
Abstract
Clinical data show that estrogen levels are inversely associated with the production of sclerostin, a Wnt antagonist that recently attracted great attention over the use of its antibody in the anabolic treatment of osteoporotic conditions. However, the molecular link between sclerostin expression and estrogen signaling is not yet known. We investigated the mechanisms by which estrogen modulates sclerostin (SOST) gene expression at the cellular level in human osteoblast cells in association with bone morphogenetic protein (BMP)2 signaling given that BMP2 is a potential inducer of SOST in human mesenchymal stromal cells (hMSCs). 17β-Estradiol (E2) alone had no effect on SOST expression, which was significantly induced by treatment with BMP2 in hMSCs and osteoblasts derived from the mandibles of female donors. However, E2 suppressed the induction of SOST and other BMP2 target genes by BMP2 in hMSCs. E2 signaling was independent of the Smad pathway, which plays a critical role in SOST induction mediated by BMP2. Instead, E2 increased the transcriptional expression of β-catenin and levels of its activated form. Silencing of the gene encoding estrogen receptor (ER)α decreased E2 activity in β-catenin activation and the suppression of SOST induction by BMP2, but had no influence on BMP2-mediated SOST induction in the same conditions. Similar results were obtained after treatment with ERα antagonist as a Wnt inhibitor. In human osteoblasts, the effect of E2 on SOST expression was either suppressive or absent, depending on the cell donor. Interestingly, the SOST expression pattern after treatment with BMP2 or BMP2/E2 in human osteoblasts showing a pattern of E2 suppression on SOST induction by BMP2 correlated with the ratio of receptor activator of nuclear factor kappa-B ligand (RANKL) to osteoprotegerin (OPG) expression. These results demonstrate that estrogen signaling in osteoblasts negatively regulates SOST expression in an indirect manner through interaction with BMP2 signaling and that this regulation involves the Wnt/ERα and β-catenin pathways. This study highlights several interactions between estrogen and BMP cascades in osteoblasts that may provide a basis for therapeutic intervention for the modification of bone mass density.
Introduction
E
Sclerostin, the protein product of the SOST gene, is expressed in osteogenic cells, including hypertrophic chondrocytes, osteoblasts, and osteocytes. 8 There is abundant evidence that SOST functions as a critical negative regulator of bone formation by antagonizing the Wnt signaling pathway.9–11 It has been suggested that sclerostin signaling in bone is induced as part of the mechanical adaptive response of osteocytes.8,12 Mechanical loading induces downregulation of SOST expression in osteocytes, 13 leading to either systemic or local regulation of bone formation in a paracrine manner, which could in turn antagonize canonical Wnt signaling in osteoblasts. 14 Furthermore, SOST knockout mice have a high bone mass phenotype and have been shown to resist bone loss in a hind limb-unloading model.14,15
Recent reports demonstrated that SOST is induced by bone morphogenetic protein (BMP) in cells of the osteoblast lineage, where it acts as a BMP antagonist. 16 In contrast with the known role of BMPs as potent osteoinducers,17,18 a few studies have reported that mice with conditional knockout (cKO) of BMP receptor type 1A (Bmpr1A) exhibit a unique phenotype with increased bone mass resulting from severely suppressed bone resorption, despite little change in the rate of bone formation. 19 This research group demonstrated that SOST plays a critical role in the phenotype of Bmpr1A cKO mice. SOST is upregulated by BMP and inhibits Wnt/β-catenin signaling by binding to the coreceptors, low-density lipoprotein receptor-related proteins 5 and 6.9,20 Sclerostin eventually enhances resorption by triggering receptor activator of nuclear factor kappa-B ligand (RANKL)–osteoprotegerin (OPG) pathway-induced osteoclastogenesis, leading to a decrease in bone mass.9,10,20
These observations led to the idea of therapeutic intervention using sclerostin inhibitors to increase bone mineral density. In preclinical studies, a sclerostin-neutralizing monoclonal antibody (Scl-Ab)II increased bone mass and bone strength in aged ovariectomized rats and aged male rats to levels greater than those found in control rats. 21 In addition, an investigation using humanized Scl-AbIV in normal female primates showed clear anabolic effects, with marked dose-dependent increases in bone formation. 22 Clinical trials using an antisclerostin antibody series have been performed with encouraging results for efficacy and safety.23,24
Taken together, these findings indicate that estrogen and BMP signaling converge on SOST expression and Wnt pathways in bone cells.4,10,25,26 However, studies performed to date very rarely provide any molecular evidence for a direct effect of estrogen on SOST expression or whether the effect of estrogen on SOST expression involves BMP signaling. Therefore, we hypothesized that estrogen modulation of SOST expression might be indirectly mediated by BMP signaling at the level of the bone cell given the potent induction of SOST by BMP2. We expect that this study will be helpful in understanding the molecular mechanism underlying estrogen regulation of SOST expression, which will also provide clues to the potential mechanism of the bone catabolic effect of BMP2.
The major aims of this study were threefold: (i) to characterize the effect of estrogen on the induction of SOST by BMP2 and the expression of other BMP-responsive genes using undifferentiated primary human mesenchymal stromal cells (hMSCs) from female donors; (ii) to investigate the molecular mechanism of estrogen action involved in the modulation of SOST expression in association with BMP2 signaling by examining the involvement of Wnt/β-catenin/estrogen receptor (ER)α and the Smad pathway in hMSCs; (iii) to investigate the estrogen effect on BMP2-mediated gene expression, including SOST, and determine markers that correlate with SOST expression in osteoblasts derived from the mandibles of female donors.
