig. 1C). The effect of WSC expansion was clearly distinguishable in the CFU assay. The CFE was significantly increased in WSC-treated hMSCs compared with their nontreated counterparts. The doubling time was gradually increased in later passages of all groups, and treatment with WSC decreased the doubling time in both hBMSCs and hPDLSCs (p<0.05; Fig. 1D). Double-label immunofluorescence staining was performed to test the cell adhesion activity of the MSCs. The intensity of integrin alpha 5 and E-cadherin expression was greater for WSC-treated hMSCs than for nontreated hMSCs and implied that the cell adhesion was increased by the treatment of WSC (p<0.05; Fig. 1E).
Multilineage differentiation of hBMSCs and hPDLSCs, and the effect of WSC application
Calcium deposits were observed after 4 weeks of osteogenic induction in all cell groups, as visualized by alizarin-red staining (Fig. 2A). The total area of mineralized nodules was significantly larger for hPDLSCs than for hBMSCs, and WSC-treated hBMSCs and hPDLSCs exhibited a further significant increase in nodule formation (p<0.05; Fig. 2B). This finding was corroborated by an additional assessment of osteogenic differentiation by exploring the representative mRNA expression of runt-related transcription factor 2 (Runx-2) using quantitative real-time PCR (p<0.05; Fig. 2C).
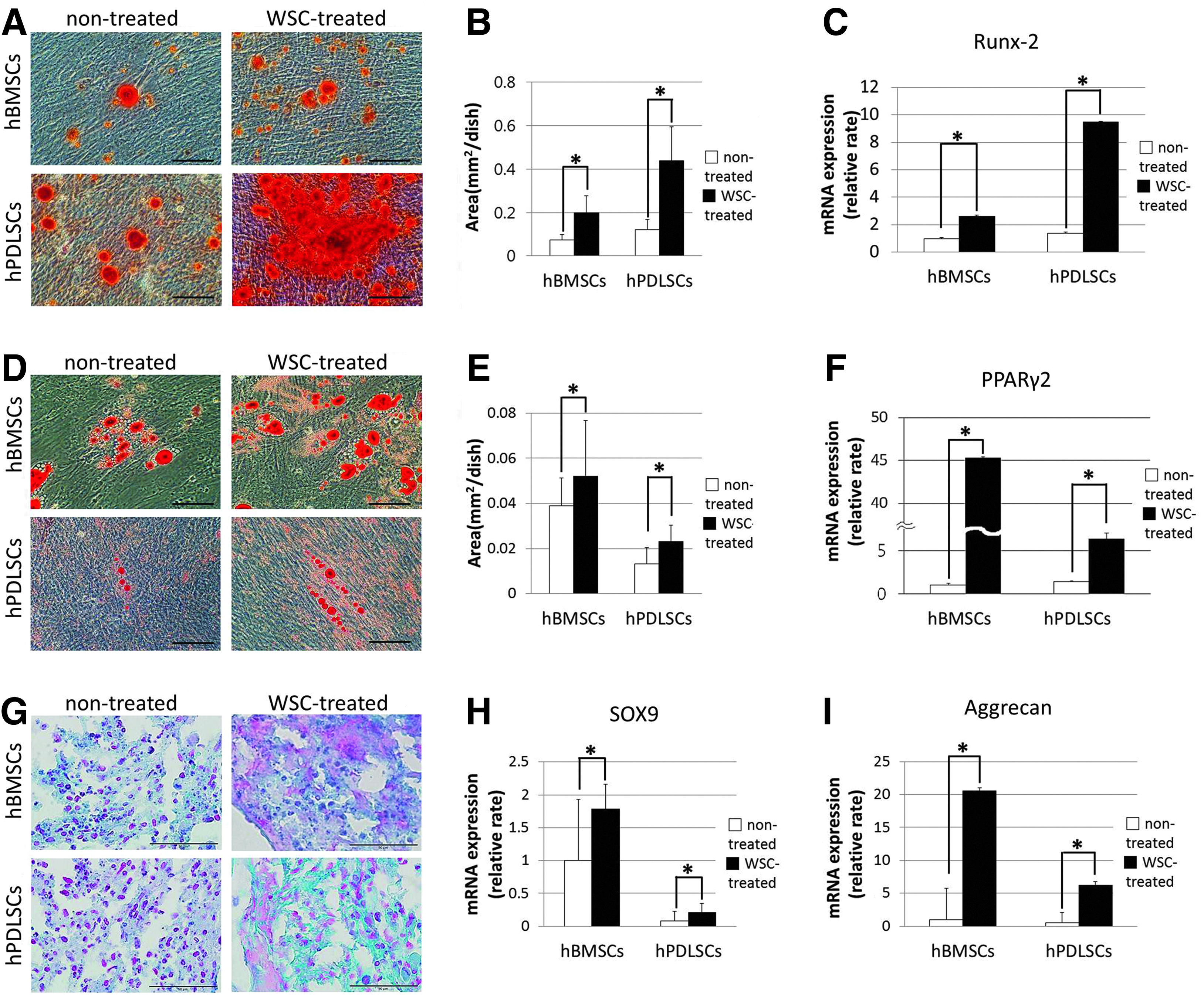
Comparison of in vitro osteogenic, adipogenic, and chondrogenic differentiation of hBMSCs and hPDLSCs, and the effect of WSC on these properties.
