
Abstract
The formation of blood vessels is a complex tissue-specific process that plays a pivotal role during developmental processes, in wound healing, cancer progression, fibrosis, and other pathologies. To study vasculogenesis and vascular remodeling in the context of the lung, we developed an in vitro microvascular model that closely mimics the human lung microvasculature in terms of three-dimensional architecture, accessibility, functionality, and cell types. Human pericytes from the distal airway were isolated and characterized using flow cytometry. To assess their role in the generation of normal microvessels, lung pericytes were mixed in fibrin gel and seeded into well-defined microcompartments together with primary endothelial cells (human umbilical cord vein endothelial cells). Patent microvessels covering an area of 3.1 mm2 formed within 3–5 days and were stable for up to 14 days. Soluble signals from the lung pericytes were necessary to establish perfusability, and pericytes migrated toward endothelial microvessels. Cell–cell communication in the form of adherens and tight junctions, as well as secretion of basement membrane were confirmed using transmission electron microscopy and immunocytochemistry on chip. Direct coculture of pericytes with endothelial cells decreased the microvascular permeability by one order of magnitude from 17.8×10−6 to 2.0×10−6 cm/s and led to vessels with significantly smaller and less variable diameter. Upon phenylephrine administration, vasoconstriction was observed in microvessels lined with pericytes, but not in endothelial microvessels only. Perfusable microvessels were also generated with human lung microvascular endothelial cells and lung pericytes. Human lung pericytes were thus shown to have a prominent influence on microvascular morphology, permeability, vasoconstriction, and long-term stability in an in vitro microvascular system. This biomimetic platform opens new possibilities to test functions and interactions of patient-derived cells in a physiologically relevant microvascular setting.
Introduction
T
Advanced in vitro systems that closely mimic the conditions of the human lung are one strategy among many to increase our understanding of pulmonary vasculogenesis and vascular remodeling. Recently, several in vitro models that reproduce vascular structures have been developed. The group of Stroock micropatterned collagen I gel and lined the 100 μm wide lumens with endothelial cells. 8 These vascular structures were stable and perfusable, but vascular remodeling was limited due to a high gel stiffness. A different approach was taken by Moya et al., 9 who used the vasculogenic properties of endothelial cells under shear stress to initiate vessel formation. Yet another method exploited the natural microvessel formation of endothelial cells inside fibrin gel by providing appropriate growth factors and signals from fibroblasts.10,11
These novel models mimic the cellular microenvironment much better than standard in vitro assays performed in static conditions. Endothelial cells attach to and modify a three-dimensional (3D) meshwork to form perfusable microvessels and with the addition of perivascular cells or physiological interstitial flow further approach in vivo-like conditions. Specifically, perivascular support cells, either fibroblasts9,10 or mesenchymal stem cells, 12 were shown to positively impact vessel orientation and stabilization, representing inherent functions performed by mural cells in vivo. However, in the micovasculature of the lung and organs such as the brain and kidney, pericytes are the main mural cells. In the lung, they are also present around pulmonary capillaries. 13 Apart from their location in the basal membrane around endothelial microvessels, pericytes are identified by surface markers such as NG2, CD146, 3G5, α smooth muscle actin (αSMA), and platelet-derived growth factor receptor β (PDGFRβ). 14 Pericytes are key players during vessel morphogenesis and in controlling vessel stabilization, remodeling, and tightness. Two recent studies have linked abnormal pericyte coverage to the progression of human PAH 4 and pulmonary fibrosis, 15 respectively, underlining their functional importance in the lung microvasculature.
In this study, our objective was to identify and assess the functional role of primary human lung pericytes and endothelial cells in a microfluidic platform for perfusable microvasculature. In a first phase, the various parameters of the in vitro model were established using primary human lung pericytes cocultured with endothelial cells from the umbilical vein. In a second step, primary human lung microvasculature endothelial cells and pericytes were used to more faithfully mimic the lung microvascular environment. Vasculogenesis and pericyte recruitment were observed over days and direct cell–cell interactions were imaged in high resolution. The effects of the pericytes on endothelial cells were investigated using two coculture strategies, by seeding these cells in direct contact or in spatially separated but communicating compartments. Our results show that the direct presence of pericytes around the microvasculature has a dramatic effect on vessel morphology, permeability, and vasoconstriction.
Materials and Methods
Device design and fabrication
A 100 μm high microfluidic device with five compartments separated by trapezoidal micropillars with 100 μm spacing was designed, based on a design reported by Kim and colleagues. 10 Unlike the aforementioned system, the central chamber of the chip was round and wider (2 mm diameter with a central pillar for stability), the adjacent flow channels were 1 mm wide and the outermost chambers 0.5 mm wide. Standard photolithography was used to pattern the microstructures with negative photoresist (GM-1070; Gersteltec) on Si-wafers. Polydimethylsiloxane (PDMS; Sylgard) was mixed at a 10:1 ratio with curing agent, casted on the mold, and cured for at least 2 h at 80°C. Chips were cut, access ports for gel loading punched with 0.5 mm biopsy punches (Shoney Scientific), and reservoirs with 5 mm punches (AEP Medicalcare). Finally, cleaned chips and standard glass cover slips were activated in oxygen plasma (Harrick Plasma Cleaner) at 650 mTorr for 25 s, bonded together, and baked overnight at 60°C before use.
Cell culture
Primary human umbilical cord vein endothelial cells (HUVEC PCS-100-010; ATCC) were cultured on 0.1% gelatin-coated flasks in endothelial growth medium 2 (EGM-2) cell culture medium (Lonza). Primary human lung microvascular endothelial cells (HMVEC-L; Lonza) were cultured on 0.1% gelatin-coated flasks in EGM-2 MV cell culture medium (Lonza). Cells between passages 3 and 6 were used for experiments.
