
Abstract
The limited regenerative capacity of articular cartilage contributes to progressive joint dysfunction associated with cartilage injury or osteoarthritis. Cartilage tissue engineering seeks to provide a biological substitute for repairing damaged or diseased cartilage, but requires a cell source with the capacity for extensive expansion without loss of chondrogenic potential. In this study, we hypothesized that decreased expression of the cell cycle inhibitor p21 would enhance the proliferative and chondrogenic potential of differentiated induced pluripotent stem cells (iPSCs). Murine iPSCs were directed to differentiate toward the chondrogenic lineage with an established protocol and then engineered to express a short hairpin RNA (shRNA) to reduce the expression of p21. Cells expressing the p21 shRNA demonstrated higher proliferative potential during monolayer expansion and increased synthesis of glycosaminoglycans (GAGs) in pellet cultures. Furthermore, these cells could be expanded ∼150-fold over three additional passages without a reduction in the subsequent production of GAGs, while control cells showed reduced potential for GAG synthesis with three additional passages. In pellets from extensively passaged cells, knockdown of p21 attenuated the sharp decrease in cell number that occurred in control cells, and immunohistochemical analysis showed that p21 knockdown limited the production of type I and type X collagen while maintaining synthesis of cartilage-specific type II collagen. These findings suggest that manipulating the cell cycle can augment the monolayer expansion and preserve the chondrogenic capacity of differentiated iPSCs, providing a strategy for enhancing iPSC-based cartilage tissue engineering.
Introduction
A
A major obstacle to using many of the proposed cell types for treating cartilage injury is the loss of chondrogenic capacity with monolayer cell expansion. Expansion is required to achieve necessary cell numbers for autologous chondrocyte implantation (ACI), 13 but primary chondrocytes rapidly progress to a de-differentiated phenotype during monolayer culture.14–16 Under certain circumstances, expanded chondrocytes can be grown in three-dimensional (3D) culture with defined conditions to promote redifferentiation to a chondrocyte phenotype, 17 although these cells may not regain the ability to synthesize sufficient matrix. 18 Certain adult stem cells such as MSCs also demonstrate a limited capacity for expansion before loss of chondrogenic potential, 19 whereas other cell types such as ASCs retain chondrogenic ability even after numerous passages. 20 Even iPSCs, which have virtually unlimited self-renewal capability in the undifferentiated state, exhibit a loss of chondrogenic potential with expansion once they have been differentiated toward the chondrogenic lineage. 21
Among the factors that influence the phenotypic change associated with prolonged culture are cell cycle inhibitors such as p21Waf1/Cip1 (hereafter referred to as p21). 22 p21 regulates proliferation by binding cyclin and cyclin-dependent kinase complexes and preventing G0/G1 and G1/S phase progression, 23 and a reduction of p21 levels is a shared mechanism by which growth factor treatment and hypoxic culture mediate enhanced proliferation of MSCs while maintaining differentiation potential.24–26 Evidence from mouse strains with enhanced healing capabilities support these findings, as reduced levels or a complete loss of p21 expression results in increased cell proliferation and recapitulation of native tissue architecture after injury. 27 Thus, the modulation of p21 provides a potential mechanism that could be used to prevent the loss of chondrogenic potential during extensive cell expansion.
Chondrocytes display very limited proliferation during normal tissue homeostasis, but immature growth plate chondrocytes undergo a phase of both proliferation and abundant matrix synthesis. 28 Studies on the chondrogenesis of MSCs support the concept of coordinated cell growth and matrix synthesis, suggesting that proliferation may be important to recapitulate the developmental paradigms of cartilage. 29 We hypothesized that knockdown of p21 expression in iPSC-derived chondrocytes would lead to increased cell proliferation in monolayer expansion while maintaining robust chondrogenic potential. To test this hypothesis, we used short hairpin RNA (shRNA) to silence the expression of the cell cycle inhibitor p21 in differentiated iPSCs and investigated the proliferative capacity and potential for utilizing these cells as a source for cartilage tissue engineering.
Materials and Methods
iPSC culture and differentiation
Murine iPSCs were derived and differentiated toward the chondrogenic lineage as previously described
21
and outlined in Figure 1. Briefly, pluripotency was initiated through the doxycycline-inducible expression of Oct4 (Pou5f1), Sox2, Klf4, and c-Myc.
30
iPSCs were maintained in an undifferentiated state through culture on mouse embryonic fibroblasts (Millipore) in the presence of 20% fetal bovine serum (FBS; Atlanta Biologicals) and mouse leukemia inhibitory factor (Millipore). Cells were differentiated toward the chondrogenic lineage using a multi-step process that resulted in a purified population of iPSC-derived chondrocytes. Differentiation included 15 days of high density micromass culture with 50 ng/mL BMP-4 (R&D Systems) and 100 nM dexamethasone (Sigma-Aldrich) provided during days 3–5 only. After micromass culture, chondrocyte-like cells were sorted with flow cytometry based on the presence of green fluorescent protein (GFP) driven by a type II collagen promoter.
31
These cells were expanded in monolayer on gelatin-coated plates at a density of 10,000 cells/cm2 in chondrogenic expansion medium consisting of Dulbecco's Modified Eagle Medium-high glucose (DMEM-HG, Sigma-Aldrich), 10% FBS, 100 nM nonessential amino acids (Gibco), 55 μM β-mercaptoethanol (Gibco), ITS+ (Becton Dickinson), 50 μg/mL
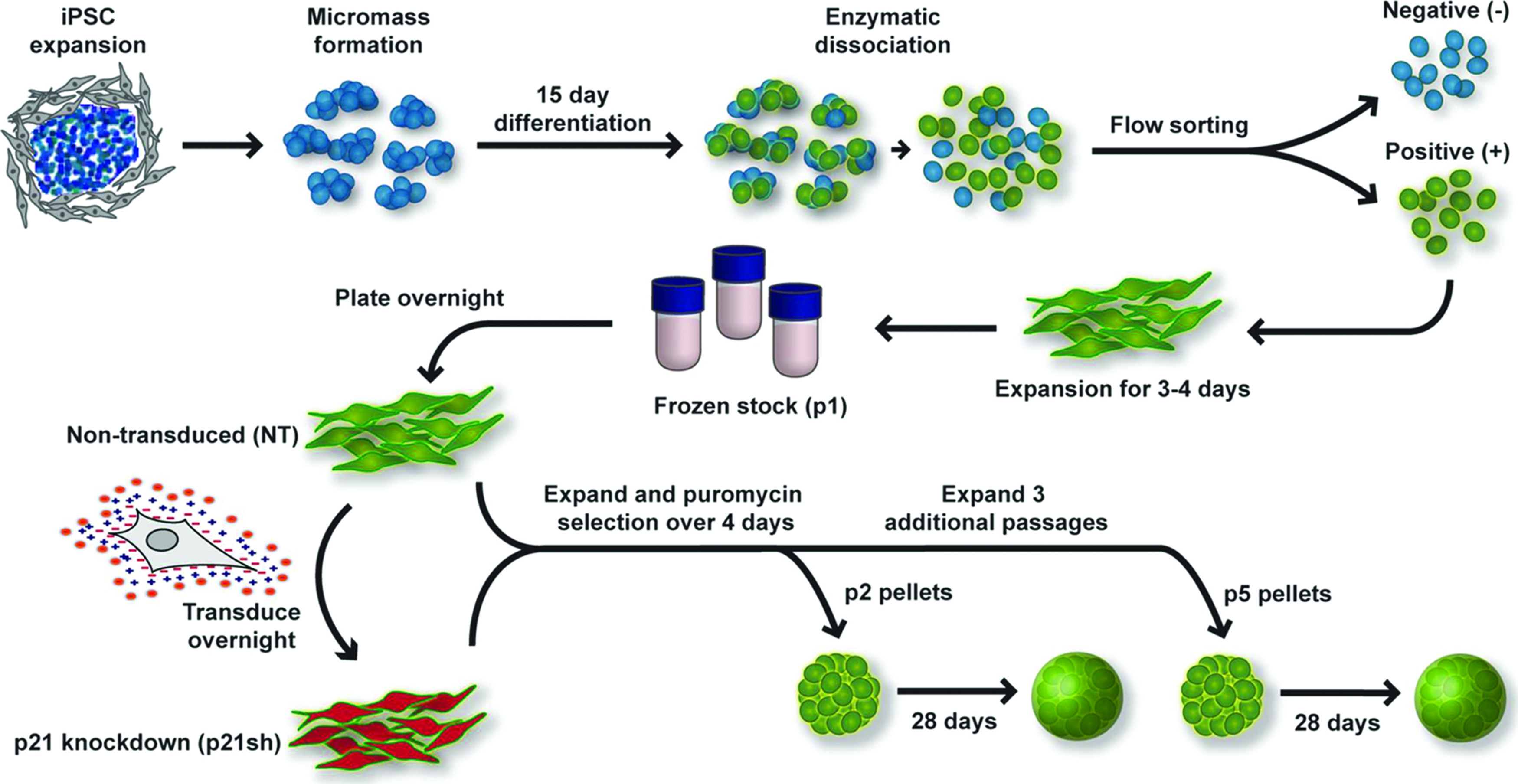