Materials and Methods
Chemicals
17β-Estradiol (E2) was purchased from Sigma-Aldrich (St. Louis, MO). TCF/LEF reporter for Wnt and Smad signaling was obtained from Qiagen (Hilden, Germany). The Dual-Luciferase Reporter Assay system was purchased from Promega (Fitchburg, WI). ERα and Smad4 siRNAs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Transfections for the reporter assay and siRNA were performed using Lipofectamine® 2000 reagent (Invitrogen, Carlsbad, CA). Antibodies for total/active β-catenin, ERα, and Smad4 were obtained from Cell Signaling Technology® (Danvers, MA), Abcam (Burlingame, CA), and Santa Cruz Biotechnology, respectively. Enzyme-linked immunosorbent assay (ELISA) reagents for sclerostin, Opg, and insulin-like growth factor-1 (IGF1) were obtained from R&D Systems (Minneapolis, MN). The Smad inhibitor, dorsomorphin, was obtained from Calbiochem® (Gibbstown, NJ), and the Wnt inhibitor, ICI 182,780, was purchased from R&D Systems. Recombinant human BMP2 (rhBMP2), prepared from Escherichia coli, was obtained from Novosis®-Dent, BioAlpha, Inc., (Gyeonggi-do, South Korea); we demonstrated its osteogenic activity in a previous study. 27
Isolation and culture of hMSCs and human osteoblasts
Bone marrow was obtained from the iliac crest of three nonosteoporotic, healthy female donors (19–25 years old) who provided informed consent. Procedures were approved by the local ethics committee of Seoul National University Dental Hospital, according to the legal regulations for human tissue and organs in Korea (CRI05008). hMSCs from bone marrow were cultured as described previously.28,29 Nucleated cells that concentrated at the interface after Ficoll-Paque (Amersham Biosciences, Uppsala, Sweden) treatment were collected and washed with phosphate-buffered saline (PBS). Collected cells were plated at a density of 2×106 cells/100 mm and cultured in expansion medium comprising low-glucose Dulbecco's modified Eagle's medium (DMEM; Welgene, Inc., Daegu, South Korea), 100 U/mL penicillin, 100 mg/mL streptomycin, and 10% heat-inactivated fetal bovine serum (HI-FBS) under a humidified atmosphere of 5% CO2 at 37°C, with medium changes every 3 or 4 days. Cells were passaged when they reached 70% confluence and reseeded in a new culture plate at a density of 3×105 cells/cm2. Cells from the second to fifth passages were used for all experiments. The ability of hMSCs to differentiate into osteoblasts, chondrocytes, or adipocytes was confirmed according to previously published protocols. 29
Primary human osteoblasts were derived from mandible biopsy specimens of four healthy, female human donors (19–25 years old) who provided informed consent. To obtain bone-derived cells, bone samples were minced and washed twice with PBS. The samples were incubated for 40 min with gentle shaking in a solution containing 0.1% type I collagenase (Sigma-Aldrich) and 0.2% dispase (Roche, Indianapolis, IN) in serum-free DMEM at 37°C. This process was performed twice, and the cells that were obtained were combined for each case and cultured separately. To investigate the effect of estrogen on SOST expression in association with BMP signaling and osteoblast differentiation, hMSCs were treated with E2 or a combination of E2 and BMP2 in osteogenic differentiation medium with DMEM supplemented with ascorbic acid (50 μg/mL) and β-glycerophosphate (10 mM) without dexamethasone (DEX), added at 1 or 2 days postplating when the cells were confluent.
Transient transfection and luciferase reporter assays
Transient transfections for luciferase reporter assays were performed using Lipofectamine 2000 reagent (Invitrogen). hMSCs were transfected with 1 ng/μL of TOPFlash luciferase reporter plasmids or Smad reporter plasmids in serum-free DMEM without antibiotics. Positive and negative reporters, corresponding to each reporter assay, were also included according to the manufacturer's instruction. After 5 h of transfection, the cells were stabilized by culturing overnight in DMEM with 10% HI-FBS, and then plated in 96-well plates (5000 cells/well). The next day, the cells were treated with E2 (100 nM), BMP2 (200 ng/mL), E2/BMP2, or no treatment for 24 h. The cells were washed once with PBS and lysed with 1× passive lysis buffer. Luciferase activity was measured using a Turner 20/20 luminometer with a Dual-Luciferase Reporter Assay kit (Promega) according to the manufacturer's instructions. Luciferase activity was calculated as the ratio of firefly luciferase activity to that of Renilla luciferase.
Gene silencing using siRNA targeting ERα or SMAD
Endogenous ERα or SMAD4 expression was knocked down by siRNA transfection using human ERα siRNA (sc-29305) or Smad4 siRNA (sc-29484), which refers to a pool of four target-specific 19 to 25 oligonucleotide siRNAs. siRNA-A, a nontargeting 20 to 25 oligonucleotide (sc-37007), was used as the control. hMSCs were transfected with human sequence-specific ERα/Smad4 siRNA or control siRNA-A at a final concentration of 30 pM using Lipofectamine 2000 reagent in serum-free DMEM without antibiotics for 5–6 h, according to the manufacturer's instructions (Invitrogen). After transfection, the cells were stabilized by culturing overnight in DMEM with 10% HI-FBS, and then the medium was replaced with DMEM with 1% HI-FBS. After incubation for 24 h, the cells were treated with E2 (100 nM), BMP2 (200 ng/mL), or E2/BMP2 or left untreated.