Isolation of lung pericytes
To prospectively isolate lung pericytes, we used lung specimens obtained from patients following surgical resection for lung cancer. Normal tissue was procured from the lung specimen at a distant site of the tumor foci. All patients gave informed written consent for usage of surgical material for research purposes, which was approved by the Ethics Commission of the Canton of Bern, CH.
Preparation of lung tissue and cell sorting was performed as previously described with modifications. 16 Briefly, normal-appearing lung tissue was resected from the tumor foci at a distance >5 cm and digested using a solution of collagenase I and II (Worthington Biochemical Company). Digestion of lung tissue was halted following the addition of 10% fetal bovine serum (FBS; Invitrogen). Single cells were stained with a panel of fluorescently conjugated human monoclonal antibodies directed at the following epitopes: CD45-PB (eBioscience), CD14-PB (eBioscience), CD31-PB (eBioscience), CD235a-PB (eBioscience), CD73-APC (eBioscience), and CD90-FITC (eBioscience). To exclude dead cells, 7-AAD was added before sorting. Cells were sorted using a BD FACS Aria III and were directly cultured in a six-well tissue culture plate precoated with 0.1% gelatin in alpha-minimum essential medium (Sigma) supplemented with 1% FBS (Gibco), 20 ng/mL of recombinant human fibroblast growth factor 2 (Gibco), 25 ng/mL of recombinant human epidermal growth factor (Gibco), 1.25 mg of human insulin (Sigma), and 1% penicillin–streptomycin (Sigma). Cells were grown at 37°C, 5% CO2, and low O2 (3%) until reaching confluence and used up to passage 6. Bone marrow-derived mesenchymal stem cells (BM-MSCs) were isolated using the same antibody and panel as described above.
Cell characterization
Following expansion, FACS-sorted lung pericytes or BM-MSCs were harvested and resuspended in a FACS staining buffer. Cells were incubated with antibodies on ice and protected from light for 30 min. Following staining, cells were washed and resuspended in phosphate-buffered saline (PBS). Cells were incubated with the following fluorescently conjugated human monoclonal antibodies: PDGFRα-biotin (eBioscience), CD105-FITC (eBioscience), NG2-FITC (eBioscience), CD146-PE (eBioscience), CD61-APC (eBioscience), and CD44-APC (eBioscience). For analysis of PDGFRα, cells were stained with streptavidin-labeled APC-Cy7 (eBioscience). 7-AAD was added to exclude dead cells and debris. Cell acquisition was performed using a BD FACS LSRII. For analysis, 30,000 events were collected and analyzed using FlowJo software version 10.7 (Treestar).
For transforming growth factor (TGF)-β1 treatment, lung pericytes and BM-MSCs were seeded in eight-well chamber slides. Cells were treated with 10 ng/mL of TGF-β1 (eBioscience) for 3 consecutive days. Thereafter, the cells were washed twice with PBS, fixed with 4% paraformaldehyde (Sigma) for 15 min, washed with PBS, and blocked in a 2% bovine serum albumin (BSA) solution (Sigma) for 1 h. Then, the cells were stained with phalloidin-TRITC (Invitrogen) and αSMA-FITC (Sigma) and nuclei were counterstained with DAPI for 2 h at room temperature. After washing with PBS, the chamber slides were imaged on a Leica DMI 4000 microscope (10× and 20× objectives).
Chip loading and maintenance
Chips were sterilized under UV for 2 h or in an ozone chamber (CoolCLAVE) before loading. Endothelial cells and lung pericytes were resuspended in 2 U/mL thrombin from bovine plasma (Sigma) in EGM-2 at a final concentration of 2×107 cells/mL (HUVEC), 1×108 cells/mL (HMVEC-L), and 1×107 cells/mL, respectively. For combined coculture, the EC and lung pericyte–thrombin suspensions were mixed at a 1:1 ratio, resulting in 1×107 HVUEC/mL, 5×107 HMVEC-L/mL, and 0.5×107 lung pericytes/mL. Fibrinogen from bovine plasma (Sigma) was dissolved in deionized PBS (Gibco) to a concentration of 5 mg/mL. In a separate tube, a clotting test with 2 U/mL thrombin in EGM-2 and 5 mg/mL fibrinogen solution was carried out to confirm clotting of the mixture after 5 min at room temperature. The HUVEC–thrombin suspension was then mixed with 5 mg/mL fibrinogen at a ratio of 1:1 and immediately pipetted into the central chamber. The solution stayed in the central chamber due to liquid pinning between micropillars. Five minutes later, the lung pericyte–thrombin suspension was mixed at a 1:1 ratio with the fibrin solution and injected into the outer chambers. After complete gelation in all chambers (10 min), EGM-2 was pushed through the flow channels and reservoirs were filled. The chips were incubated at 37°C and 5% CO2 in Petri dishes with a moist tissue for humidification. Cell culture medium was exchanged after 24 h and then every 48 h by emptying all four reservoirs. The reservoirs were filled with medium to create a transient pressure drop across the central chamber with an initial pressure head of 1.3 mm. This hydrostatic pressure enhanced medium supply to the cells in the central chamber. Chips were kept in culture for up to 14 days.
Immunostaining
The chips were washed twice with PBS, fixed with 4% paraformaldehyde (Sigma) for 15 min, and washed three times with PBS. If intracellular markers were used, 0.1% Triton X-100 (Sigma) was applied for 10 min and washed away 3× with PBS. After 1 h of blocking in a 2% BSA (Sigma) solution, primary antibodies VE-cadherin (Santa Cruz), collagen IV (Abcam), PECAM-1 (Santa Cruz), αSMA (Novus Biological), and ZO-1 (Invitrogen) were incubated overnight at 4°C. Subsequently, devices were washed three times with PBS and incubated with Alexa fluorophore-coupled secondary antibodies (Molecular Probes), as well as phalloidin (1:100; Invitrogen) and Hoechst (1:1000; Invitrogen) for 2 h at room temperature. After final washings with PBS, the chips were imaged on a Zeiss confocal LSM 710 microscope (10×, 20×, and 40× objectives).