Adipogenic potential was also evaluated by measuring the area of stained lipid vesicles that formed after adipogenic stimulation (Fig. 2D). The total area of lipid vacuoles was significantly larger for hBMSCs than for hPDLSCs (p<0.05), and treatment of WSC further increased the area of lipid vacuoles in these two cell types (p<0.05; Fig. 2E). The expression of peroxisome proliferator-activated receptor γ2 (PPARγ2), which is considered a master gene for adipogenesis, was also observed, with an explosive increase in WSC-treated hBMSCs compared with WSC-treated hPDLSCs (p<0.05; Fig. 2F).
The degree of proteoglycan synthesis was greater for WSC-treated cells than for their nontreated counterparts after 21 days of chondrogenesis (Fig. 2G). Moreover, the expressions of SOX9 and aggrecan (ACAN) mRNA in hBMSCs and hPDLSCs under chondrogenic induction were significantly increased in the presence of WSC (p<0.05; Fig. 2H, I).
Comparison of the effect of WSC on in vitro collagen synthesis by hBMSCs and hPDLSCs
The Sircol assay for soluble and insoluble collagen revealed that, as shown previously, the amount of soluble collagen synthesized was significantly lower in WSC-treated hPDLSCs than in the nontreated group. 28 In contrast, the synthesis of soluble collagen was increased in WSC-treated hBMSCs. The insoluble collagen deposits on the WSC-treated hBMSC and hPDLSC culture dishes were increased compared with their nontreated counterparts (p<0.05; Fig. 3A). The content of hydroxyproline exhibited a similar pattern as found for soluble and insoluble collagen by the Sircol assay (p<0.05; Fig. 3B). Each content was normalized according to the final cell number. The real-time PCR performed to determine the changes in collagen expression at the mRNA level revealed that the expressions of mRNA for collagen types I–V, HAS1, HAS2, and HAS3, and the LOX family were higher in WSC-treated hPDLSCs than in the nontreated group. In contrast, the mRNA expressions of collagen types I and IV were decreased in WSC-treated hBMSCs. In this study, the expressions of HAS2 and HAS3 were significantly greater in WSC-treated hBMSCs than in their nontreated counterparts. Treatment with WSC increased the expressions of LOX family genes in both hBMSCs and hPDLSCs (p<0.05; Fig. 4).