Quantitative real-time reverse transcription–polymerase chain reaction
Total RNAs were extracted by the addition of 0.5 mL of TRIzol reagent (Life Technologies, Waltham, MA) to cells on a plate as described in the manufacturer's instructions. One microgram of RNA was subjected to cDNA synthesis with SuperScript™ Reverse Transcriptase II and oligo12–18 primers (Invitrogen). We used SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) to detect the accumulation of PCR product during cycling with the ABI Prism 7700 Sequence Detection System (Applied Biosystems). The thermocycling conditions were as follows: predenaturation at 95°C for 10 min; 40 cycles of denaturation at 95°C for 15 s, and annealing and extension at 60°C for 1 min, followed by a final dissociation cycle at 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. Real-time reverse transcription–polymerase chain reaction (RT-PCR) was carried out in triplicate in at least three independent experiments (n>3). Oligonucleotide primers were designed using real-time RT-PCR system sequence detection software v1.3 (Applied Biosystems) and their sequences are provided in Table 1. Fold differences in the expression level of each gene were calculated for each treatment group using CT values normalized to transcript levels of the housekeeping gene, 18S rRNA, according to the manufacturer's instructions.
ALP, alkaline phosphatase; BMP2, bone morphogenetic protein-2; IGF1, insulin-like growth factor-1; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor kappa-B ligand; SOST, sclerostin; 18S rRNA, 18S ribosomal RNA.
Alkaline phosphatase assay
We assayed alkaline phosphatase (ALP) activity by measuring the amount of ρ-nitrophenol produced using ρ-nitrophenol phosphate substrate. Cell lysates were mixed with alkaline buffer solution and gently shaken for 10 min. ALP substrate was added at room temperature for 30 min. After the reaction was stopped with 0.05 N NaOH, the absorbance at 405 nm was read and compared with a standard curve prepared with ρ-nitrophenol standard solution. Enzyme activity was normalized to the protein concentration of the cell layer determined using a protein assay (Bio-Rad, Hercules, CA).
Enzyme-linked immunosorbent assay
Sclerostin or Opg levels in the culture supernatants were determined using an ELISA kit (R&D Systems). After centrifugation, cell culture supernatants (n=4–5) were added to 96-well ELISA plates. Standards for cytokines (0–1000 pg/mL) were run in each series. After incubation, aspiration, and washing, a human conjugate of each protein (100 μL/well) was added according to the manufacturer's instructions. The OD of each well was determined within 30 min using a microplate reader set to a 450-nm wavelength with correction for optical imperfections in the plate.
Western blotting
Total cell lysates of cultured hMSCs were prepared by lysing cells in RIPA buffer containing 50 mM Tris buffer (pH 7.5), 50 mM NaCl, 1% Triton X-100, 1 mM ethylene glycol tetraacetic acid, 10 mM Na4P2O9, 5 mM Na3VO4, 50 mM NaF, and protease inhibitors (10 μg/mL leupeptin, 10 μg/mL aprotinin, 10 μg/mL pepstatin A, 0.1 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol). Nuclear and cytoplasmic fractionation of cultured hMSCs was performed using NE-PER™ nuclear and cytoplasmic extraction reagents (Thermo Scientific, Rokford, IL) according to the manufacturer's instructions. Total cell lysates (20 μg/lane) or nuclear/cytoplasmic fractions (15 μg/lane) were subjected to electrophoresis through a 10% sodium dodecyl sulfate–polyacrylamide gel and transferred to PolyScreen polyvinylidene difluoride membranes (PerkinElmer Life Sciences, Hopkinton, MA). Membranes were blocked with 5% nonfat dry milk in TBST buffer (0.1 M Tris-buffered saline [pH 7.5]/0.1% Tween) and probed with antibodies. The following antibodies were used in this study: polyclonal rabbit anti-total β-catenin (Abnova, Taipei, Taiwan); polyclonal rabbit anti-nonphospho (active) β-catenin antibody, diluted 1:2000; polyclonal rabbit anti-ERα antibody, diluted 1:200; monoclonal mouse anti-Smad4 antibody, diluted 1:200; and monoclonal mouse anti-α-tubulin antibody, diluted 1:200. Primary antibodies were detected with horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG; Santa Cruz Biotechnology) for total/active β-catenin and ERα or with HRP-conjugated anti-mouse IgG (Dako, Glostrup, Denmark) for Smad4 and α-tubulin. Finally, blots were stained with an enhanced chemiluminescence detection system (Amersham Biosciences) according to the manufacturer's instructions.
Immunofluorescent staining
Sclerostin or β-catenin expression in hMSCs was detected at an average cell density of 3000 cells per coverslip. Cells were washed thrice with PBS, fixed in 4% paraformaldehyde for 30 min, and blocked in blocking solution for 30 min. Cells were incubated with antibody against sclerostin (anti-rabbit, 1:100; Abnova) or active β-catenin (anti-rabbit, 1:400; Cell Signaling Technology, Inc.) overnight at 4°C, washed twice with PBS, and then incubated with secondary antibody labeled with fluorescein isothiocyanate (anti-rabbit IgG, 1:500; Biomeda) for 1 h. Primary antibody controls were processed in parallel using only the secondary antibody. Slides were washed in PBS for 10 min and incubated with PBS-buffered 4′,6-diamino-2-phenylindole solution (1 mg/mL; Santa Cruz Biotechnology) using a dilution ratio of 1:1000 for nuclear staining. Section images of the stained cells were captured using an Olympus Fluoview FV300 confocal laser scanning microscope and Fluoview software (Olympus Optical Co., Ltd.).