Overview of experimental approach. Undifferentiated induced pluripotent stem cells (iPSCs) were expanded on a feeder layer of mouse embryonic fibroblasts, plated in high density micromass cultures for 15 days of differentiation, which included the growth factor bone morphogenetic protein 4 during days 3–5. After differentiation, chondrocyte-like cells expressing green fluorescent protein (GFP) driven by a type II collagen promoter were sorted by flow cytometry. Sorted cells were expanded in primary passage and then stored as frozen stocks for future experiments. Cells were subsequently plated into passage 1 (p1) and transduced the following day with virus encoding short hairpin RNA (shRNA) to reduce the expression of p21 (p21sh) or kept as nontransduced (NT) cells. Both cell groups were expanded and then trypsinized to obtain passage 2 (p2) cells. Cells were either replated for continued expansion or used for quantitative real-time polymerase chain reaction (qRT-PCR) analysis and the formation of chondrogenic pellet cultures. After three additional passages, the same analysis was performed on passage 5 (p5) cells. See Materials and Methods section for additional details and media formulations. Color images available online at www.liebertpub.com/tea
p21 knockdown with shRNA
shRNA targeting the mouse p21 transcript was expressed in a modified pLVTHM lentiviral vector (Addgene #12447). 32 The GFP reporter in the pLVTHM vector was replaced with a dsRedExpress2-IRES-puromycin resistance cassette using PacI and SpeI restriction sites. shRNA specific to the p21 transcript (p21sh) had the following target sequence: 5′-GGAGCAAAGTGTGCCGTTG-3′ as reported. 33 A vector delivering a “scrambled” shRNA sequence (scr) to control for the effects of the transduction process was also generated using the following target sequence: 5′-GATTACCGTATGGGCTGTC-3′. The scrambled sequence was confirmed to have no homology to the mouse transcriptome by an NCBI BLAST search and contained fewer than 15 base pair identities with any predicted mRNA sequence. To produce vesicular stomatitis virus glycoprotein pseudotyped lentivirus, HEK293T cells were plated at a density of 5.1e 3 cells/cm2 in DMEM-HG (Gibco, cat #11995) supplemented with 10% FBS. The next day, cells in 10-cm plates were co-transfected with the shRNA lentiviral expression plasmid (20 μg), the second-generation packaging plasmid psPAX2 (Addgene #12260, 15 μg), and the envelope plasmid pMD2.G (Addgene #12259, 6 μg) by calcium phosphate precipitation. 34 After 12–14 h, transfection medium was exchanged for 10 mL of fresh 293T medium. Conditioned medium containing lentivirus was collected 24 h after the first media exchange. Lentiviral supernatant was cleared of producer cells by filtration through 0.45 μm cellulose acetate filters and then concentrated 20-fold by centrifugation through a 100 kDa molecular weight cutoff filter (Millipore). Concentrated viral supernatant was then snap-frozen and stored at −80°C for future use. Frozen stocks of passage 1 iPSC-derived chondrocytes were plated into chondrogenic expansion medium and were transduced the following day. For transduction, concentrated viral supernatant containing the p21 shRNA (p21sh) or scrambled sequence (scr) was diluted 1:40 with chondrogenic expansion media. The cationic polymer polybrene was added at a concentration of 4 μg/mL to facilitate transduction. Nontransduced cells did not receive virus but were treated with polybrene as a control. After 16 h of transduction, the medium was exchanged to remove the virus, and 2 μg/mL puromycin was used to initiate selection for transduced cells ∼36 h after transduction. Cells were passaged once they reached ∼90% confluence for further expansion or used for chondrogenic pellet cultures as described below.
Expansion rates and cell cycle analysis
To determine cell expansion rates, cells were seeded at ∼10,500 cells/cm2 (100,000 cells in 9.5 cm2 wells of Corning plates) at passages 2, 3, and 4. Cells were passaged every 3 days and an automated cell counter (Countess; Invitrogen) was used to determine the cumulative fold-expansion for each well over the three passages. Additionally, cells at the end of passage 2 were used to analyze the cell cycle distribution with propidium iodide staining based on a previously described method. 35 Briefly, cells were fixed in cold 70% ethanol and stained with a solution of 0.1% (v/v) Triton X-100 (Sigma-Aldrich), 10 μg/mL propidium iodide (Biolegend), and 100 μg/mL DNase-free RNase A (Sigma-Aldrich) for at least 30 min before analysis by flow cytometry. The percentage of cells in S phase was determined from the region between the two maximal peaks. For comparison, undifferentiated iPSCs and expanded murine chondrocytes isolated from the hips of 12-day-old mice were analyzed in the same fashion.
qRT-PCR
At the end of passages 1 and 4 (denoted as passage 2 and passage 5 to indicate analysis was performed after trypsinization), cells were harvested for total RNA isolation using the RNeasy Plus RNA isolation kit (Qiagen). cDNA synthesis was performed using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Quantitative real-time polymerase chain reaction (qRT-PCR) using SsoFast EvaGreen SYBR Green Supermix was performed with the CFX96 Real-Time PCR Detection System (Bio-Rad) with the oligonucleotide primers reported in Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/tea. The results are expressed as fold-increase mRNA expression of the gene of interest normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh) expression by the ΔΔCt method.
Pellet culture for chondrogenic differentiation
Cells were used to establish chondrogenic pellet cultures at the end of passages 1 and 4 (denoted as passage 2 and passage 5 pellets). The chondrogenic differentiation medium contained the same components as chondrogenic expansion medium but with the addition of 10 ng/mL transforming growth factor β3 (R&D Systems) and 100 nM dexamethasone (Sigma) and without the FBS or bFGF. Cells were distributed to individual wells of 96-well round bottom plates (BD Falcon) with 150,000 cells in each well. Plates were centrifuged twice at 200×g for 5 min, and samples were cultured for up to 28 days with media changes every 2–3 days to maintain 250 μL of media per well. Pellets were harvested at days 9, 18, and 28 for biochemical and histological analyses.