Time-lapse observation
For time-lapse observations and imaging of endothelial–pericyte interactions, the cells were labeled with lipophilic cell tracker dyes PKH67 green (HUVECs) and PKH26 red (lung pericytes) before seeding. The devices were loaded into the Nikon Biostation CT and imaged every hour with the 4× magnification in brightfield, red and green fluorescence. Cell culture medium was replaced every 48 h.
Vascular area and perfusability quantification
To characterize the vascular networks, z-stacks (10 slices covering a 100 μm depth) of tile scans (2.4×2.4 mm) of the central chamber were analyzed using the open source image analysis software, Fiji (http://fiji.sc/Fiji). The vascularized areas were defined as being phalloidin-positive (separated cocultures in Fig. 5A, B) and PECAM-1-positive (direct cocultures in Fig. 7A). The phalloidin or PECAM-1 signals were quantified from the entire central chamber. The z-stacks were projected on a single plane and binarized with the automatic triangle thresholding method. The binary operation “close” and outlier removal were applied to remove small unconnected particles. “Analyze particles” was used to quantify the vascularized area inside the central chamber. Only areas larger than 100 μm in diameter were taken into account to determine the vascularized area percentage. The number of perfusable entrances was assessed visually by checking openings in 3D images obtained with the confocal microscope. Each cell culture condition experiment was repeated three to six times. Average values with standard deviations are reported. Results were compared using a two-tailed Student's t-test.
For measurements of the minimal and maximal diameters, the width of three of the smallest and three of the largest vascular segments (perpendicular to the vessel wall) was measured on tile scans of PKH26-labeled microvessels by using the line and measurement command in Fiji. The mean value and standard deviation of six chips are reported.
Electron microscopy
The chips were fixed in 2.5% glutaraldehyde (Agar Scientific) in 0.15 M HEPES buffer overnight (total osmolarity: 696 mOsm, pH 7.35), and postfixed for 1 h in a 1% solution of osmium tetroxide (Electron Microscopy Sciences) in 0.1 M sodium cacodylate buffer (total osmolarity: 369 mOsm, pH 7.40). After rinsing in 0.05 M maleate buffer (pH 5.0) the chips were dehydrated in increasing concentrations of ethanol (70%, 80%, 96%, 100%), passed through acetone, and left in a 1:1 mixture of ethanol:epoxy embedding medium (Sigma) overnight. On the following day the chips were filled with epoxy embedding medium and left to polymerize at 60°C for 4 days. Ultrathin sections (70 nm) were cut on a Reichert-Jung Ultracut E microtome, put on formvar-coated 2×1 mm single-slot copper grids and double stained with 1% uranyl acetate (Sigma) and 3% lead citrate (Leica Microsystems). Micrographs were taken in a Philips EM 400 transmission electron microscope.
Microbead tracking
Fifty microliters of a yellow fluorescent 1 μm polystyrene bead suspension (Polysciences) was added to one of the four emptied chip reservoirs, generating a pressure head of 1.3 mm. To track individual beads over time, time-lapse images were acquired using a Zeiss confocal LSM 710 at one image per second for 5 min and were analyzed with the TrackMate v2.5.4 plugin in the software Fiji. Bead velocities were added as color map to the images.
Microvessel permeability
Seventy kilodaltons rhodamine isothiocyanate (RITC)-coupled dextran (Sigma) at 1 mg/mL in PBS was added to one reservoir. Real-time movies (Panasonic Lumix DMC-LX7) or time-lapse videos (one image every 15 s; Leica DMI 4000) were taken for several minutes. The vascular permeability was calculated as described previously,
17
by measuring fluorescent intensities across vascular segments over time:
where ΔI is the initial intensity increase when RITC-dextran is added, (dI/dt) the change of intensity over time due to leakage, and r/2 the volume to area ratio for a cylindrical segment. Three chips (n=3) were measured per condition and six to eight regions were quantified per chip. A two-tailed Welch-corrected Student's t-test was applied.
Vasoactive response quantification
Phenylephrine (Sigma) was dissolved in water (1 mM) and diluted to a concentration of 10 μM in PBS. As control, the equivalent amount of water in PBS was used. After staining the microvasculature with CellMask Orange (Invitrogen) on day 7, 100 μL of the diluted phenylephrine was added into one empty reservoir. A vascular segment was imaged for 5–10 min (Cy3, one image every 10 s; Leica DMI 4000) following drug exposure. The vessel width was assessed over time by measuring the size of a vascular segment (perpendicular to the vessel wall), and plotting a kymograph in Fiji (pixel intensities along the line as a function of time). The vessel walls were identified as the two intensity maxima closest to the image border. The vessel width at each time point was determined as the distance between the two maxima, and normalized to the initial width. Three chips were measured per condition (n=3) and five vessel widths were quantified per chip. Results were compared at each time point with a two-tailed Student's t-test.
Results
Primary human lung pericyte isolation and characterization
To address the immunophenotypic identity of human lung pericytes in vivo, we combined multiple surface markers to prospectively identify putative lung pericytes. As shown in Figure 1, we identified a cluster of nonhematopoietic (negative for CD45 and CD14), nonendothelial (negative for CD31), and nonepithelial cells (negative for EpCAM) that were positive for key mesenchymal markers CD73 and CD90. When prospectively sorted, these lineage−EpCAM−CD73+CD90+ cells were spindle shaped resembling pericyte morphology and formed typical colony-forming units (Fig. 1 lower right). Following expansion, the cultured cells were analyzed by multiparameter flow cytometry (Fig. 2A) and shown to highly express the cell adhesion marker integrin beta-3 (ITGB3, CD61), which is involved in binding extracellular matrix proteins. Interestingly, the lung cells demonstrated minimal expression for CD146, a marker found to be highly upregulated on BM-MSCs. Further analysis revealed that the ITGB3+ lung cells expressed other key pericyte markers NG2 and PDGFRα, whereas on BM-MSCs, PDGFRα was minimally expressed. Moreover, putative lung pericytes were positive for CD105 receptor (endoglin) and CD44, the receptor for hyaluronic acid. To investigate whether these cells are responsive to the pleiotropic cytokine TGF-β1, they were exposed to TGF-β1 for 3 days in culture. TGF-β1-treated cells showed an upregulation in F actin stress fibers using the high affinity probe phalloidin and αSMA compared to untreated cells (Fig. 2B). Together, these results show that the Lin−EpCAM−CD73+CD90+ cells isolated from human lungs match the criteria of pericytes, as demonstrated by morphology, the cell surface markers NG24 and PDGFRα18,19 and response to TGF-β1. These cells are thus a suitable choice as perivascular supporting cells for an in vitro lung microvascular model.

