
Abstract
Extensive investment on research and development of cell therapy products (CTPs) and tissue-engineered products (TEPs) has been made in Korea, and various commercial products are born in market. The Ministry of Food and Drug Safety (MFDS) in Korea regulates CTPs and TEPs as biological products under the authority of the Pharmaceutical Affairs Act. The Korean MFDS approved 16 CTPs and 4 stem CTPs and authorized 135 clinical trials, including 60 sponsor-investigator trials. Currently, the advent of stem cell technology and new biomaterials gives an impetus to develop more innovative CTPs and TEPs to treat intractable and serious diseases. This article deals about the regulatory process for approving CTPs and TEPs in Korea. Regulatory policies of the MFDS for supporting the development of novel products and ensuring the safety of the CTPs and TEPs are reviewed.
Biopharmaceutical-Related Offices in the Ministry of Food and Drug Safety
I
Figure 1 shows biopharmaceutical-related offices in the MFDS. In the Headquarter, the Biopharmaceutical Policy Division and the Quality Management Division are responsible for policy development, safety evaluation, and safety management after marketing. In the NIFDS, Cell and Gene Therapy Products Division is responsible for the review of quality, safety and efficacy of data, and guideline development. Six regional offices coordinate to take MFDS responsibility to regulate importing, marketing, and distributing the approved products.
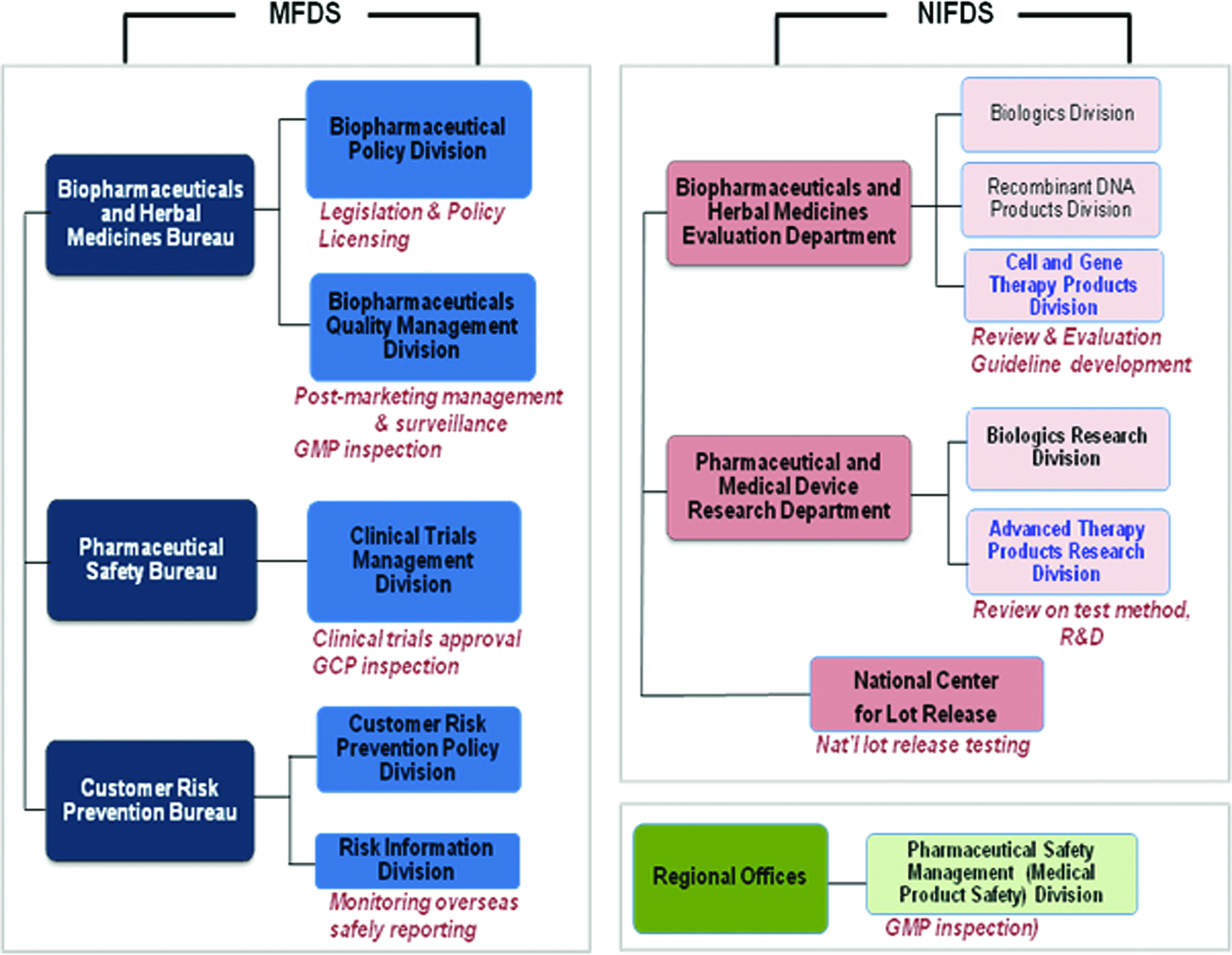
Biopharmaceutical-related offices in the Korean Ministry of Food and Drug Safety (MFDS). Color images available online at www.liebertpub.com/tea
Overview of Regulatory System
Well-established regulatory framework for drugs and biopharmaceuticals is applied to control cell therapy products (CTPs) and tissue-engineered products (TEPs), such as other drug and DNA recombinant products. At the stage of development, good clinical practice and good laboratory practice are required for nonclinical and clinical studies, respectively. In addition, the evidences must be fully validated to ensure the quality, safety, and efficacy of the products at the product licensing step. After marketing authorization, the products meet regulatory requirements, including reevaluation and reexamination (Figure 2).
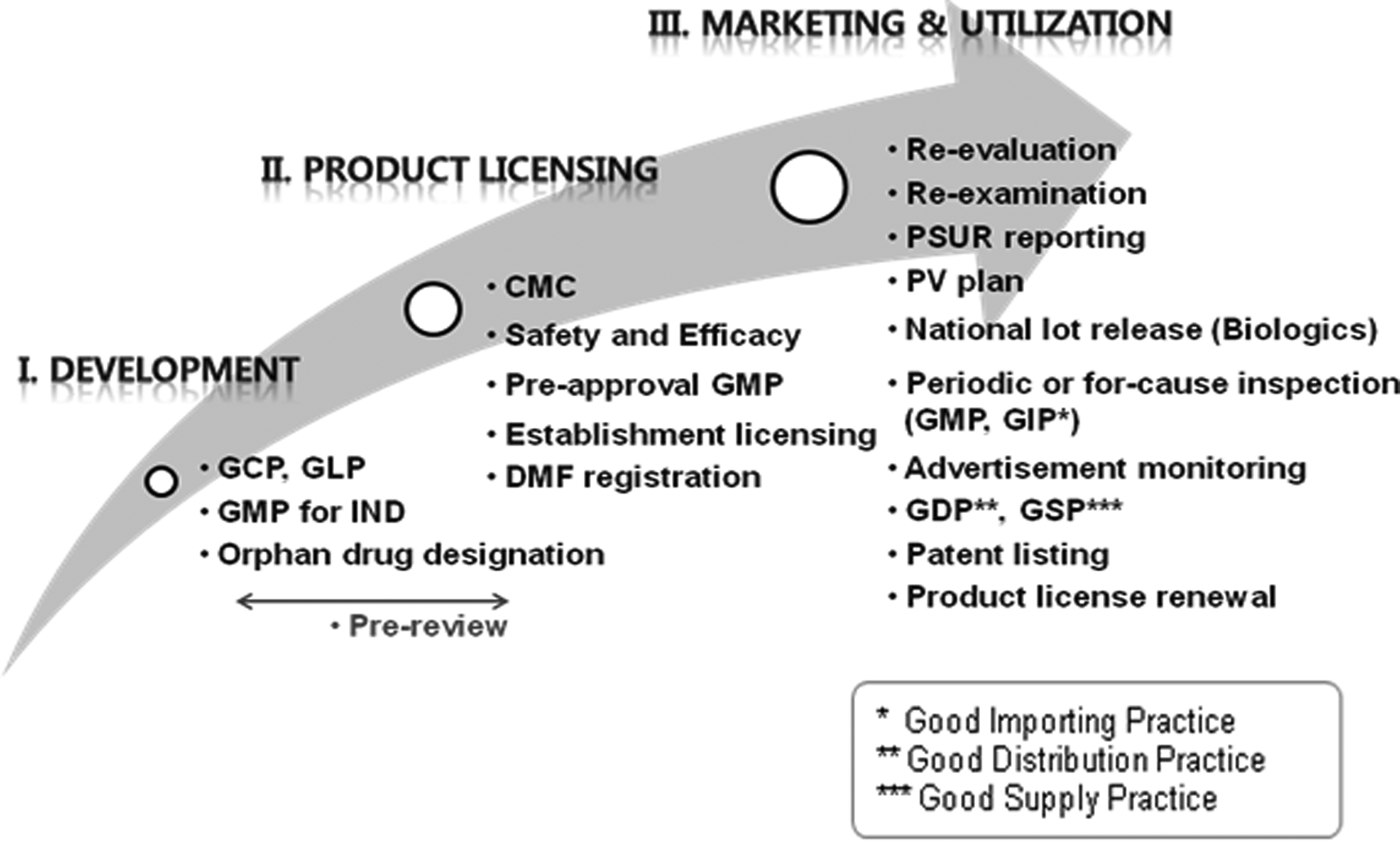
Overview of regulatory system.