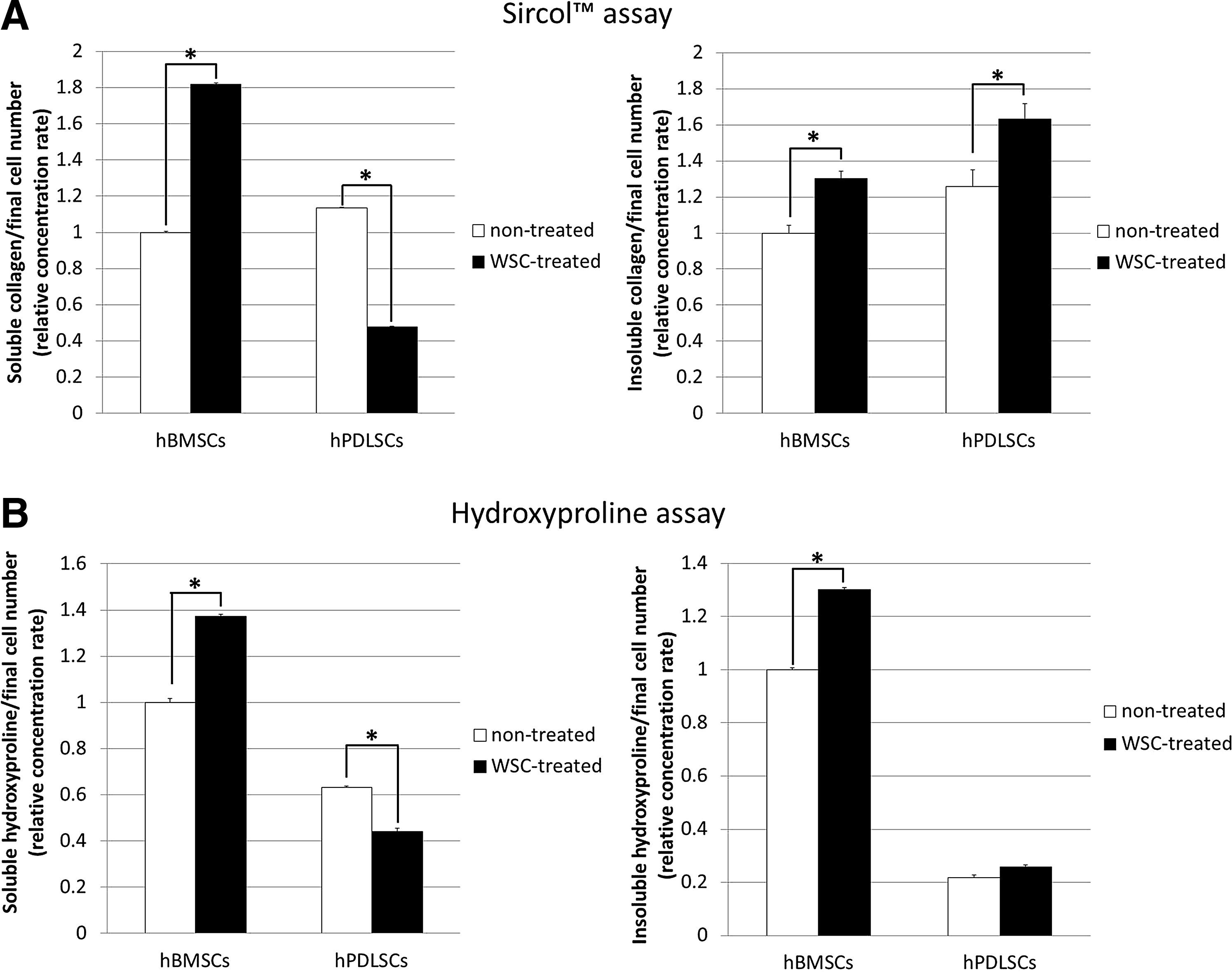
Enhanced collagen synthesis in WSC-treated hBMSCs compared with WSC-treated hPDLSCs in vitro.
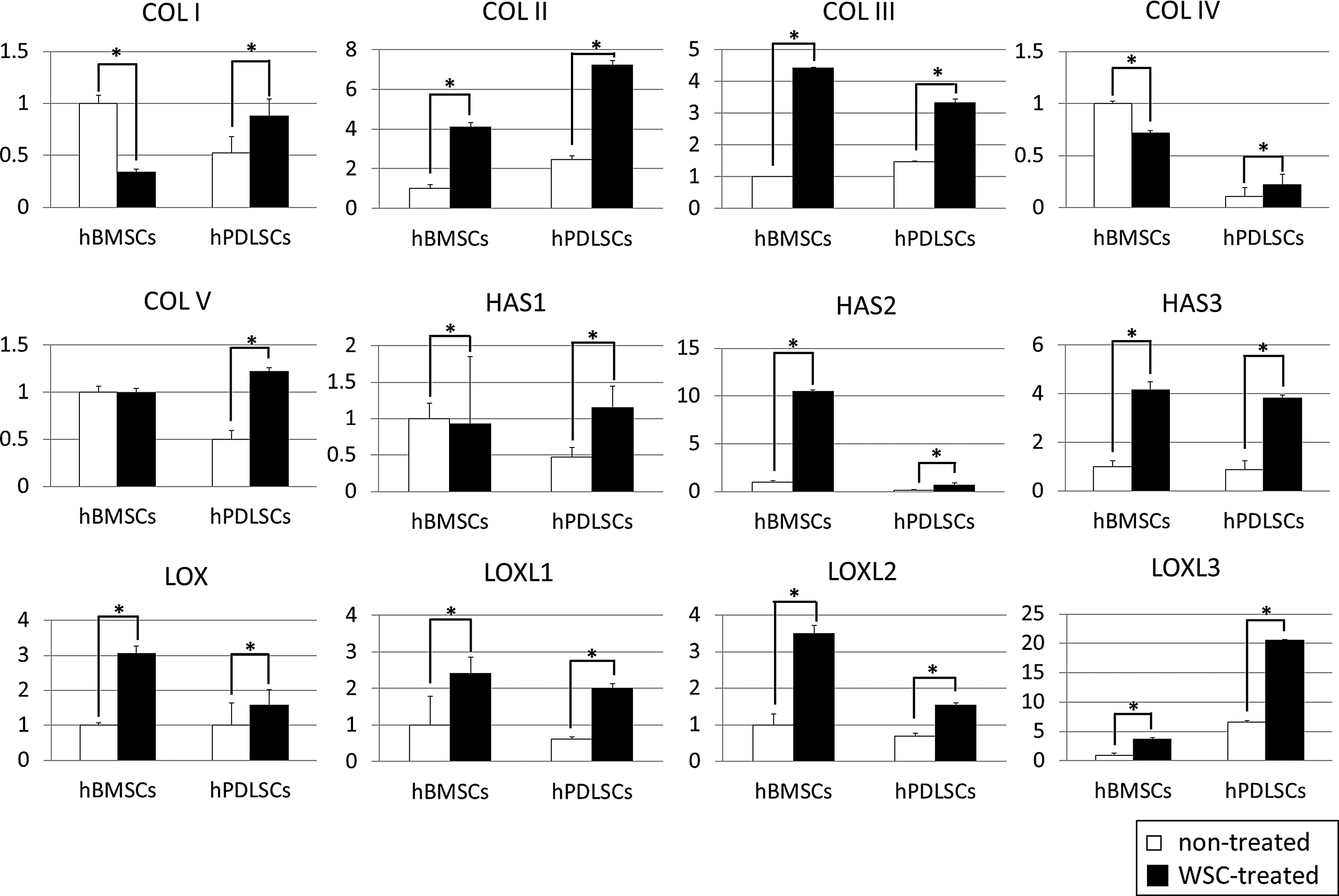
Results of the real-time polymerase chain reaction (PCR) assay. The mRNA expressions of collagen types I–V, hyaluronic acid synthase 1 (HAS1), HAS2, HAS3, lysyl oxidase (LOX), LOX-like 1 (LOXL1), LOXL2, and LOXL3 were significantly enhanced by treatment of hPDLSCs with WSC. In contrast, the mRNA expressions of collagen types I and IV were decreased in WSC-treated hBMSCs. Each experiment was performed at least in triplicate (*p<0.05).
Comparison of the effect of WSC on the in vivo collagen tissue formation
Histology analysis was performed at 8 weeks after transplantation of either WSC-treated or nontreated MSCs. Each transplant maintained its shape, with no signs of inflammation at the point of injection or within the transplants after removal from the mice after 8 weeks (Fig. 5A). The explants from the WSC-treated groups proved to be slightly heavier than those from the nontreated groups, and the difference was not statistically significant (Fig. 5B). Masson's trichrome staining revealed that the amount and density of collagen fibers and collagenous ECM synthesized by the WSC-treated hBMSCs and hPDLSCs were significantly greater than those synthesized by their nontreated counterparts (p<0.05; Fig. 5C, D).