Isolation of putative pericytes from human lung tissue. A subset of small cells was isolated using a six antibody/viability marker panel and polychromatic fluorescence-activated cell sorting. Unfractioned lung cells were initially displayed and gated (R1) on a side scatter-forward scatter pseudocolor density plot, which was subgated to discriminate doublets (R2). Single cells (R2) were further gated to identify live cells that were negative for the dye 7-AAD (gate R3). These cells were subgated onto an antigen plot to display a cluster of lineage-negative (Lin−; CD45, CD14, CD31 and CD235a) and EpCAM-negative cells (gate R4) and Lin−EpCAM+ cells (gate R5). Lin−EpCAM− cells (R4) were then displayed as a quadrant gate to identify CD73 and CD90 subset of cells. Lower right: A representative image of a typical colony-forming unit generated from sorted Lin−EpCAM−CD73+CD90+ cells obtained from gate R6 and higher power image of single colony (4×). Color images available online at www.liebertpub.com/tea
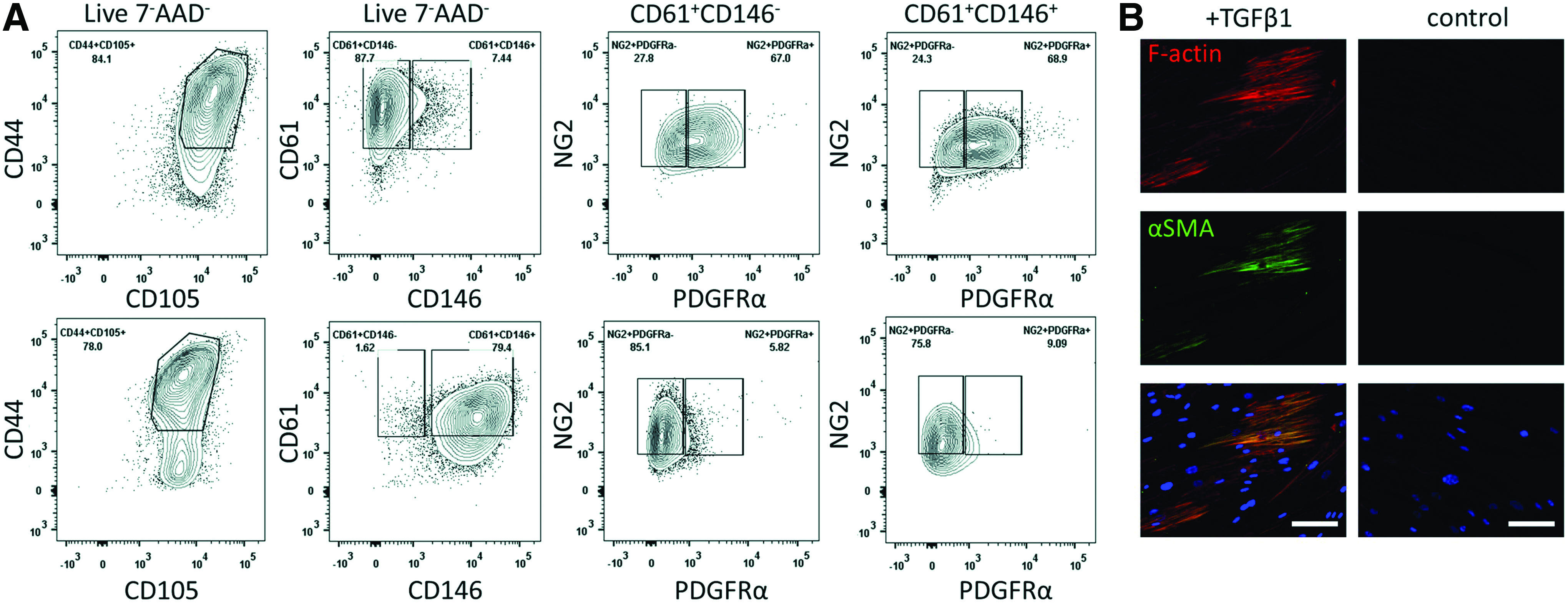
Lin−EpCAM−CD73+CD90+ lung pericyte characterization.
Direct and indirect cocultures in the microfluidic chip
The specific design of the microfluidic chip enabled perfusable microvasculature to form and to investigate the effects of the pericytes on the endothelial vessels (Fig. 3). The microvasculature was formed inside a 100 μm high central chamber of the chip that was filled with cells and fibrin hydrogel. After fibrin polymerization, the two flow channels adjacent to the central chamber were filled with physiological medium. The hydrogel/physiological medium interface enabled molecular diffusion of nutrients, oxygen, and growth factors required for the cells to rearrange and create perfusable vessels. Micropillars situated at the perimeter of the central chamber stopped the spreading of the fibrin gel by holding the hydrogel in place due to liquid pinning by surface tension (Fig. 3C). Two additional compartments situated at 1 mm from the central chamber across both flow channels were filled with fibrin gel and pericytes. To examine the vasculogenic potential of the lung pericytes, two coculture strategies were investigated. The first strategy consisted of an indirect coculture approach, in which endothelial cells were spatially separated from the lung pericytes. Endothelial cells suspended in fibrin solution were seeded into the central chamber, whereas the outer chambers were filled with lung pericytes suspended in fibrin solution. The second strategy was based on a direct coculture of endothelial cells and pericytes in the central chamber. In this approach, pericytes were also cultured in the outer chambers. In both strategies, cells could migrate from the outer chambers toward the central chamber through the hydrogel/physiological medium interfaces or vice versa.