Assessment of proliferation in pellet cultures with BrdU
Separate pellet cultures were incubated with 10 μg/mL of the nucleotide analog 5-bromo-2′-deoxyuridine (BrdU; Sigma) for the duration of days 0–9, 9–18, or 18–28. Pellets were harvested immediately after the end of the treatment window. Cryosections were denatured in 2N HCl for 20 min at room temperature and then washed thrice for 20 min in neutralizing buffer containing 50 mM NaCl and 100 mM Tris–HCl. Samples were blocked for 20 min in phosphate buffered saline (PBS) supplemented with 1% bovine serum albumin, 5% FBS, and 0.1% Triton X-100 (Invitrogen) and then incubated overnight at 4°C in mouse anti-BrdU (Sigma) at 1:1000 in blocking solution. Sections were then labeled with anti-mouse secondary conjugated to AlexaFluor 594 (Molecular Probes) at a 1:500 dilution in blocking serum for 60 min. Sections were incubated with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) before mounting in ProLong Gold Antifade reagent (Life Technologies). Images were analyzed using a custom ImageJ macro. The percentage of BrdU-positive nuclei was calculated by normalizing the number of BrdU-positive particles over a threshold size to the number of DAPI-positive nuclei.
Biochemical analysis for DNA, GAG, and collagen content
Pellets were washed with PBS and stored frozen until digestion with papain for subsequent analysis of DNA and glycosaminoglycan (GAG) content as previously described. 36 Briefly, the PicoGreen assay (Molecular Probes) was used to measure DNA content and GAGs were analyzed using the 1,9-dimethylmethylene blue assay with a 525 nm wavelength. Aliquots of the cell suspension used to establish pellet cultures were harvested at day 0 and analyzed in parallel to provide a reference value for the starting DNA content of each group. Total collagen was determined by measuring the hydroxyproline content using a previously described assay. 37
Histology and immunohistochemistry
Pellets were harvested for cryosectioning by washing with PBS, embedding in optical cutting temperature compound (Tissue-Tek), and snap freezing in liquid nitrogen before storage at −20°C. Cryosections of 8–10 μm were collected on glass slides. For histology, slides were fixed in 10% neutral buffered formalin and then stained with safranin-O/fast-green/hematoxylin as described. 36 For immunohistochemistry, unfixed sections were digested with pepsin for 5 min (10 min for type I collagen), treated with primary antibody (II-II6B3 from Iowa hybridoma bank for type II collagen at a dilution of 1:3, 8D4A1 from Chondrex for type I collagen at 1:200, c7974 from Sigma for type X collagen at 1:200), incubated with secondary antibody for type II and type X (ab97021; AbCam), and stained with Histostain Plus kit and AEC Single (Life Technologies). Appropriate positive and negative controls confirmed specificity of staining.
Western blots
Cells were lysed in 50 mM Tris–Cl (pH 7.4), 150 mM NaCl, 0.5% Triton X-100, and 0.1% sodium dodecyl sulphate (SDS). Total protein was quantified using the BCA assay (Pierce). Lysates were mixed with loading buffer and boiled for 5 min; equal amounts of total protein were run in NuPAGE Novex 10% Bis-Tris polyacrylamide gels (Life Technologies) and transferred to nitrocellulose membranes. Nonspecific antibody binding was blocked with 5% nonfat milk in TBS-T (50 mM Tris, 150 mM NaCl and 0.1% Tween-20) for 30 min. The membranes were then incubated with primary antibody in 5% milk in TBS-T: mouse anti-p21 (clone F-5; Santa Cruz) diluted 1:1000 overnight at 4°C or anti-GAPDH (clone 14C10; Cell Signaling) diluted 1:5000 for 60 min at room temperature. Membranes labeled with primary antibodies were incubated with anti-mouse (Santa Cruz) or anti-rabbit horseradish peroxidase-conjugated antibody (Sigma-Aldrich) diluted 1:5000 for 30 min and washed with TBS-T for 30 min. Membranes were visualized using the Immun-Star WesternC Chemiluminescence Kit (Bio-Rad) and images were captured using a ChemiDoc XRS+ system and processed using ImageLab software (Bio-Rad).
Statistical analysis
Samples were analyzed using student's t-test or two-way ANOVA with Fisher's PLSD post hoc test when applicable (α=0.05).
Results
Knockdown of p21 expression
Expression of the shRNA targeting p21 caused a reduction in p21 mRNA levels of ∼80% as assessed by qRT-PCR (Fig. 2A, p<0.05) and also caused a reduction in p21 protein as determined by western blot (Fig. 2B). This reduction in p21 protein was maintained throughout the 28 days of chondrogenic pellet culture (Fig. 2C). A scrambled shRNA sequence showed a small increase in p21 protein expression (Fig. 2D) and a decrease in the size of chondrogenic pellet cultures (Fig. 2E) compared with nontransduced (NT) cells that was potentially an effect of viral transduction. To ensure that effects of p21 knockdown were significant relative to the unaltered state, subsequent experiments compared differentiated iPSCs with silenced p21 expression (p21sh) to NT cells.
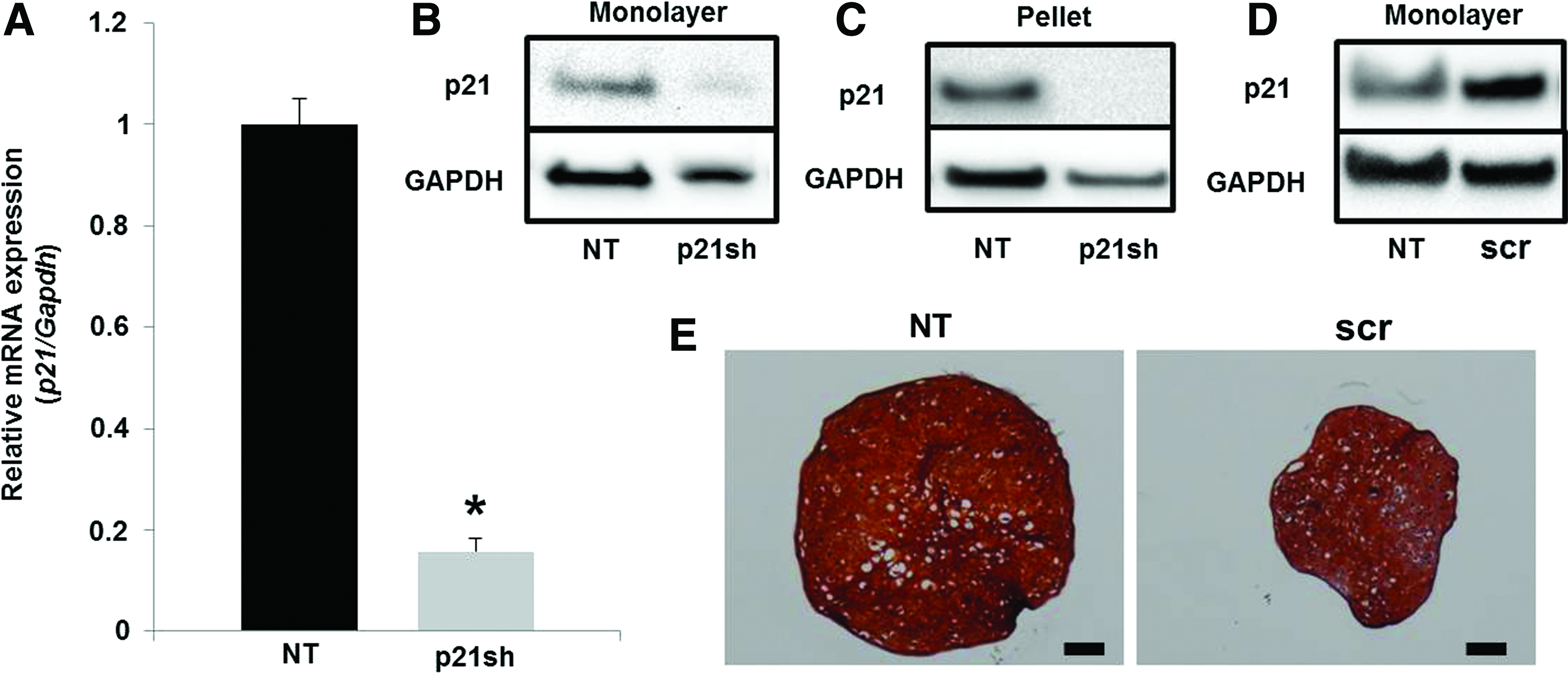
p21 knockdown.