Regarding the legislative basis, CTPs and TEPs are regulated under the Pharmaceutical Affairs Act and its Enforcement Regulations. In each phase of development, approval, and postmarketing management, people developing CTPs or TEPs should comply with laws and related regulations as shown in Figure 3.

Legislative basis for cell therapy products (CTPs) and tissue-engineered products (TEPs).
In Korea, CTPs are considered as biologics. The definition of CTPs is “A medicinal product manufactured through physical, chemical, and/or biological manipulation, such as in vitro culture of autologous, allogeneic, or xenogeneic cells.” However, when a medical doctor performs minimal manipulation, which does not cause safety problems such as using autologous or allogeneic cells in the course of surgical operation or treatment at a medical center, it is an exemption case.
Figure 4 shows the regulatory territories for the cell, tissue, and organ. Most of the CTPs except minimal manipulation in medical center are approved under the Pharmaceutical Affairs Act. However, Human Tissue Safety and Control Act is the legislative basis for the human tissues for transplantation. The TEPs, including substantially manipulated cells and scaffolds, are categorized into biologics or medical devices.
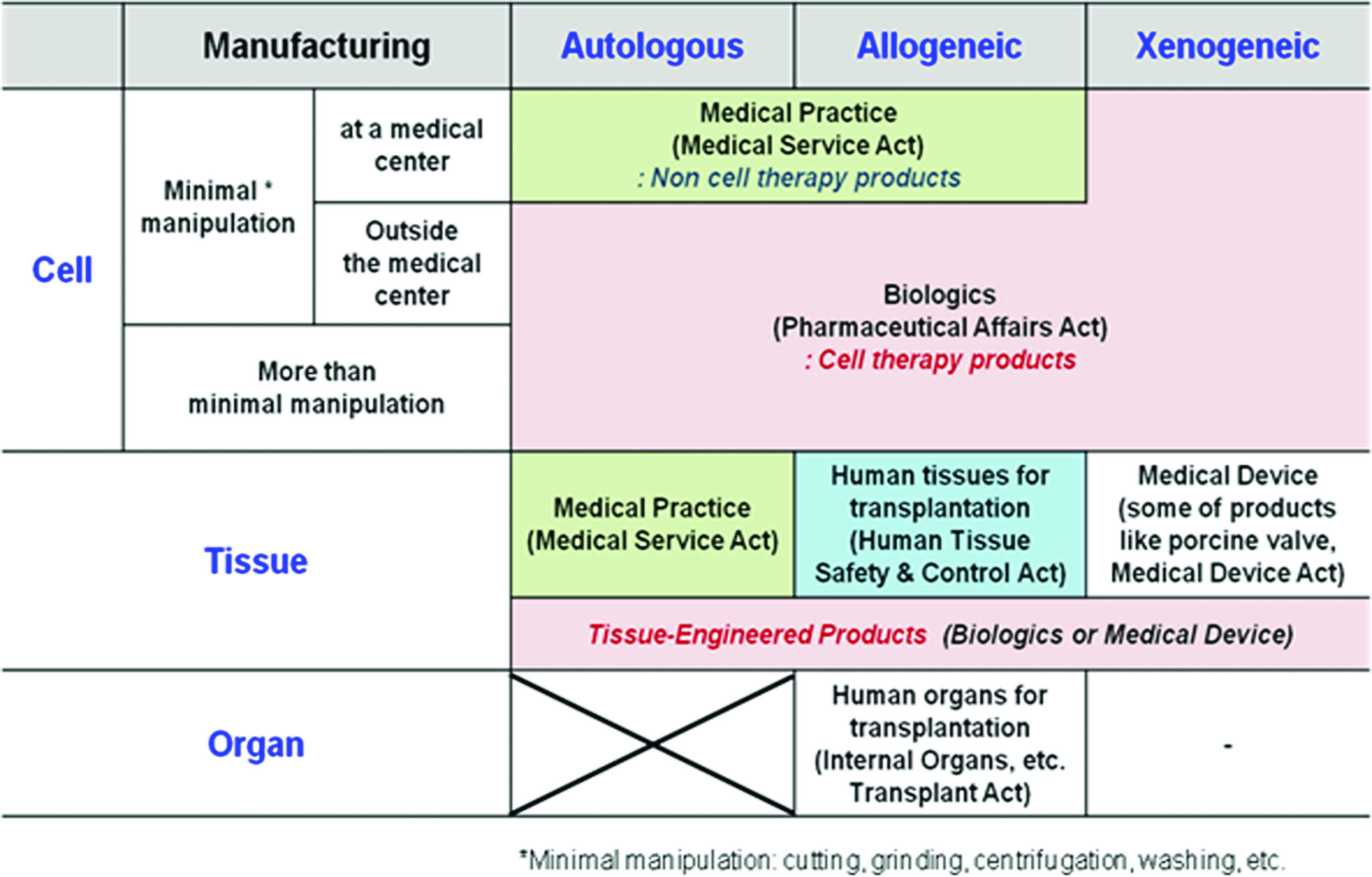
Regulatory territories for cell and tissue. Color images available online at www.liebertpub.com/tea
In the Korean MFDS, there is no regulatory definition on TEP yet, and TEPs have been approved as CTPs. Combination products containing cells and devices are classified as biologics or medical devices through evaluating the primary mode of action. Depending on cases, offices of biologics and medical devices cooperate to approve the marketing authorization of products. The MFDS is considering TEP as an engineered tissue used to repair the recipient's tissue. TEPs are manufactured from cells or tissues of human or animal through substantial manipulation, such as cell culture and/or combination with scaffold, and acquire new biological characteristics and structural properties.5–6
Approved CTPs and TEPs in Korea