Microfluidic chip specifications.
Lung pericytes support vasculogenesis by paracrine signals
Following seeding in the central chamber, HUVECs extended toward each other (Fig. 4 and Supplementary Movie S1; Supplementary Data are available online at www.liebertpub.com/tea) resulting in vacuole formation, a hallmark of vasculogenesis20,21 (Fig. 4B top). The process was dynamic with cells fusing, branching, and parting from each other within hours. Vasculogenesis started independently from the spatially separated lung pericytes. After 3–5 days, the overall layout of the vascular segments stabilized, that is, the vessel shape did not dramatically change anymore. However, sprouting events still occurred (Fig. 4B center). Intravascular lumen grew wider and opened up toward the flow channels between days 4 and 7. In this setting, vascular structures were kept in culture for at least 14 days (Supplementary Fig. S1).
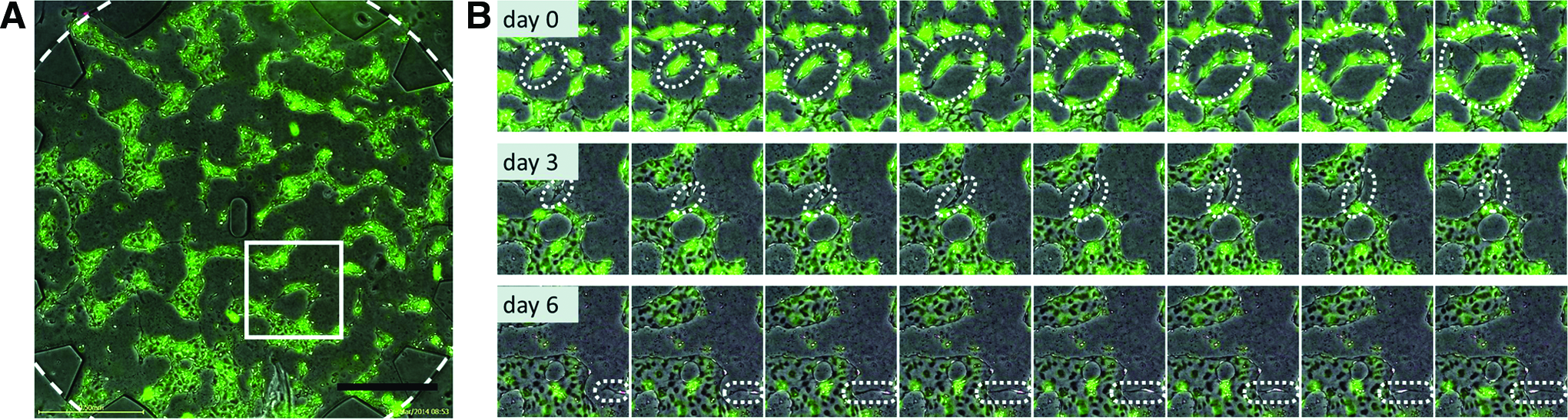
Vasculogenesis in the microfluidic chip. PKH67 (green)-labeled HUVECs were seeded into the central chamber, and PKH26 (red)-labeled pericytes were seeded into the side chambers, and vasculogenesis was observed with time-lapse imaging for 7 days.
Lung pericytes, originally seeded in a separate outer microcompartment, migrated across the flow channel toward the central chamber and invaded it within 3–5 days (Fig. 4C bottom and Supplementary Movie S1). They slowly migrated through the fibrin gel of the central chamber and aligned along the microvasculature. Importantly, vascular structures only opened up toward the flow channels in the presence of lung pericytes in the outer channel (Supplementary Fig. S2A, B). Cell migration also took place in the opposite direction, whereby HUVECs migrated from the central chamber and moved into the flow channels, thus extending the vascular structure (Supplementary Fig. S3).
In vitro microvessels show typical vascular features and are perfusable
After 7 days of coculture, patent, interconnected, and perfusable microvessels had formed throughout the 2 mm wide central chamber. Vessel widths ranged between 20 and 220 μm. On average, 55.4%±11.0% of the central chamber was covered by vascular structures, and 73.5%±13.3% entrances were opened, connecting the microvascular network with the flow channels (n=17) (Fig. 5A, B). The mean vascularized area percentage obtained from three independent experiments was 52.0%±4.3% (n=6 chips), 61.1%±16.2% (n=6), and 52.7%±7.5% (n=5). The variation between experiments was not significant (p=0.31), thus the vascular structures formed in a reproducible manner. Likewise, the percentage of perfusable entrances was comparable between experiments with 72.2±8.6% (n=6), 73.6%±19.3% (n=6), and 75.0%±11.8% (n=5) (p=0.95). When EGM-2 was replaced only every 48 h as compared to every 24 h, the vascularized area percentage decreased from 63.1%±3.2% (n=5) to 52.0%±4.3% (n=6) (p=0.001), and there were less perfusable entrances (83.3%±5.9% (n=5) compared to 72.2%±8.6% (n=6) (p=0.037) (Fig. 5A).