Effect of knockdown on cell expansion
p21sh cells demonstrated an enhanced potential for cell expansion, with approximately double the level of total expansion compared with NT cells over three passages (Fig. 3A, p<0.05). The three additional expansion passages with p21sh cells generated almost 150 times the number of cells available for tissue engineering at passage 5 as compared to passage 2 (Fig. 3A). To determine whether p21 knockdown altered the cell cycle profile, the percentage of cells in S phase was determined with propidium iodide analysis. p21sh cells showed a significant increase in the percentage of cells in S phase compared with NT cells at the same passage (Fig. 3B, p<0.05). Mouse primary chondrocytes and undifferentiated iPSCs of the same line as the differentiated cells were used as controls to establish the dynamic range expected for the assay. With knockdown, iPSC-derived chondrocytes had a slightly higher percentage of cells in S phase compared with mouse chondrocytes (Fig. 3B, p<0.05), but without knockdown this percentage was lower than mouse chondrocytes (Fig. 3B, p<0.05). All iPSC-derived chondrocytes showed an S phase percentage that was more similar to chondrocytes than to undifferentiated iPSCs, nearly half of which were in S phase.
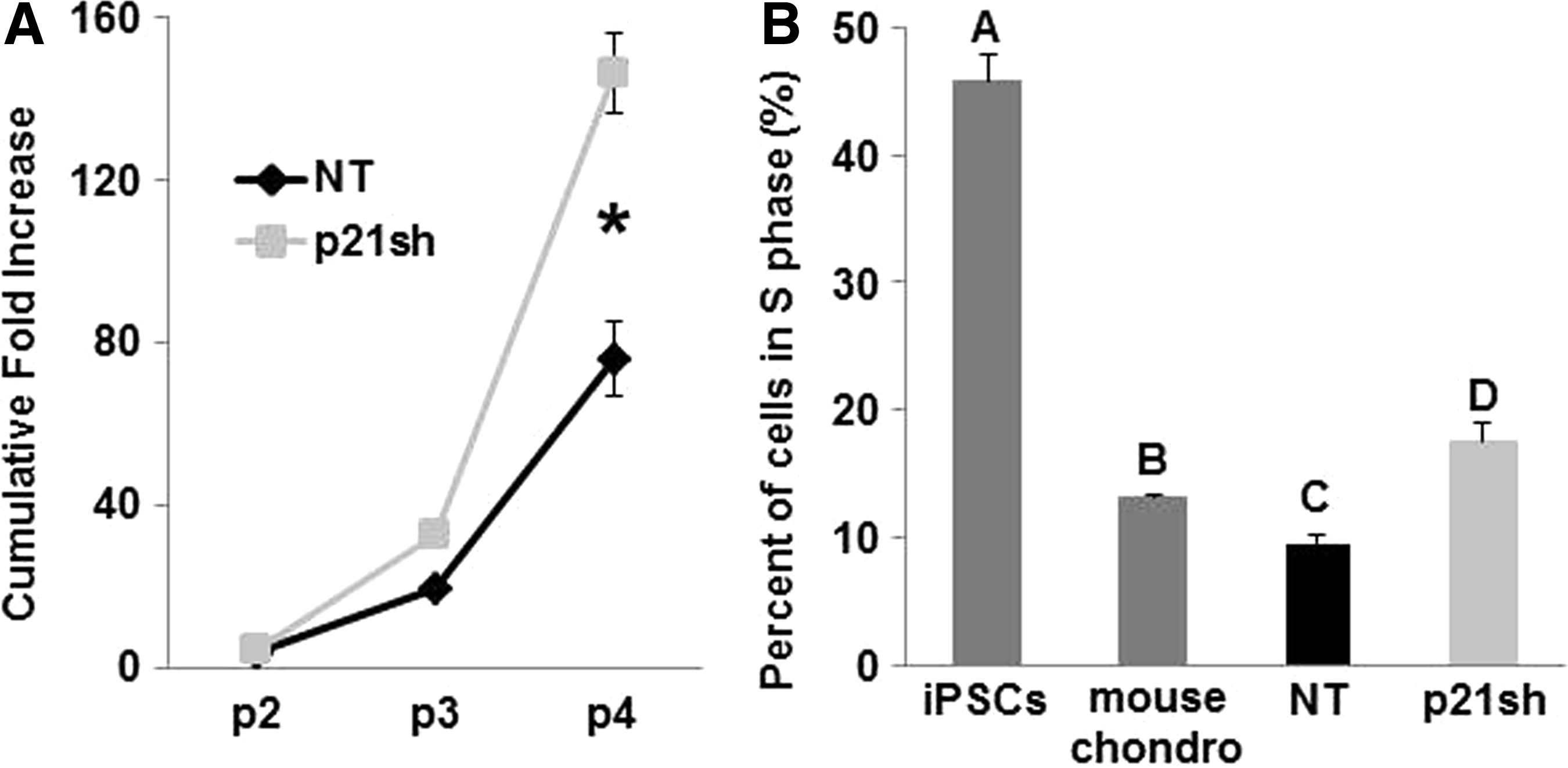
Cell expansion.
Gene expression during monolayer expansion
Analysis of gene expression with qRT-PCR showed that knockdown of Cdkn1a (p21) expression was maintained through passage 5 (Fig. 4, p<0.05). This effect appears to be specific to p21, as a cell cycle inhibitor from a different family, Ink4a (p16), did not show reduced expression (Fig. 4, p>0.05). In NT cells, the expression of p21 decreased (p<0.05) and p16 was unaffected (p>0.05) with continued passaging, suggesting that iPSC-derived chondrocytes do not upregulate G1/S phase cell cycle inhibitors under the expansion conditions used in this study. The chondrogenic markers type II collagen (Col2a1) and aggrecan (Acan) decreased with monolayer expansion (p<0.05), as expected, and p21 knockdown did not preserve expression of these chondrogenic markers (Fig. 4). The expression of collagens that are not typically expressed by chondrocytes, types I (Col1a1) and X (Col10a1), were also monitored over passage. Knockdown of p21 significantly reduced the expression of Col1a1 at passage 2 (p<0.05), and caused nearly a three-fold reduction in Col10a1 at passage 5 (p<0.05).