Statistical analyses
All data are presented as mean±standard error of the mean. Statistical analyses were performed using SPSS 20 (IBM Co., Armonk, NY). Differences between two groups were evaluated using two-tailed Student's t-test, and the comparison of data for more than two groups was performed using two-way analysis of variance post hoc through the Bonferroni method. Differences with p<0.05 were considered significant.
Results
Induction of SOST by BMP2 in hMSCs
Treatment of hMSCs and human osteoblasts with the BMPs, BMP-2, -4, and -6, has been reported to induce SOST. 16 We confirmed that BMP2 treatment (200 ng/mL) induced SOST expression in primary hMSCs prepared from three female donors. In contrast to SOST induction in murine cells, 9 SOST induction by BMP2 in human cells gradually increased with time, consistent with a previous report. 16 Although SOST upregulation differed among the donors, it showed a time-dependent increase in all cases (Fig. 1). In two of the tested hMSC cultures (#1 and 2), SOST expression was significantly increased by BMP2 from day 3, and a noticeable increase in SOST expression was generally observed at day 7 after treatment in all tested cells (Fig. 1). However, in untreated hMSCs, the basal level of SOST did not increase over time from day 1. BMP2 treatment also enhanced the expression of the bone-forming markers, ALP and IGF1, which are BMP2-responsive genes, in all three hMSC cultures.
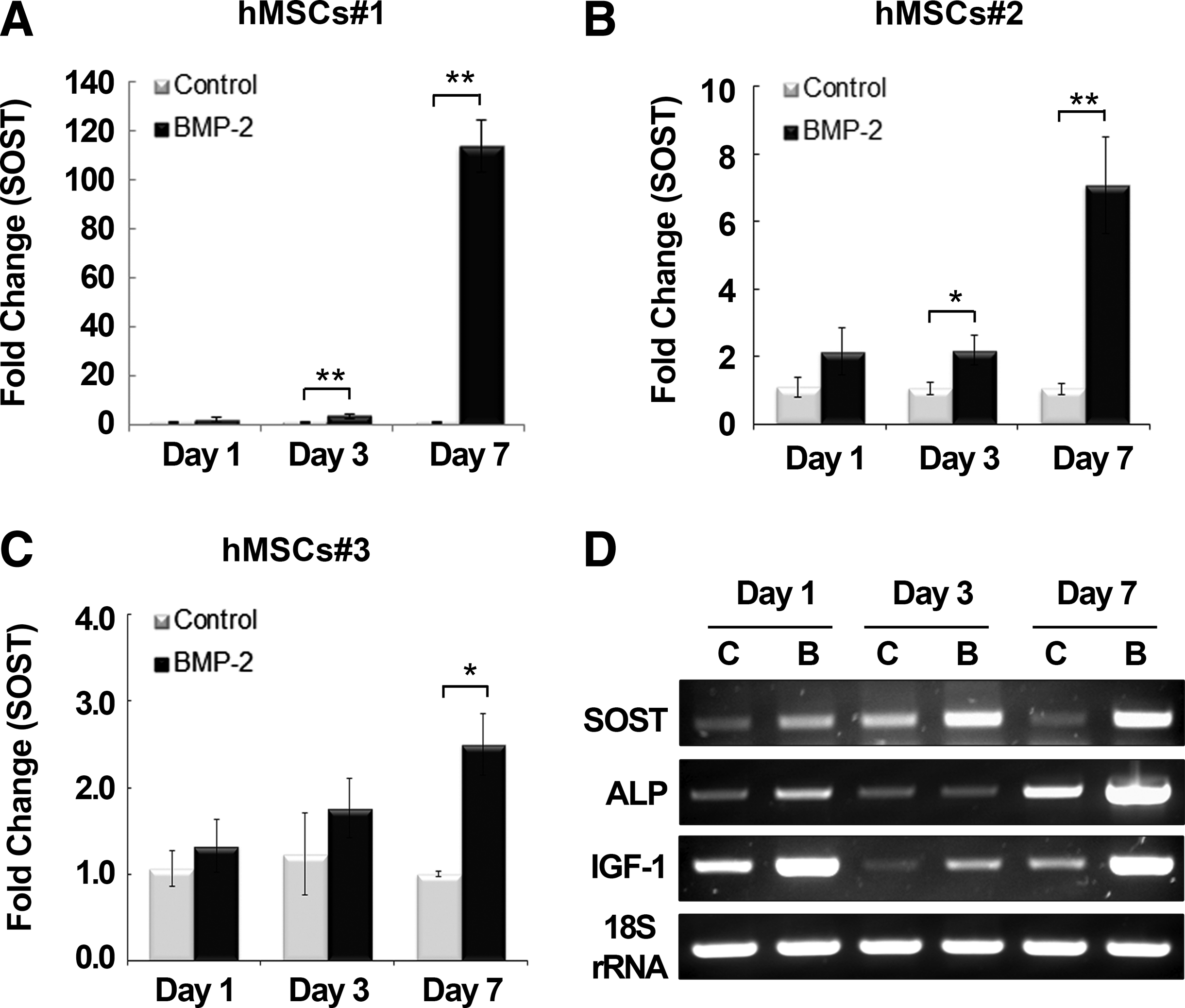
Time course of sclerostin (SOST) expression in human mesenchymal stromal cells (hMSCs) treated with bone morphogenetic protein (BMP)2. Three different cell lines of primary hMSCs (hMSCs#1, 2, 3) were prepared from the bone marrow of healthy female donors. SOST expression was examined using real-time reverse transcription–polymerase chain reaction (RT-PCR) at days 1, 3, and 7 after treatment with BMP2 (200 ng/mL). Values are expressed relative to those of nontreated hMSCs at day 1 with normalization to 18S rRNA for hMSCs#1
Effects of estrogen on BMP2-mediated SOST induction
The effect of estrogen on BMP2 signaling was investigated at day 7 because all cultures of hMSCs showed SOST upregulation at this point. We used 100 nM of E2, a concentration that was shown in a previous report to stimulate the greatest extent of osteoblast differentiation in hMSCs. 30 SOST expression was examined at day 7 after treatment of cells with BMP2 (200 ng/mL), E2 (100 nM), or both BMP2 and E2, and the results were compared with those of nontreated cultures (Fig. 2). E2 alone had no effect on the basal expression of SOST in hMSCs, whereas BMP2 increased its expression (p<0.01) in all three hMSC lines (#1, 2, and 3). Interestingly, there was a marked decrease in the level of SOST expression (p<0.01 compared with BMP treatment) when hMSCs were cotreated with E2 and BMP2 (Fig. 2A).