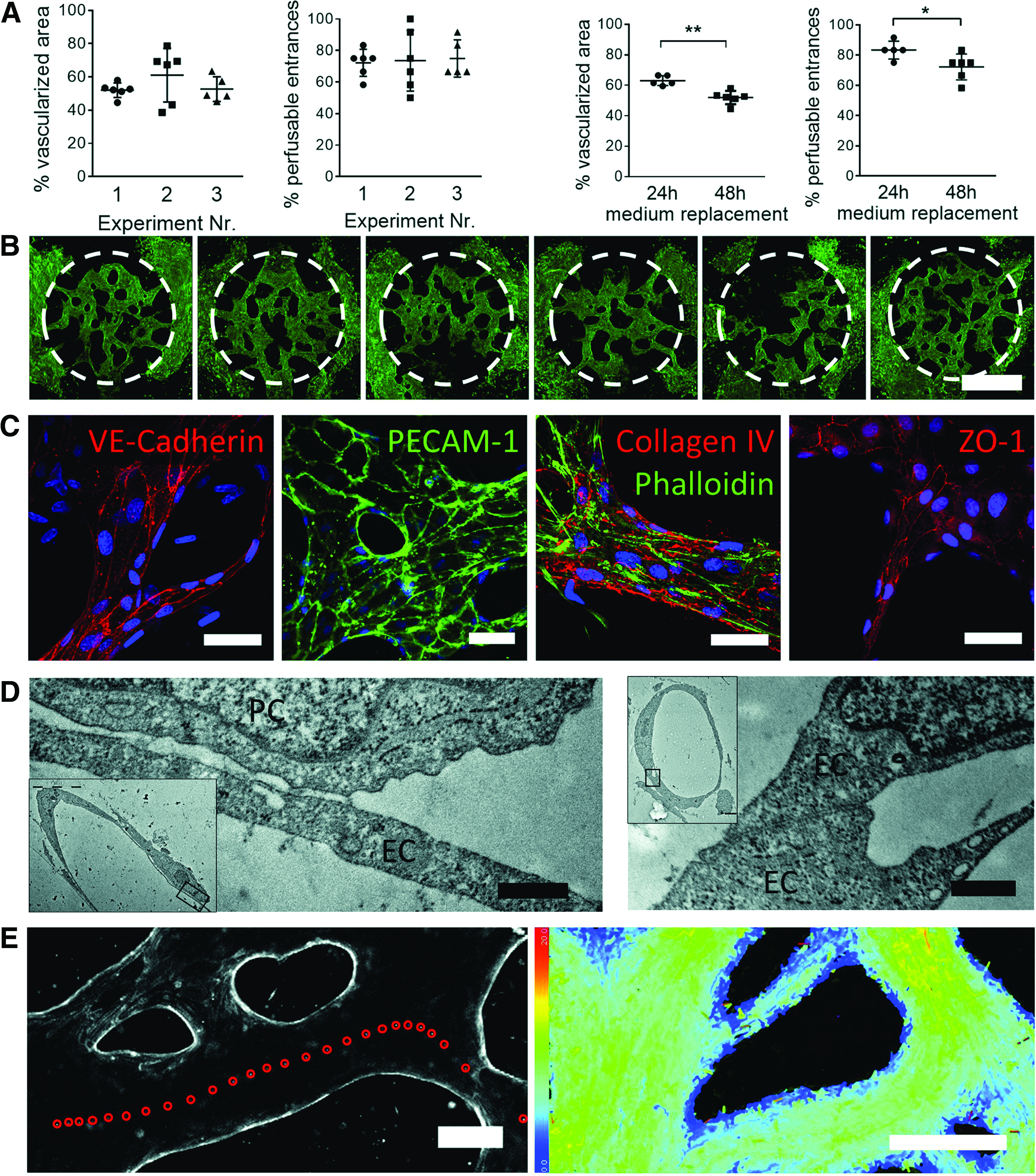
Microvessel characterization on day 7.
Immunostainings for VE-cadherin and PECAM-1/CD31 confirmed the presence of adherens junctions between the endothelial cells building the microvessels (Fig. 5C). Furthermore, ZO-1 confirmed the presence of tight junctions along all vessels. Most importantly, microvessels secreted basement membrane components on the basolateral side, as confirmed by collagen IV staining. Tight association between the endothelial cells of a microvessel was also confirmed with transmission electron microscopy (Fig. 5D right). Furthermore, pericytes were only found on the abluminal side, often in close contact with the microvasculature (Fig. 5D left).
Perfusability is one of the key features of a functional vasculature. To confirm accessibility of the microvessels from the flow channels, a suspension of fluorescent polystyrene microspheres (1 μm diameter) was loaded in one reservoir, generating an initial pressure drop of 1.3 mm across the central chamber. Indeed, microbeads flowed through accessible vascular segments (Fig. 5E and Supplementary Movie S2). Under these flow conditions the mean bead velocity was 6.8 μm/s with minimum and maximum speeds comprised between 0 μm/s at the border of the vessels and up to 20 μm/s in the center (Supplementary Fig. S4). The flow path through the microvasculature was also visualized by adding RITC-dextran (Supplementary Movie S3).
Direct coculture with lung pericytes alters vascular morphology, permeability, and vasoactive response
To observe the effects of direct coculture between lung pericytes and HUVECs, both cell types were mixed and seeded in the central chamber. Lung pericytes were also seeded in the outer chambers to enable vessel opening. The resulting vessel morphology was strikingly different to the separated coculture setting. In direct coculture (Fig. 6A top), vascular diameters were smaller and more streamlined as compared to the separated coculture that had more tortuous vessels (Fig. 6A bottom). Indeed, the measurements of the smallest and largest diameters were significantly smaller for the direct coculture than the separated coculture (p=0.005 for the smallest and p<0.001 for the largest diameter, Table 1). Vessel diameters were also more narrowly distributed in the direct coculture (difference from smallest to largest diameter 116.7 μm) than the separated coculture (198.6 μm). Furthermore, immunostaining revealed that some pericytes lining the abluminal side of the microvessels highly expressed αSMA (Fig. 6C). In addition to the morphological differences, the vascular integrity was also altered. Perfusion with 70 kDa RITC-coupled dextran filled the vascular structures, but leaked into the interstitial space in the separated coculture setting (Fig. 6B, arrow), whereas no leakage from vessels was obvious in the direct coculture. The calculated permeability coefficients were 17.8×10−6 and 2.0×10−6 cm/s for the separated and direct coculture, respectively (Table 1). Thus, the direct presence of pericytes lowers the permeability by a factor of 8 (p=0.057).