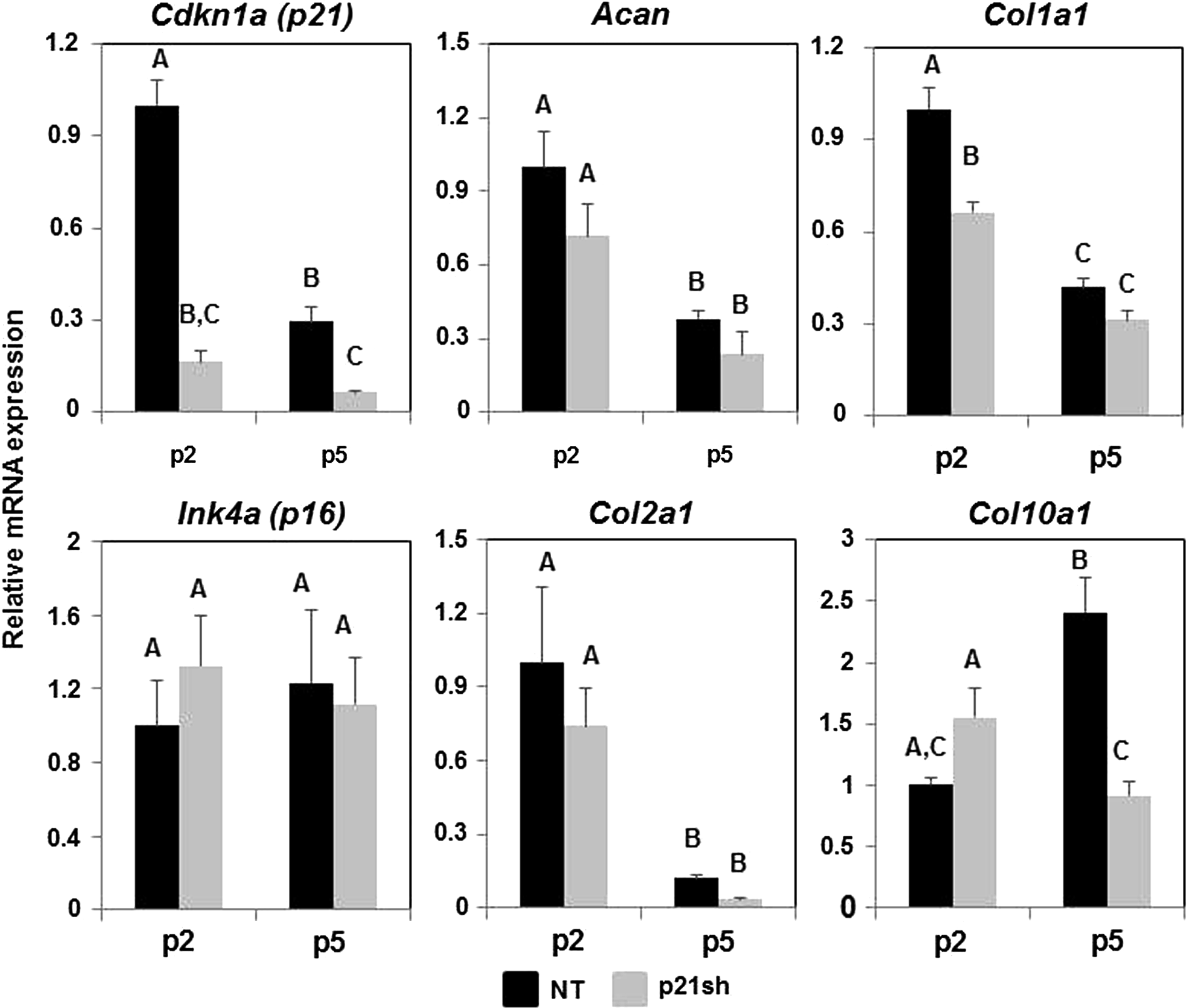
qRT-PCR during cell expansion. mRNA expression of cells either NT (black bars) or expressing p21 shRNA (p21sh, gray bars) was analyzed at passages 2 and 5. Gene expression was normalized to GAPDH and to passage 2 NT cells. Data from n=6 total combined from two independent cell preparations. Groups not sharing a letter indicate p<0.05.
DNA synthesis and cell content during pellet culture
To determine whether the knockdown of p21 affected the synthesis of new DNA under chondrogenic differentiation conditions, we analyzed the incorporation of BrdU into newly replicated DNA during distinct time periods. When BrdU was provided from the time of pellet formation until day 9, pellets from p5 p21sh cells had an increased percentage of BrdU-positive cells compared with NT cells (representative images in Fig. 5A, quantitative results in 5B, p<0.05). When BrdU was added during days 9–18 or 18–28 of culture, BrdU analysis indicated less proliferation in pellets from p5 p21sh cells compared with days 0–9 (p<0.05), and no significant difference to corresponding NT pellets was found at later time points (p>0.05). Additionally, BrdU incorporation showed no significant differences with time in pellets from p5 NT cells (p>0.05). The BrdU analysis was corroborated by measurements of total DNA content, which was significantly higher in p21sh cells compared with NT cells at day 9 in both p2 and p5 pellets (Fig. 5C, p<0.05). For both p2 and p5 cells, the difference between DNA content in p21sh and NT pellets was maintained at the days 18 and 28 time points (p<0.05), but DNA content decreased in p21sh pellets at day 28 compared with day 9 (p<0.05). Together, these results indicate that p21 knockdown increases the total cells present during the earlier stages of pellet culture. However, DNA synthesis is slowed and cell loss eventually occurs in later stage pellet cultures even with p21 knockdown.