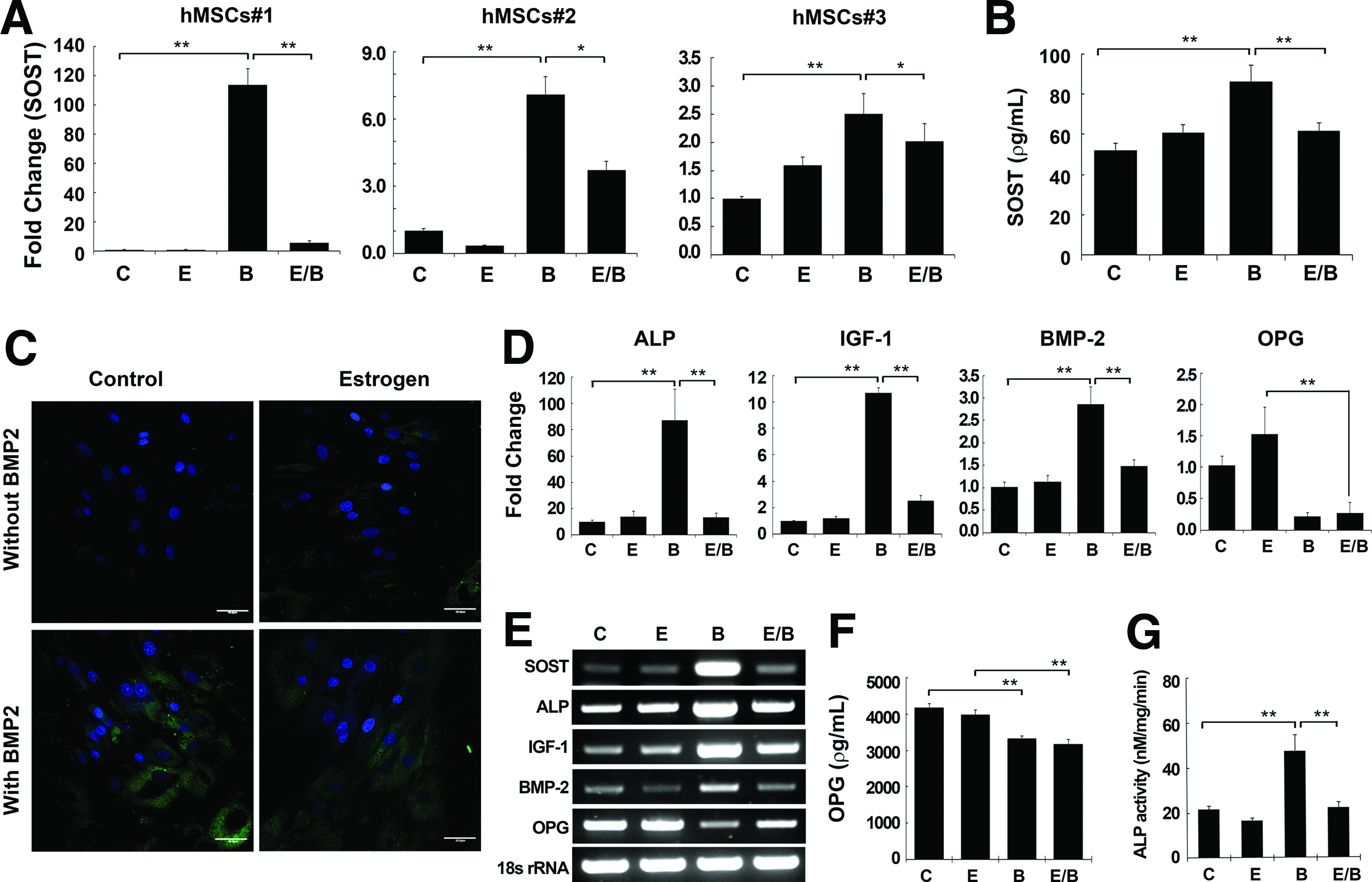
Suppressive effect of estrogen on BMP2 signaling in hMSCs. The three different lines of primary hMSCs (hMSCs#1, 2, 3) used in Figure 1 were treated with E2 (E; 100 nM), BMP2 (B; 200 ng/mL), or E2 and BMP2 (E/B) for 7 days.
SOST expression was further examined at the protein level using ELISA and immunofluorescent staining in hMSC#1 and #2 (data are presented for hMSCs#1), which showed a similar phenotype after treatment with E2, BMP2, or E2/BMP2. Extracellular release of sclerostin was not affected by E2 alone, but was increased by 65.7% by BMP2 compared with the nontreated control group (Fig. 2B). Moreover, consistent with the transcriptional expression of SOST, treatment of cells with a combination of E2 and BMP2 decreased Sost protein expression by 28.3% relative to BMP2 treatment alone. Immunofluorescent detection of intracellular sclerostin expression at day 7 was intense in the cytosol, but not in the nuclei, with a similar pattern to the ELISA results (Fig. 2C).