The direct presence of pericytes affects vessel morphology, permeability, and response to phenylephrine. Experiments were carried out on day 7 on direct cocultures (top row) or separated cocultures (lower row).
To assess the physiological functionality of the pericytes on in vitro microvasculature, we perfused the vasoactive α1 adrenergic receptor agonist phenylephrine through microvessels in direct and separated cocultures. The direct presence of pericytes influenced the response to phenylephrine. Microvessels with pericyte lining constricted immediately upon phenylephrine administration (p<0.05 between 120 and 300 s, n=3 chips per condition) compared to administration of the vehicle (Fig. 6E and Supplementary Fig. S5). No significant difference between phenylephrine and the vehicle was observed in the separated coculture.
Microvasculature with primary human lung-derived cells only
To mimic the microvasculature of the lung more closely, we replaced HUVECs with HMVEC-Ls. Similarly to HUVECs, HMVEC-Ls assembled to form continuous microvascular networks, surrounded by and supported with lung pericytes. In contrast to direct coculture of HUVEC and lung pericytes, the seeding ratio for HMVEC-L and lung pericytes needed to be increased from 2:1 to 10:1 to promote perfusable microvessels. At lower seeding ratios, the pericytes were more prone to block openings toward the flow channel and impede perfusability. Microvasculature made of primary lung-derived endothelial cells and pericytes covered 47.74%±6.05% of the central chamber area on day 5, and 62.5%±14.43% of entrances were perfusable (n=4). The vessels expressed PECAM-1 and VE-cadherin, showed patent and perfusable lumen, and pericytes were found on the abluminal vessel side exclusively, in close proximity to the endothelial cells (Fig. 7).
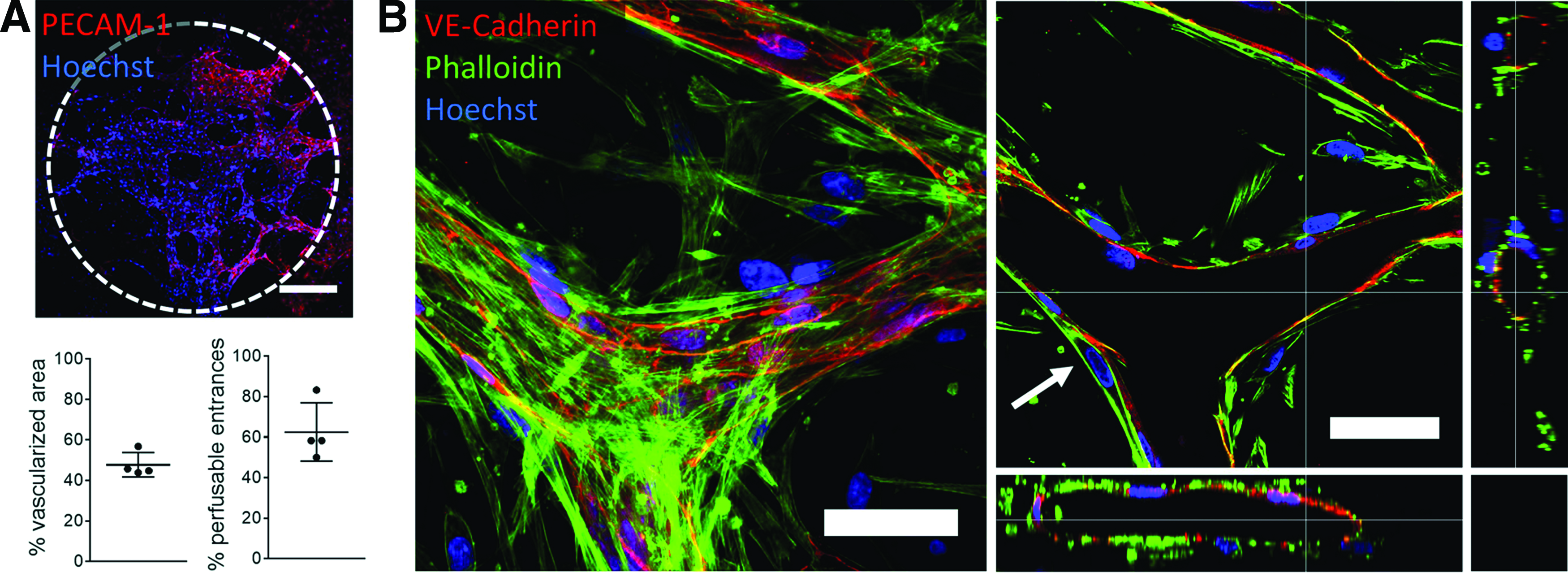
In vitro human lung microvasculature.
Discussion
Microfabrication technologies applied to vascular biology open new possibilities to study the fascinating processes of vasculogenesis, angiogenesis, and vascular remodeling. Not only can such new experimental platforms address fundamental questions in vasculogenesis, but they may also be used in the future to vascularize tissues before implantation 22 or optimize drug treatments on personalized vasculature models.
Although several studies recently reported on vasculogenesis on chip,9,10 none focused on reproducing tissue-specific microvasculature with well-defined cell types. To mimic the microvasculature of the lung parenchyma, primary pericytes from human lung parenchyma were used in this study. These cells were prospectively sorted from human lung samples using mesenchymal surface markers while being devoid of known hematopoietic, endothelial, and epithelial markers. The selected cells expressed pericyte-associated cell surface markers and upregulated αSMA upon stimulation with TGF-β1. Interestingly, pericyte surface markers4,14,18 were more highly expressed in the lung pericytes than the BM-MSCs, underscoring their specialized function as mural support cells. Taken together, these results confirm that the sorted cell type possess phenotypic and functional characteristics known to be found in pericytes.