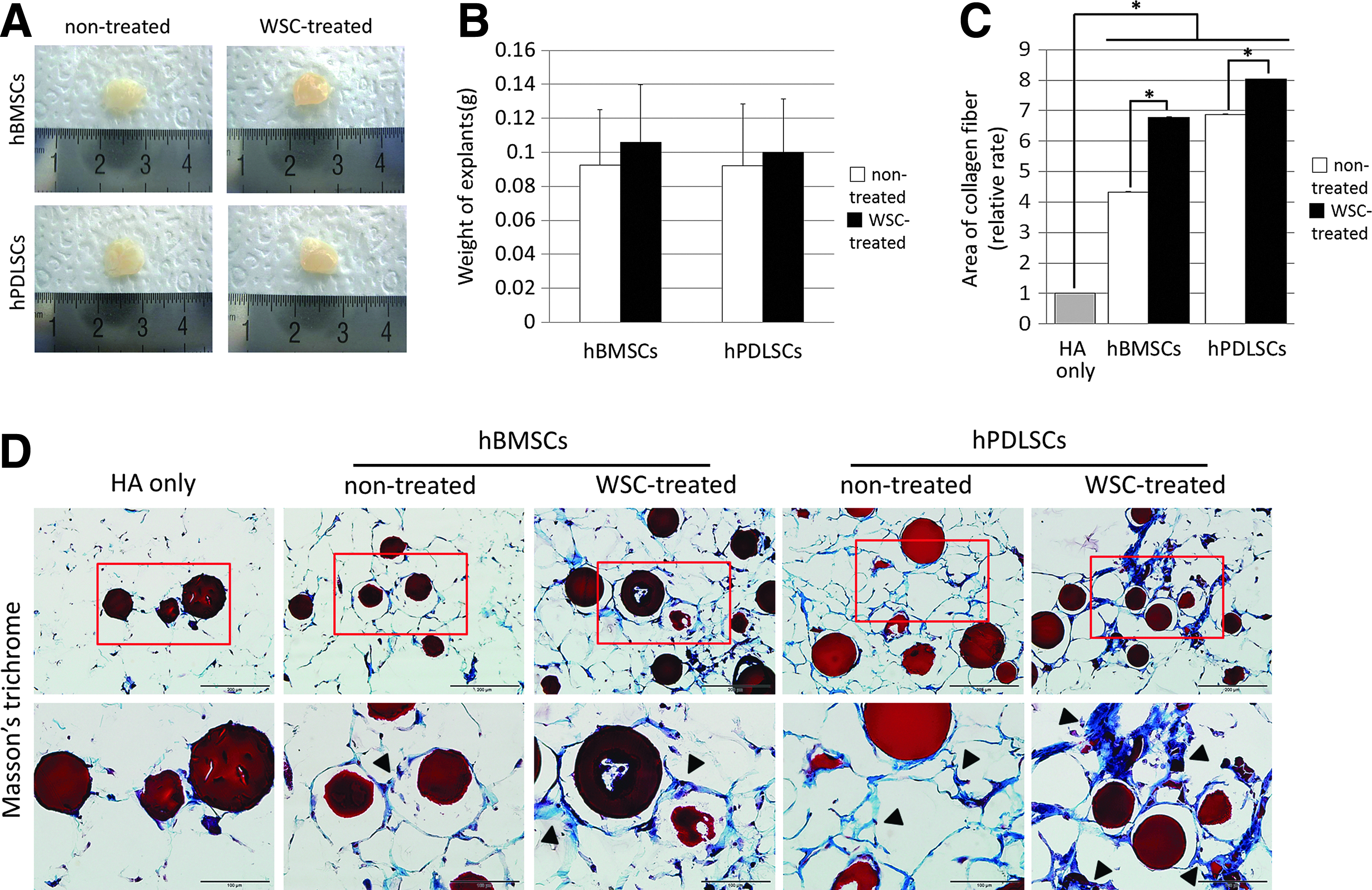
Regeneration of collagen fibers by WSC-treated hBMSCs at 8 weeks post-transplantation compared with WSC-treated hPDLSCs.
Quantitative analysis of the collagen synthesized by the MSCs was achieved via immunohistochemistry. Confirmation that the synthesized tissues originated from the transplanted hBMSCs and hPDLSCs and not from host cells came from the finding that the explant tissue in all except the HA carrier-only (negative control) group was stained darkly for hMito. Exposure of the newly formed collagen tissue to types I and III anticollagen antibodies in vivo revealed that the cells in the WSC-treated groups were more positively stained than those in the nontreated groups. The expression pattern of hydroxyproline was similar to that of collagen types I and III, and it was expressed along the newly formed collagen tissue (Fig. 6A). The hPDLSCs in the WSC-treated group were more positively stained than the nontreated group as previously shown. 28 However, the number of collagen type I-positive cells was decreased in WSC-treated hBMSCs (Fig. 6B), which was corroborated by the in vitro data of real-time PCR.