We also examined the expression of other BMP2-responsive genes after E2 treatment (Fig. 2D). Similar to SOST expression, E2 alone had no effect on the basal expression of ALP, IGF1, or BMP2 in hMSCs, whereas BMP2 treatment significantly upregulated the expression of these markers (data shown for hMSC line#1). Likewise, expression of the ALP, IGF1, and BMP2 genes decreased when the cells were treated with both E2 and BMP2, as observed for SOST expression. The expression of OPG, a presumed target of estrogen, was not influenced by E2 treatment, but was downregulated by BMP2 (p<0.01). However, the real-time RT-PCR analysis to detect RANKL expression was the same as that of the negative control without cDNA in all test groups, indicating that RANKL expression in hMSCs is too low to be detected. Cotreatment of hMSCs with E2 and BMP had no effect on OPG expression, which was maintained at a reduced level after BMP2 treatment. This pattern was consistent in all tested hMSCs. Consistent with real-time RT-PCR data, Opg expression by ELISA was decreased by 20.0% (p<0.01) by BMP2 or by 23.9% (p<0.01) by E2/BMP2 treatment compared with the nontreated control group, whereas there was no change in the E2 only group (Fig. 2F). ALP activity showed the same trend as expression by real-time RT-PCR (Fig. 2G).
Involvement of Wnt/β-catenin/ERα signaling in the E2-mediated suppression of SOST induction by BMP2
Both the Smad and Wnt signaling pathways have been implicated in modulating the suppressive effect of E2 on BMP2-mediated SOST induction. 31 First, we investigated the effect of the Wnt pathway on E2- and BMP2-mediated SOST induction in hMSCs#2. We tested whether E2 increases the transcriptional activity of β-catenin using a TCF/LEF-responsive vector (Fig. 3A). There was a significant (17.8-fold, p<0.01) increase in TOPFlash luciferase activity in E2-treated cells compared with nontreated controls, whereas cells treated with BMP2 alone did not show any change. Combined treatment with E2 and BMP2 resulted in a 3.7-fold increase over the control (p<0.01), which corresponded to a reduction compared with E2 alone (p<0.01), but an increase compared with BMP2 alone (p<0.01). Positive and negative controls functioned as expected in the TOPFlash luciferase assay.

E2 activation of the Wnt/β-catenin/estrogen receptor (ER)α signaling pathway.
The effect of the Wnt pathway on E2- and BMP2-mediated SOST induction was assessed by treatment with the Wnt inhibitor, ICI 182,780 (100 nM), as an antagonist of ERα. 32 Treatment with ICI 182,780 partially recovered the E2-mediated suppression of SOST to levels similar to those after BMP2 treatment alone, but did not affect BMP2-mediated SOST induction (Fig. 3B). We confirmed sclerostin expression at the protein level using ELISA (Fig. 3C). Consistent with the results of real-time RT-PCR, ICI 182,780 treatment recovered sclerostin expression of cells cotreated with BMP2 and estrogen similar to the level of sclerostin observed in BMP2-treated cells, but had little effect on sclerostin induction by BMP2. These results implicate involvement of the Wnt pathway or ERα in the action of E2 on sclerostin suppression through cross talk with BMP signaling.
Effect of ERα gene silencing on E2 signaling in association with BMP2 signaling
Because the Wnt inhibitor used in this study targets ERα, we investigated the involvement of ERα in the Wnt pathway during estrogen signaling. Blocking of ERα expression using ERα siRNA was confirmed by western blotting using anti-ERα antibody (Fig. 3E). In cells transfected with control siRNA, there was a noticeable difference in the level of active β-catenin after E2 treatment, whereas total β-catenin level showed little difference from the nontreated control. However, the enhanced expression of active β-catenin induced by E2 was slightly decreased by combined treatment with BMP2, consistent with results of the TOPFlash reporter assay.
Transfection with ERα siRNA resulted in lower expression of β-catenin after E2 treatment (alone and with BMP2) compared with the same groups transfected with control siRNA. Real-time RT-PCR showed that transcriptional SOST expression after transfection with ERα siRNA was consistent with the effects of the Wnt inhibitor, ICI 182,780 (Fig. 3D). BMP2 still increased SOST expression 3.9-fold, but the suppressive effect of E2 on BMP-mediated SOST induction was not evident in cells transfected with ERα siRNA, indicating that ERα plays a role in SOST regulation through interaction between estrogen and BMP2.
Additionally, we observed nuclear localization of activated β-catenin after estrogen treatment. Immunofluorescent staining using an antibody specific for active β-catenin on subcellular fractions confirmed that estrogen increased the level of cytoplasmic β-catenin compared with that of control cells (Fig. 3F). Active β-catenin expression in the cytosolic fraction was more apparent after estrogen treatment, compared with the control. Nuclear localization of active β-catenin was clearly detected at 1 hr after estrogen treatment, while it was diminished at 3 h.