To study their role in the context of microvessel assembly and function, the lung pericytes were seeded into the microfluidic platform together with endothelial cells. The different seeding options allowed to separately observe paracrine and direct contact interactions of lung pericytes and ECs. Not surprisingly, the presence of lung pericytes in the chip was necessary for long-term vessel stabilization.21,23,24 Of note, it was sufficient to seed the two cell types at 1 mm distance from each other to obtain stable vascularization. Over time, both cell types migrated across the separating channel and lung pericytes started to wrap around newly formed vascular segments. The influence of lung pericyte signals on vessel orientation was evident, because vascular segments only opened up toward the flow channel if pericytes were present on the opposing side. This observation shows not only pericyte recruitment during vascular formation, but also reorientation of endothelial neovessels according to pericyte location. The vascular development was also influenced by the growth factors contained in the EGM-2 medium, as shown by the increased area and opened segments when new media were supplied every day compared to every second day.
In addition to the pericyte recruitment toward the vasculature, we also observed dramatic differences between the two coculturing strategies. When lung pericytes and ECs were mixed and coseeded from the outset, pericytes spread around microvascular segments but were never observed inside vessel lumens. In this direct contact setup, microvessels were narrower and less tortuous as compared to spatially separated seeding. In addition, vessels with a pericyte lining also showed significantly decreased vascular permeability and no leaks in the vessel walls. In fact, pericyte-lined vessel permeability was similar to values measured in mammalian venules, 25 and eight times lower than permeability of vessels with ECs only. This effect may be linked to increased basement membrane secretion in the direct contact setting, 26 but this needs to be verified in subsequent studies. This finding is also in line with the observation that vessels lacking pericytes are wider, have a more tortuous morphology, and are more prone to rupture.27,28 Moreover, some pericytes in close contact with the microvessels highly expressed αSMA, showing a functional response upon direct contact with endothelial cells. Further investigation is needed to determine the molecular interaction that leads to this upregulation.
Administration of phenylephrine demonstrated the vasoconstrictive activity of pericytes, and is consistent with reports of pulmonary microvascular contraction upon phenylephrine administration in vivo. 29 We observed slightly smaller values of constriction (3% change in diameter) compared to the animal model (6% change in diameter). However, the reported values were measured in small pulmonary arterioles (46 μm in diameter) whereas our measurements were performed in larger endothelial vessels (120 μm in diameter) covered by pericytes only, thus the two values are not directly comparable. The versatile seeding strategy allowed us to compare the response of vessels made of endothelial cells only and of pericyte-covered endothelial vessels. Interestingly, the former had a slight tendency to enlarge upon fluid loading (both with control and phenylephrine administration), in contrast to the vessels in direct coculture. This observation suggests that pericyte-lined microvessels may possess a higher stiffness compared to endothelial microvessels alone. Taken together, the pericyte functions on microvascular morphology, permeability, and vasoconstriction known from in vivo observations were reproduced in our in vitro platform and demonstrate the physiological relevance of our system.
In the present study, the final microvasculature was perfusable and covered an area of 3.1 mm2. Importantly, the vascular network formation was highly repeatable and resulted in comparable network architecture, despite spontaneous vascular self-assembly. Furthermore, we considered the entire vascular area for analyses, not only selected regions of interest. The longevity, reproducibility, and high area coverage of the perfusable microvasculature may be of interest for future vascularization studies in tissue engineering. In contrast to our results, recent in vitro vasculogenesis assays using spatially separated cocultures did not describe recruitment and direct contact of supporting cells with microvessels.10,11 This might be due to the shorter experimental duration used in these studies. It is also possible that the primary cells used here are more migratory and physiologically relevant as compared to the lung fibroblasts reported earlier.9,11 In addition, lung resident pericytes might be more specialized in stabilizing microvessels than the less differentiated MSCs. 30
Traditional in vitro assays can measure either permeability (cell seeding on a porous insert) or vasculogenesis (e.g., cell seeding on matrigel), however, there is no readout for vessel morphology of perfusable vessels. In contrast, we can compare vessel morphology, permeability, and marker expression in the same experiment. This platform is also suitable to perform functional assays that are difficult to carry out in animal models. Recording vasoactive responses of the lung microvasculature in vivo is very delicate, and only possible with advanced equipment such as a thoracic window combined with intravital videomicroscopy. 29 In contrast, this readout was straightforward and applicable to human cells in the current platform using fluorescent staining and time-lapse microscopy.
A limitation in our study was the choice of endothelial cells, because HUVECs differ from lung microvascular endothelial cells in terms of gene expression, growth, permeability, and morphology.31,32 To further increase resemblance of the current in vitro model to pulmonary microvessels and capillaries, we therefore used cells from human lung parenchyma exclusively (HMVEC-L and lung pericytes). We demonstrated successful vasculogenesis and the establishment of patent, perfusable microvessels with patient-derived endothelial cells and pericytes from the distal airway. In the future, it is conceivable to test drugs on such models to optimize individual drug treatments for patients suffering from diseases affecting the microvasculature.
Conclusion
To understand the complex interactions of vascular biology in health and disease, in vitro microvascular models must be simple but not reductionist. In this study, we developed an in vitro lung microvasculature with prospectively sorted primary pulmonary pericytes and endothelial cells in a microfluidic coculture system. Our setup enabled the formation of microvessels that resembled human lung microvasculature in terms of 3D morphology, vascular marker expression, ultrastructure, permeability, and vasoactive response. To further increase the biological similitude to pulmonary microvessels, human microvascular endothelial cells from the lung were seeded together with lung pericytes and built stable and perfusable microvascular networks. This simple platform enables building tissue- and patient-specific microvasculatures amenable to drug testing or modeling of diseases such as PAH.
Footnotes
Acknowledgments
Special thanks go to Matthieu Delincé for help with photolithography at the Center of MicroNanoTechnology (CMi) at EPFL, Franziska Graber and Adolfo Odriozola for help with transmission electron microscopy, and Prof. Dr. Robert Rieben for discussions and kindly providing endothelial cells. The authors also acknowledge the Microscopy Imaging Center and the FACSLab at the University of Bern.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.