By June 30, 2014, 16 CTPs have been approved and 90 sponsor-initiated clinical trials have been approved. In addition, 74 investigator-initiated trials are approved by the MFDS. Table 1 demonstrates cell types and indication of the approved CTPs, which include chondrocytes, immune cells, fibroblasts, or osteoblasts. Lately, four stem cell products are approved and introduced into market, such as autologous bone marrow-derived mesenchymal stem cell (MSC) for the patients with acute myocardial infarction, allogeneic umbilical cord blood-derived MSC for the treatment of articular cartilage defects, and autologous adipose-derived MSC as orphan drug to treat Crohn's disease, and as autologous bone marrow-derived MSC for the patients with amyotrophic lateral sclerosis.
ALS, amyotrophic lateral sclerosis; AMI, acute myocardial infarction; CTPs, cell therapy products; MSC, mesenchymal stem cell.
Approximately 10 clinical trial protocols per year are approved with indications for a wide range of target diseases, as shown in Table 2.
The four TEPs are approved as CTPs, and all of them are skin products derived from keratinocyte or fibroblast. Eight clinical trials have been approved for TEP, which include clinical trials such as the artificial liver study using pig hepatocytes applied to liver failure patients.
Regulatory Considerations of CTPs and TEPs
The characteristics of CTPs are highly research-driven and innovative products, which are often developed by bioventures, universities, research institutions, or hospitals whose personnel usually have limited understanding of the regulations. CTPs are also manufactured in small batch size, and the final products are living cells in common. Sometimes, they are not fully characterized for their mechanism of action. Furthermore, they are combined with surgical procedure and have limited clinical records, such as long-term effect, assay for safety, and efficacy evaluation. One of the major issues on the use of cells includes cell banking, which usually applies to the amplification of allogeneic cells. In addition, drug substance can be introduced in the course of stopping point in cell manufacturing process. From the early phase of development, the well-defined characterization data for cells are required to set up the specification and to ensure quality management of TEPs.