Western blotting at the time points of 1 and 24 h under the same conditions as in Figure 3E showed a similar trend to immunofluorescent staining (Fig. 3G). In cells transfected with control siRNA or ERα siRNA, estrogen alone enhanced cytosolic expression of active β-catenin at 1 and 24 h compared with the nontreated control. However, the nuclear fraction of active β-catenin was slightly increased at 1 h, and then decreased with time, showing little expression at 24 h in all treatments (estrogen, BMP, estrogen+BMP). Transfection with ERα siRNA resulted in less expression of active β-catenin in the cytosolic and nuclear fractions after E2 treatment (alone and with BMP2), especially at 24 h, compared with the same groups transfected with control siRNA. Total catenin expression was mainly observed in the cytosolic fraction with little difference between treatments, which was also independent of blocking ERα. We confirmed the absence of cross-contamination between nuclear and cytoplasmic fractions (data not shown).
Noninvolvement of the Smad pathway in the modulation of SOST expression by E2 coupled with BMP signaling
BMP2 activates intracellular mediators such as Smad proteins to induce SOST; however, it is unclear whether the interaction between E2 and BMP2 signaling involves the Smad pathway. The transcriptional activity of SMAD4 was not affected by E2 treatment in the Smad-luciferase reporter assay, whereas it was significantly increased by BMP2 treatment (BMP2 alone and E2/BMP2) compared with the nontreated control, with no difference between BMP2 alone and BMP2 plus E2 (Fig. 4A). SOST expression was assessed after treatment with dorsomorphin (10 μM), a Smad inhibitor, using real-time RT-PCR and ELISA, which led to a complete blockage of SOST induction by BMP2 treatment, with or without E2 (Fig. 4B, C). 33 Further investigation using Smad4 siRNA showed a similar trend to treatment with dorsomorphin. Silencing of the SMAD4 gene using anti-Smad4 antibody was confirmed by western blotting, which showed a definite reduction in BMP2-induced expression after transfection with Smad4 siRNA transfection compared with the same group with control siRNA (Fig. 4D). There was little difference in the levels of active β-catenin between cells transfected with control siRNA or Smad4 siRNA. There was a complete blockage of SOST induction in cells transfected with Smad4 siRNA after BMP2 treatment, with or without E2, as seen after treatment with the Smad inhibitor, dorsomorphin (Fig. 4E).
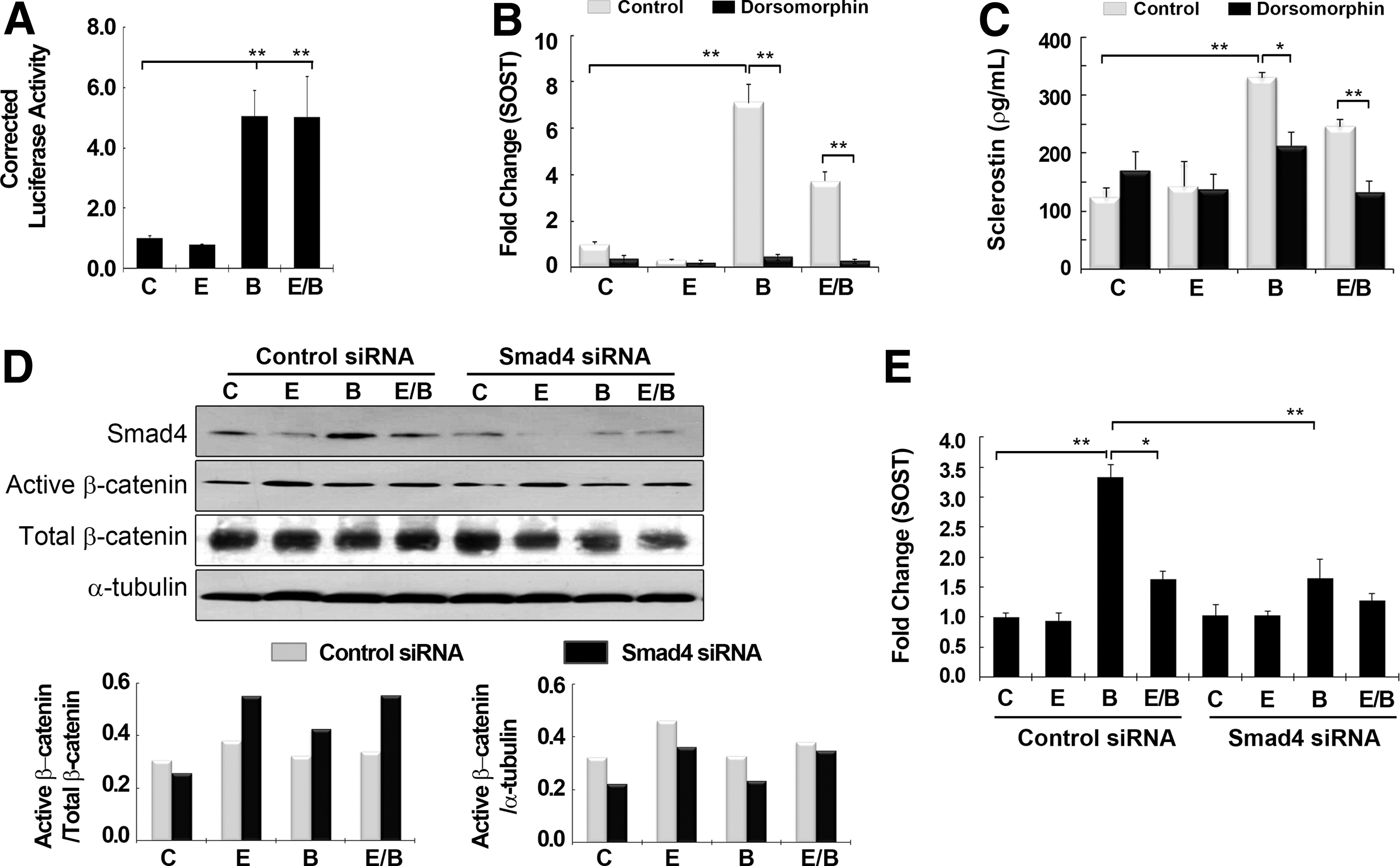
E2 signaling is independent of the Smad pathway.