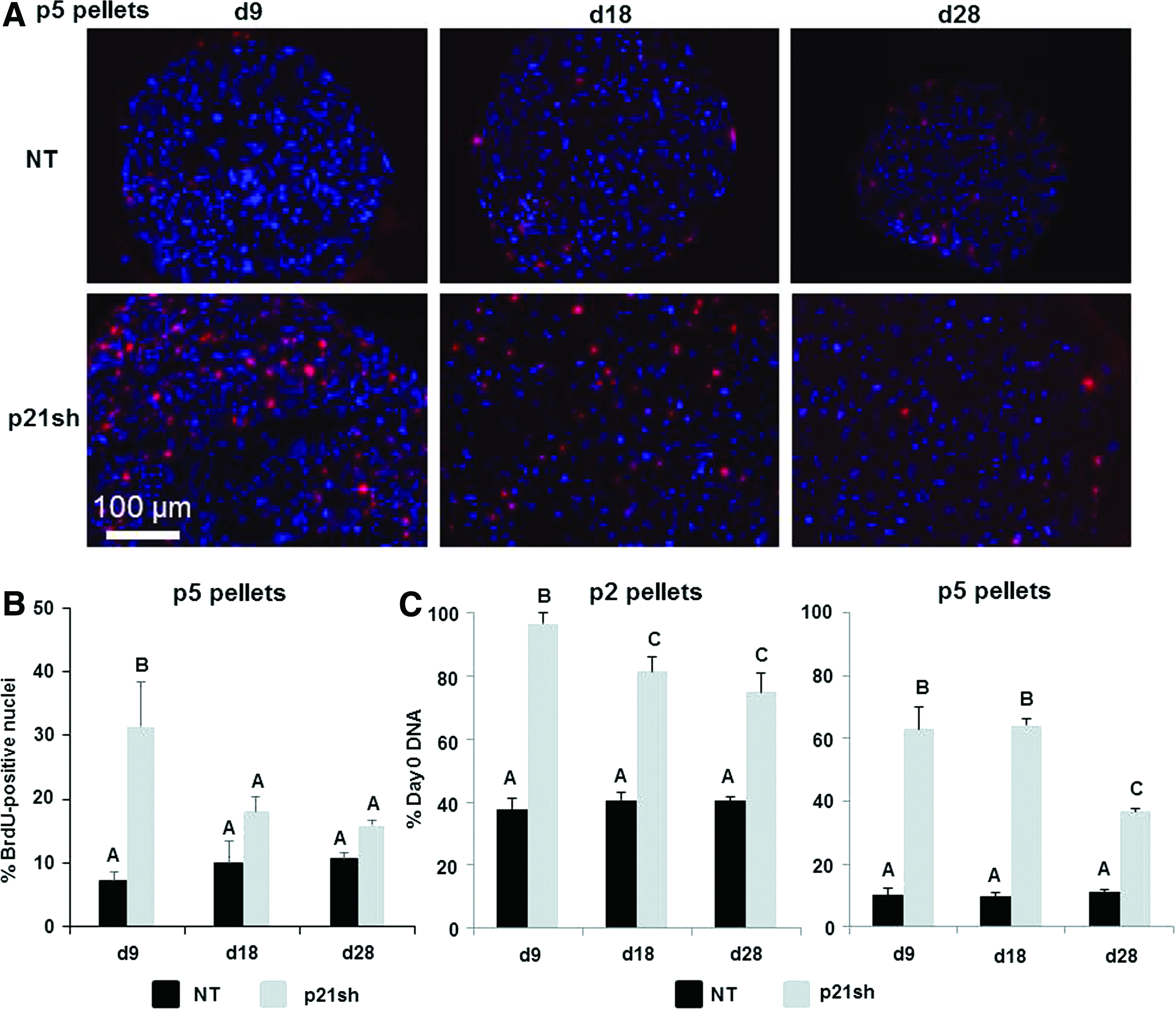
BrdU incorporation during chondrogenic pellet culture.
GAG production
Pellets made with passage 2 NT cells and pellets made with passage 2 p21sh cells contained sulfated GAGs that stained positive for safranin-O, although p21 knockdown enhanced staining at the day 9 time point (Fig. 6A). Pellets made from passage 5 NT cells were small and showed minimal staining, while pellets made from passage 5 p21sh cells were larger and showed significant GAG staining (Fig. 6B). Pellets from passage 2 p21sh cells demonstrated a higher GAG content than pellets from passage 2 NT cells, as assessed via the DMMB assay (Fig. 7A, p<0.05). The higher total GAG content observed in p21sh pellets at the day 18 and 28 time points was likely due to higher cell content, as GAG/DNA levels were similar in the NT and p21sh groups at those latter time points (p>0.05). However, GAG/DNA values at day 9 showed that p21 knockdown did result in greater synthesis of GAGs on a per cell basis in pellets from passage 2 cells (p<0.05). In pellets made with passage 5 cells, p21 knockdown had a dramatic effect on both total GAG and GAG/DNA at all time points (Fig. 7B, p<0.05). The pellets made with passage 5 NT cells showed minimal GAG production compared with pellets made with passage 2 NT cells (p<0.05). In contrast, pellets made with passage 5 p21sh cells showed total GAG and GAG/DNA values that were even higher than levels in pellets made with passage 2 p21sh cells (Fig. 7A vs. Fig. 7B). Together, these data indicate that p21 knockdown maintains the potential for GAG production with extended passaging.

Histology for GAGs.
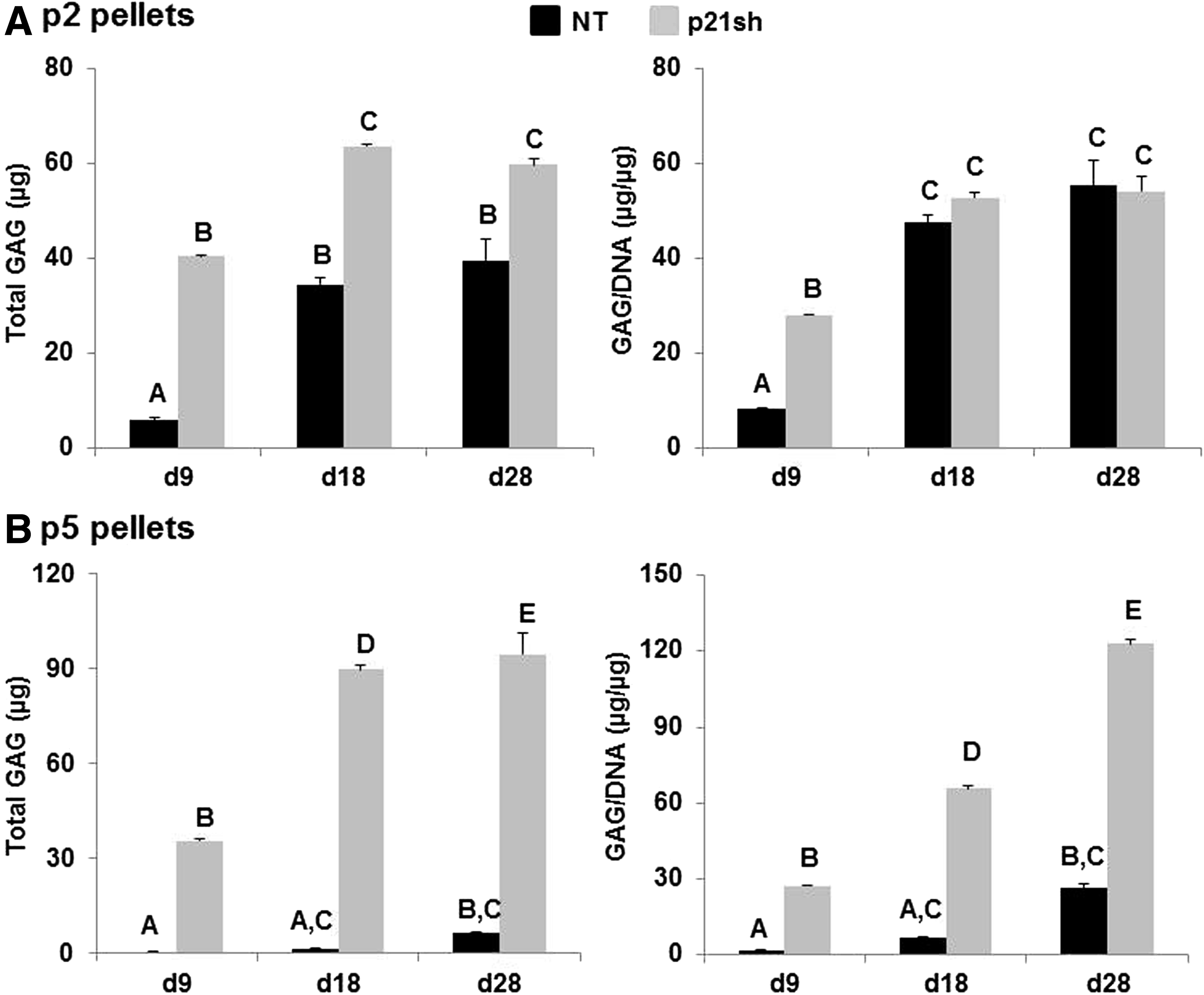
Quantification of GAG content.