The characteristics of TEPs are complex, heterogeneous, and small in manufacturing size. Manufacturing inconsistency, sterile processing, and dynamic character of the products are issues for the review of TEPs.7–8
The components of TEP are cells and scaffolds. Chemistry, Manufacturing, and Control issues on the CTP are also applied for the approval of TEPs because TEP includes cells. Cell characterization should be correlated with the specification and function of TEPs. Qualification and control of the reagents and materials used in manufacturing TEPs are very important. It is recommended to use clinical- or good manufacturing practice-grade reagents and materials and to control their qualities. International Conference on Harmonization published the Quality-by-Design (QbD) guidelines, which emphasize that quality system should be built in by design from the development stage of a drug. The QbD concept, such as critical quality attribute and critical process parameter, can be applied to the development of the TEPs for obtaining better quality.
For scaffolds, points to consider are material selection, design and manufacturing, sterilization, design control, and testing. Scaffolds are usually reviewed in the Medical Device Evaluation Department in the NIFDS of the MFDS. At the stage of combination of cells and scaffolds, data from various tests should be produced and submitted for evaluation of the process.9–11
At product assembly stage, data on manufacturability and consistent reproduction should be acquired. Regarding characterization data on cell–scaffold, there are product performance and potency issues related to topical tissue, migration out of the scaffold, in vivo remodeling, and altered interaction. Characterization can be performed through combination of both in vitro and in vivo tests.
Full sterility test is necessary and adding fast-test method is recommended. Identity test includes the cell surface markers or combined form of cell–scaffold. Potency should be related to clinical response and product performance. Product stability test is also required.
Guidelines on CTPs
Important guidelines related to CTPs published by MFDS are listed in Table 3. As CTP is quite different from the chemical drugs or recombinant DNA products, guidelines dedicated to CTPs should be established and available to facilitate the complex multistep process in the development of CTP.
CMC, Chemistry, Manufacturing, and Control; GMP, good manufacturing practice.
Recently Implemented Regulatory Policies
The Korean MFDS announced that they have implemented new regulations relating to CTP. In 2012, a pre-review process was introduced that the developer can submit documents for approval of clinical trials or marketing authorization before application of the approval of clinical trial investigational new drug and new drug application. After the pre-review of them, advice or comment on the data can be provided. In developing stage, orphan drug can be designated early with nonclinical study data. Results from investigator-initiated clinical trial can be used for the clinical phase I data, only autologous CTPs, if the clinical protocol is properly reviewed and safety is shown.
As the representative policies specific to CTPs, the manufacturer should report the safety data for every use of approved CTP for first 2 years in the re-evaluation period after the CTP is approved and marketed. MFDS designates a project manager for each developing or approved CTPs. This is for the life cycle management of the products and to help developer by arranging consulting meetings with reviewers. The MFDS always emphasizes the international activity for the regulatory harmonization especially in the advanced therapy field. One of them is the ICH Regulators Forum for Cell Therapy Group that regulatory authorities of nine countries participate in and have on-line and off-line meeting three to four times per year.
Conclusion
It takes tremendous time and efforts to get approval of a new drug. Especially, dossier for quality, preclinical, and clinical studies of the product should be sufficient to demonstrate acceptable levels of safety and efficacy for ensuring public health. CTPs and TEPs are going through the equal regulatory process, although those products are very diverse in nature. Since they are novel and complex products, the regulator and developer have limited clinical experience to review and guide. Therefore, the MFDS would take case-by-case approach, flexibility, and scientific evidence-based review. The clinicians, scientists in academia, and basic researchers who are interested in developing the new products should understand the regulations fully, and the MFDS always recommend early communication with regulatory authority through pre-review meeting.
Footnotes
Acknowledgments
I thank Dr. Tae-Gyun Kim who works in the Cell and Gene Therapy Products Division of Korea Ministry of Food and Drug Safety especially for his valuable comments on this article.
Disclosure Statement
No financial support or potential conflicts of interest are involved in the publication of this article.