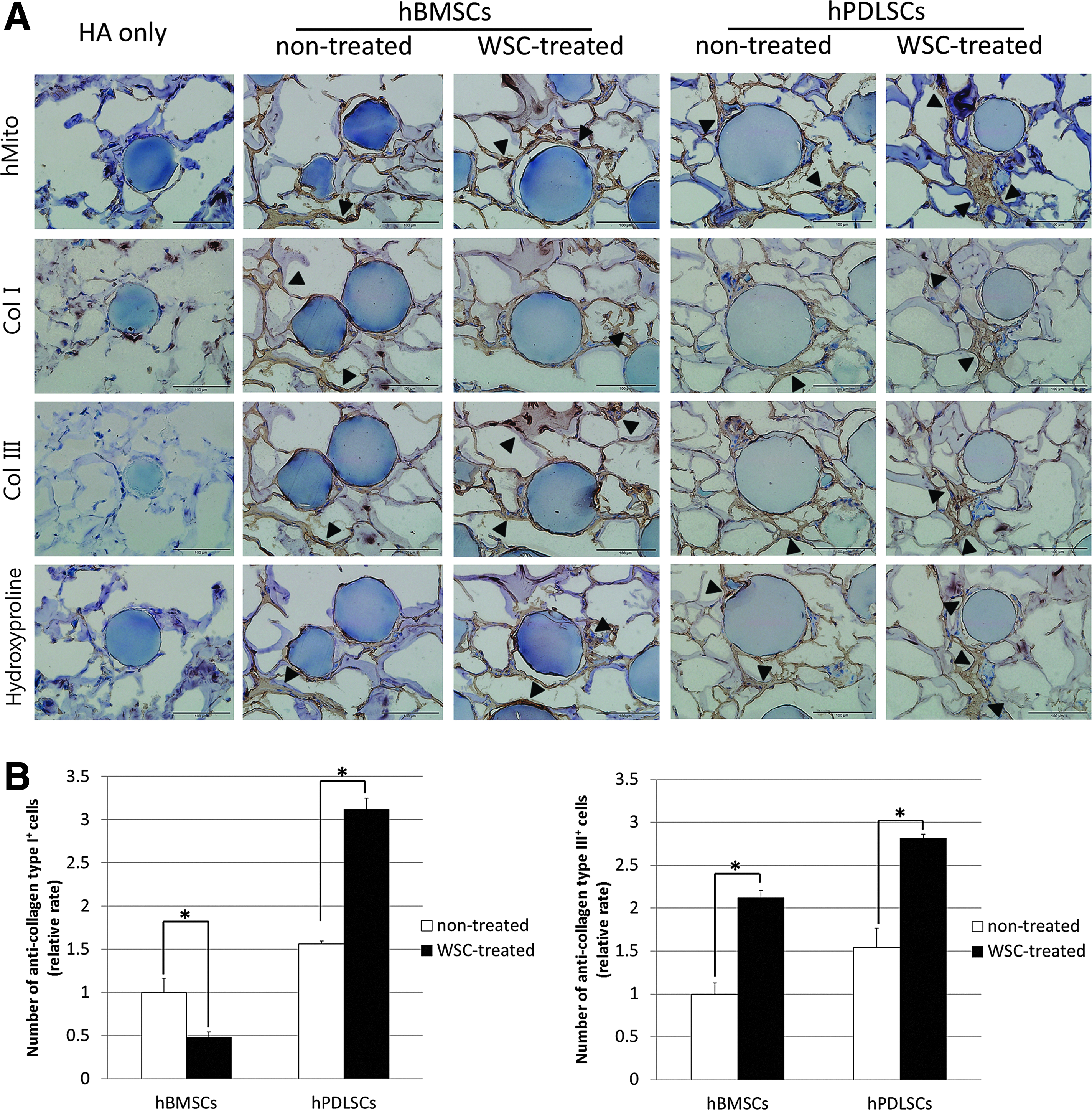
Immunohistochemistry results for collagen types I and III, and hydroxyproline.
Discussion
In our previous study, treatment of hPDLSCs with WSC enhanced the formation of insoluble collagen, which may lead to more mature and stable collagen tissue formation. 28 Interestingly, in this study, it was found that treating hBMSCs with WSC significantly increased the formation of both soluble and insoluble collagen. This increase in fresh and immature collagen synthesis by WSC-treated hBMSCs could potentially benefit the clinical outcomes of soft-tissue augmentations in the cosmetic field, while the increased amount of insoluble collagen produced by WSC-treated hBMSCs would be appropriate for application where mature and rigid collagen tissue formation is required. Consequently, the results of this study imply that WSC can be utilized in combination with MSCs which are appropriate to the type of tissue required, and that the enhancement of collagen tissue regeneration observed with WSC-treated hMSCs may be valuable in the fields of cosmetic and plastic surgery.