Collagen production
Total collagen content was assessed quantitatively with the hydroxyproline assay, while immunohistochemical labeling for types I, II, and X collagen was used to determine which collagen proteins were produced. Total collagen in the pellet was increased in passage 2 p21sh cells compared with NT cells, but p21 knockdown did not alter the amount of collagen/DNA (Fig. 8, p>0.05). At passage 5, pellets from p21sh cells had higher total collagen at all time points but lower collagen/DNA at days 9 and 18 (p<0.05). Staining for cartilage-specific type II collagen was present in all pellets made from passage 2 cells, even at the day 9 time point (Fig. 9A). Type II collagen labeling was less intense for pellets made from passage 5 NT cells, especially at the day 9 time point, but pellets from passage 5 p21sh cells retained robust staining at all time points (Fig. 9B). Additional labeling was used to assess the relative contribution of collagens indicative of either a fibrocartilage (type I) or the hypertrophic chondrocyte (type X) phenotype. Pellets did not label for the presence of type I collagen except in passage 5 pellets made from NT cells, which showed labeling at the periphery and in the center of the pellets at all time points (Fig. 10B). The presence of type X collagen was investigated at day 28 and NT pellets showed enhanced labeling as compared to pellets from p21sh cells at both passages, with greater labeling at passage 5 (Fig. 10C). The immunohistochemistry findings showed that the predominant collagen in all pellets was type II collagen, with some contribution of types I and X collagen in p5 pellets.

Quantification of total collagen content.
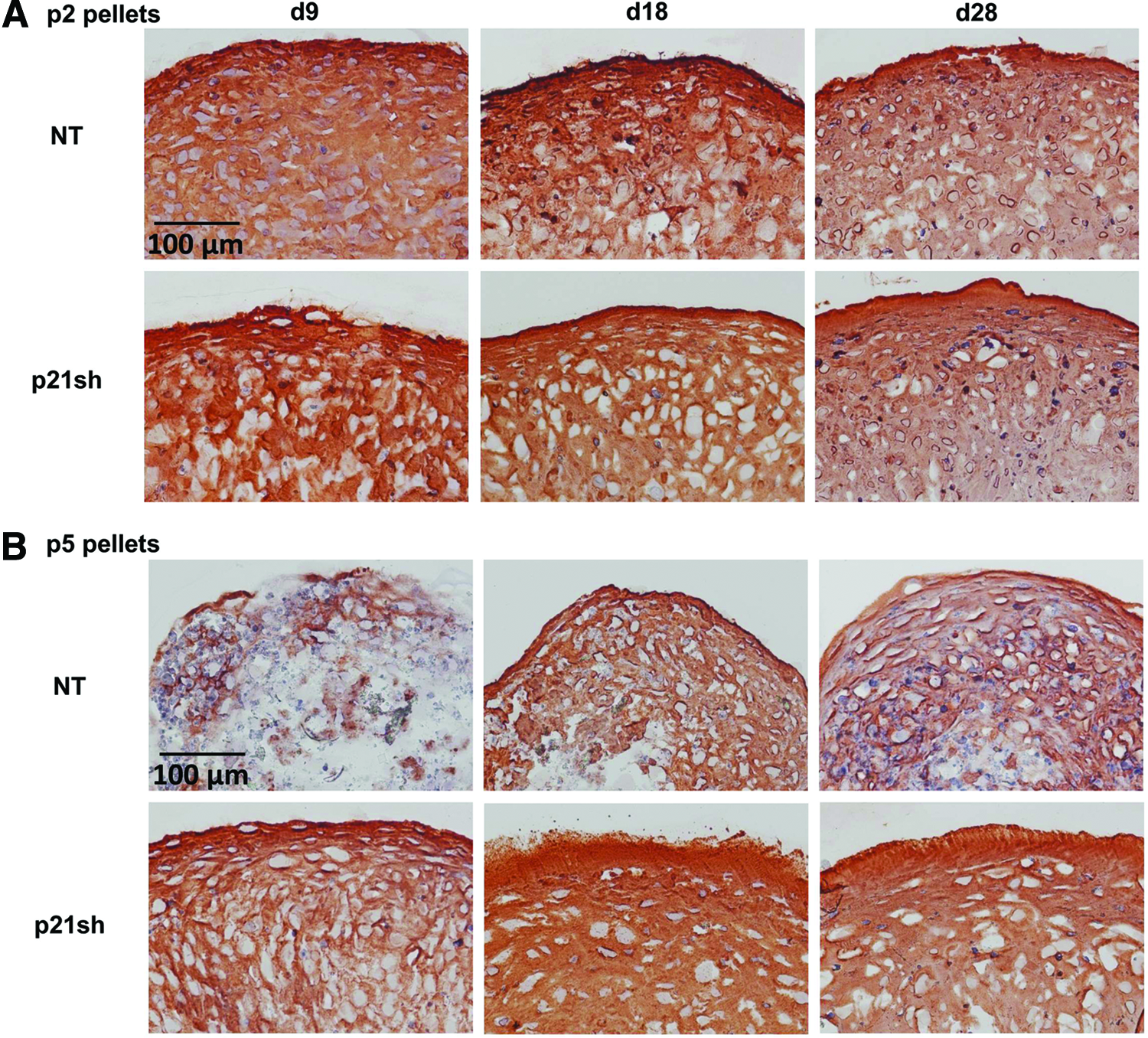
Immunohistochemistry for type II collagen.

Immunohistochemistry for type I and type X collagen.
Discussion
Cartilage tissue engineering necessitates a cell source that can maintain chondrogenic potential even after the monolayer expansion that is often required to provide sufficient cell quantities. We showed that knockdown of the cell cycle inhibitor p21 preserved the potential of iPSC-derived chondrocytes to synthesize GAGs after three additional passages that increased the cell number by nearly 150-fold. Furthermore, p21 knockdown allowed the cells to remain in the cell cycle during initial stages of chondrogenic differentiation in 3D culture without sacrificing matrix synthesis. These results support the feasibility of an approach for generating a sufficient supply of chondrogenic cells for tissue engineering strategies: (1) iPSCs provide a self-renewing starting cell population that is genetically matched to the donor without an invasive tissue harvest; (2) multi-step differentiation and purification protocols provide an intermediate cell stock that can undergo quality control characterization; (3) p21 knockdown can be utilized for significant monolayer expansion and continued proliferation without a loss of GAG synthesis in a 3D engineered construct.