In this study, the expressions of mRNA for collagen types I–V were significantly higher in WSC-treated hPDLSCs than in the nontreated group, while only the levels of collagen types II and III were increased, and the other mRNA expressions were downregulated in WSC-treated hBMSCs. The composition of the various collagen types involved in the wound-healing process is very complicated, and an age-dependent collagen-type profile is commonly observed. For instance, the skin contains mainly collagen types I and III, with fetal skin containing a higher proportion of collagen type III than adult skin, although type I is the predominant form in skin at both ages. 40 As the fetus develops, the type III/I collagen ratio changes from higher to lower as its skin approaches that of an adult, which explains the relatively scarless healing of fetal skin in comparison to the healing of adult skin.40,41 Therefore, the significantly increased formation of collagen type III and conversely decreased collagen type I for WSC-treated hBMSCs may indicate their potential of enabling fetal-like wound healing and ideal soft-tissue formation for cosmetic and plastic surgeries.
HA is one of the major carbohydrate components of the ECM, and it can be found in most tissues and organs. 42 In a recent study, biomaterials were constructed from HA with a view to applying them to wound healing and for moisturizing purposes. 42 HA is actively produced by HAS during wound healing and tissue repair to provide a framework for the ingrowth of blood vessels and fibroblasts. 43 Furthermore, HA serves various functions, including space filling and provision of matrix. 44 Therefore, the authors postulated that the increased expressions of HAS2 and HAS3 could contribute to the stabilization of regenerated collagenous tissue. 45 In addition, LOX family genes initiate collagen synthesis and elastin cross-linkage, 46 and perform an essential role in the formation of connective tissue. 47 LOX, which is expressed during wound healing, is, therefore, associated with tissue repair and induces fibroblast production in granulomatous tissue.48,49 As a result, it seems that the increased expressions of HAS and LOX family genes by hMSCs may positively influence the wound-healing process.
Interestingly, the adipogenic differentiation, as reflected by the mRNA of expressions of PPARγ2, increased explosively in WSC-treated hBMSCs. This finding is very interesting, as PPARs are one of the well-known wound-repair modulators. 38 In general, the PPAR family, as major regulators of lipid, glucose, and amino-acid metabolism, is involved in the modulation of adipose metabolism and the function of inflammatory and noninflammatory cells. 50 PPARs regulate inflammation to prevent tissue necrosis and chronic oxidative damage, and can partially mitigate or prevent these types of injury. 50 In particular, the PPARγ2 signal subdues the inflammatory reaction by immune cells or macrophages in vitro and has therapeutic effects on inflammatory disease models.50,51 Although the precise mechanism is unclear, it has been suggested that PPARs play a protective role in the wound-healing process, and that MSCs, with their potent immunosuppressive activities, are essential for tissue repair, regeneration, and immunomodulation. 52 We presume that the upregulation of the expression of PPARγ2 in hMSCs after WSC treatment will provide a beneficial effect in clinical wound healing, with increased ECM formation; however, further studies with a defined study model are required to confirm this.
hMSCs are known to be multipotent and can differentiate into multiple lineages in response to appropriate signals, 53 and as a consequence these cells have been intensively investigated for potential applications in cell therapy, wound healing, and regenerative medicine.54–56 A common assumption is that all hMSCs are similar, regardless of their original tissue source,57,58 and that they can be utilized for any type of tissue regeneration. However, their cellular and regenerative characteristics appear to differ according to the source tissue. 59 Moreover, this study has demonstrated that two types of hMSC, hBMSCs and hPDLSCs, can be distinguished from each other according to their cell proliferation characteristics, collagen formation, and related mRNA expressions. These differences in the phenotypes and functions of different MSCs types will greatly influence the outcome of regenerated tissue products in clinical applications. 60 Thus, it is important to select the most suitable cell source according to the type of tissue required.
In summary, treating hBMSCs and hPDLSCs with WSC significantly enhanced the activity of the hMSCs and increased their collagen tissue regeneration both in vitro and in vivo. The two types of cell responded similarly, but there were significant differences with regard to the degree of soluble collagen formation and mRNA expressions, which suggest that the selection of cells for regeneration therapy should be purpose driven and tissue specific.
Footnotes
Acknowledgments
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (No. 2012M3A9C6049862) and by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2012R1A2A4A01007124).
Disclosure Statement
No competing financial interests exist.