The most dramatic effect of p21 knockdown in this study was the preservation of chondrogenic potential over extensive passaging. While undifferentiated iPSCs show virtually unlimited expansion potential, our previous studies indicated that twice-passaged iPSC-derived chondrocytes generated the most robust cartilage matrix in this system, 21 and this study confirmed that pellets from passage 5 cells had reduced production of GAGs and decreased staining for type II collagen. However, p21 knockdown during expansion eliminated the loss in subsequent GAG production, with even higher levels of total GAG and GAG per cell from passage 5 p21sh cells compared with pellets from passage 2 cells with or without p21 knockdown. Our findings that altering p21 levels affects the expansion rates and GAG production of iPSC-derived chondrocytes are consistent with previous results in human MSCs showing that reduced p21 expression through the use of hypoxia, growth factors, or siRNA can increase both proliferative and differentiation potential.24,25,38
An important result of p21 silencing was attenuation of the sharp decrease in cell number associated with early stages of pellet culture in control cells, a phenomenon that also occurs in other cell types. 29 BrdU staining indicated that increased DNA synthesis was one factor in the higher DNA content of pellets from p5 p21sh cells compared with controls, but silencing p21 may also have played a role in modulating cell survival. It is also important to note that DNA synthesis did decrease in p21sh cells after the first 9 days of pellet culture and that cell loss did occur in this group by day 28. Cell density is an important variable in the process of chondrogenesis for a variety of cell types due to signaling pathways active in cartilage development (reviewed in Bobick et al. 39 ), and it is possible that the effects of p21 knockdown on GAG synthesis were at least partially due to altering the cellular dynamics during early pellet culture. Further investigation using hydrogels that can be modified to isolate variables such as cell density and cell–matrix interactions would be valuable in pursuing this question. 40
Upregulation of cell cycle inhibitors during in vitro expansion is believed to contribute to cellular senescence,25,41 and levels of p21 expression are involved in guiding cell fate decisions regarding cell cycle reentry, temporary exit, or permanent exit associated with senescence.22,42 The role for p21 in mediating the cell cycle during chondrogenic differentiation is supported by studies with mouse chondroprogenitor lines43–45 and from observations of developmental abnormalities related to cartilage growth. 46 Interestingly, we showed dramatic effects of p21 knockdown on chondrogenesis despite the observation that NT iPSC-derived chondrocytes continue to expand in monolayer and the mRNA expression of p21 or p16Ink4a did not increase with passage. Furthermore, p21 knockdown preserved GAG synthesis in subsequent pellet culture without preventing the loss of chondrogenic gene expression during monolayer expansion, as Col2a1 and Acan decreased similarly with further expansion in both groups. This observation is similar to experiments with human articular chondrocytes that show bFGF treatment does not prevent de-differentiation in culture but does enhance the ability of cells to redifferentiate when placed in appropriate conditions.47–50 These data suggest that p21 may interact with one of the many signaling pathways controlled by cell–matrix interactions during the chondrogenesis of stem cells. 51 The contrast between the effects of p21 knockdown during two-dimensional and 3D culture extends to the regulation of collagens associated with fibroblastic (type I) and hypertrophic chondrocyte (type X) phenotypes, as knockdown resulted in modest changes in monolayer gene expression but did alter the degree of immunohistochemical staining of these collagen types in p5 pellets. Additional studies are needed to define the molecular pathways by which reduced p21 levels specifically modulate chondrogenic differentiation and to determine how a single factor regulates both cell expansion and the capacity for subsequent matrix synthesis.
The use of a cell source that can be significantly expanded before subsequent differentiation presents new possibilities compared to differentiated cells or adult stem cells. One such possibility is generating individual lots of chondrogenic cells that could serve a large number of patients, with the advantage of reduced cost in terms of lot characterization. Notably, this strategy is consistent with an emerging concept that the clinical application of iPSCs will benefit from proliferative intermediate cell populations that can be frozen and stored to allow for quality control of every lot. 52 The feasibility of using allogeneic adult stem cell transplantation for musculoskeletal applications has been demonstrated in clinical trials, 53 but using iPSC-derived chondrocytes with appropriate expansion would allow for larger lots that could be immunologically matched to a significant percentage of the population. 54 A second possibility is obtaining sufficient cell numbers to move from developing tissue engineering solutions for cartilage defects to whole joint resurfacing strategies for patients with widespread OA. The average defect in ACI procedures is ∼5 cm2 and requires ∼12 million cells,13,55 but the restoration of an entire joint surface would most likely involve a large scaffold seeded at high density and would therefore require significantly more cells. 6 The features of continued cell expansion in a 3D context and rapid matrix production indicate that this cell source may be particularly well suited for use with scaffolds designed to limit or eliminate in vitro culture before implantation.56,57 Furthermore, iPSCs can be derived from OA patients58,59 and thus allow for autologous approaches to treatment of widespread joint disease.
While iPSCs have several advantages as a starting cell source, 12 the manner in which pluripotent cell lines are derived affects the potential for uncontrolled growth and some established lines display tumorigenic potential during chondrogenic differentiation. 60 Efforts to derive iPSCs from adult chondrocytes using nonintegrating methods such as mRNA delivery may address some of these concerns, 61 and methods to isolate successfully differentiated cells can add additional control over the phenotype of cells transplanted for therapy.62,63 Indeed, the multi-step differentiation strategy employed in this study uses positive selection with a Col2 reporter as an additional safeguard to exclude nondifferentiated iPSCs. The paradigm of producing differentiated cell types from human iPSCs for clinical applications is being established in the use of retinal pigment epithelium for age-related macular degeneration, 64 with encouraging preclinical safety profiles being used as the basis for clinical trials.
One potential concern with utilizing the knockdown of a cell cycle inhibitor is that this approach may increase the oncogenic risk, as spontaneous tumorigenesis has an incidence of 40% at 16 months in mice with germline knockouts of p21. 65 However, reduced p21 levels alone have not been associated with increased formation of tumors of mesenchymal origin65–67 and germline knockouts of p21 demonstrate increased tumor latency compared with similar studies performed with germline knockouts of p53 or p16Ink4a.68,69 Importantly, our study shows that the growth advantage conferred by reducing p21 expression is mostly present during monolayer expansion and lessens with time during chondrogenic pellet culture. This knowledge should help inform future strategies that may build on this work by using transient means to alter p21 levels, such as the development of small molecule inhibitors to temporarily block p21 activity. Systems designed for temporary reduction of p21 levels would alleviate concerns that p21 silencing may increase the risk of tumor formation. The use of means other than viral transduction to manipulate the cell cycle would also avoid effects of the transduction process itself, as we noted an increase in protein levels of p21 and reduced chondrogenesis after treatment with a scrambled shRNA sequence that has no homology to the mouse transcriptome.
Conclusions
This study demonstrates that the cell cycle inhibitor p21 can be targeted to augment the proliferation and tissue production by chondrogenically differentiated iPSCs. Silencing p21 with RNA interference resulted in higher monolayer expansion rates of differentiated iPSCs and limited the extent of cell loss that occurred during early cartilage pellet culture. Unlike control cells, iPSCs with reduced p21 levels maintained the ability to synthesize a high level of GAGs even after extensive monolayer expansion. These findings provide a potential strategy for enhancing the regenerative capability of donor cell sources for cartilage tissue engineering strategies.
Footnotes
Acknowledgments
Supported by the National Institutes of Health Grants OD008586, AR061042, AR50245, AR48852, AG15768, AG46927, AR48182, and AR065956; NSF grant CBET-1151035; the Nancy Taylor Foundation for Chronic Diseases; the Collaborative Research Center, AO Foundation, Davos, Switzerland,; the AOSpine Foundation; and the Arthritis Foundation. Training was supported by a National Science Foundation Graduate Research Fellowship (B.O.D., P.I.T.); an American Heart Association Predoctoral Fellowship (P.I.T.); NIH T32AI007217 (V.P.W.); an Arthritis Foundation Postdoctoral Fellowship (V.P.W.); and the Flight Attendant Medical Research Institute (N.C.).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.