Effect of estrogen on BMP2-induced upregulation of SOST in human osteoblasts
To determine whether estrogen had the same effect on BMP2 signaling in undifferentiated hMSCs and differentiated osteoblasts, we examined the effects of estrogen on primary human osteoblasts derived from the mandibles of four females. We pursued the same strategy as for hMSCs, using real-time RT-PCR and ELISA to assess the effects of E2 on BMP2-induced SOST expression. In contrast to the common response of all three hMSC cultures to E2, the effect of E2 on SOST induction by BMP2 differed among the human osteoblast lines (Fig. 5A). Of the four different cell lines, two cell lines (#1 and #2) showed the same pattern as seen in hMSCs, that is, E2 had an inhibitory effect on BMP2-mediated SOST upregulation, whereas in the other two cell lines (#3 and #4), E2 did not change the BMP2-induced level of SOST. Representative cell line #1, in which E2 had an inhibitory effect, expressed sclerostin protein by ELISA in a manner similar to the SOST levels determined by real-time RT-PCR (Fig. 5A, B).

The response of human osteoblasts to estrogen in association with BMP2 signaling. Primary human osteoblasts derived from four different donors (hOB#1, 2, 3, and 4) were treated with none (control; C), E2 (E; 100 nM), BMP2 (B; 200 ng/mL), or E2 and BMP2 (E/B) for 7 days.
An interesting finding was that combined treatment of osteoblasts with E2 and BMP2 did not suppress the upregulated expression of other BMP2-responsive genes, such as ALP or IGF1, in any of the tested osteoblasts, independent of whether estrogen was able to suppress the BMP2-mediated increase in SOST expression (Fig. 5C). In addition, we found that ALP expression in human osteoblasts was not significantly increased in response to BMP2 treatment, in contrast to hMSCs. However, OPG expression was consistent with that in hMSCs and was not changed by E2 treatment compared with the control, but was downregulated by BMP2. Cotreatment with E2 and BMP had no effect on OPG expression, which was maintained at a reduced level similar to that after BMP2 treatment. The protein levels of ALP and Opg were consistent with their transcriptional levels following treatment with E2, BMP2, or E2/BMP2 (Fig. 5D, E). Unlike hMSCs, ALP activity did not show significant increases with BMP2 treatment. Furthermore, there was no relationship between SOST and ALP activity in any of the cell lines. In vitro matrix mineralization determined by von Kossa staining showed little difference from the control group in the four osteoblast cell lines after treatment with Dex, E2, BMP2, or E2/BMP2 (Fig. 5F).
Relationship between SOST expression and the ratio of RANKL to OPG expression in human osteoblasts
Using human osteoblast cell lines #1 and #2, which showed estrogen-mediated suppression of SOST induction by BMP2, we investigated the effect of BMP2 or E2 dose on SOST expression (data shown are for osteoblast line #1). SOST expression was examined at increasing concentrations of BMP2 (100, 200, and 300 ng/mL) and was found to reach a maximum at 200 ng/mL and then decrease at 300 ng/mL (Fig. 6A). The expression level of the osteoclast marker, RANKL, differed among individuals and either followed a similar pattern to SOST expression or did not change appreciably as the dose of BMP2 increased. However, OPG expression was decreased by BMP2 treatment independent of the BMP2 dose. Nevertheless, the relative ratio of RANKL to OPG showed an identical pattern to that of SOST expression with increasing doses of BMP2, indicating that the increase in the ratio of RANKL to OPG is probably caused by the induction of SOST by BMP2 (Fig. 6B). The effect of the E2 dose was investigated by incubating cells with increasing concentrations of E2 (1, 10, and 100 nM) at a fixed concentration of BMP2 (200 ng/mL) (Fig. 6C). E2 had a suppressive effect on SOST induction regardless of the dose. The relationship between the E2 dose and expression of RANKL and OPG was not definite. However, the resulting ratio of RANKL to OPG expression showed a similar pattern to SOST expression and was significantly decreased by E2 compared with that of cells treated with BMP2 only, independent of the E2 dose.

Relationship between SOST expression and the ratio of receptor activator of nuclear factor kappa-B ligand (RANKL) to OPG expression in human osteoblasts.
Discussion
There is a growing interest in sclerostin-based treatment as a novel potential anabolic therapy for postmenopausal osteoporosis. An antibody to sclerostin dramatically increased bone formation in ovariectomized rats and intact monkeys, without affecting bone resorption.21,22 Although these data implicate a suppressive effect of estrogen on sclerostin production, the precise molecular mechanism by which estrogen regulates the transcriptional expression of SOST remains unclear. Interestingly, previous studies showed that BMPs are potential inducers of SOST expression in mouse- and human-derived MSCs or osteoblasts,9,16 whereas growth factors such as IGF1 or transforming growth factor β had no such effect in hMSCs. 16 Therefore, this study aimed to elucidate the mechanism of estrogen modulation of SOST expression in human bone cells in association with BMP2 signaling, which would provide a logical basis for recent therapies targeting sclerostin in osteoporotic patients.
BMP2 upregulates SOST expression through the Smad pathway.9,11 In contrast to other BMP antagonists such as noggin or chordin, which inhibit BMP2 signaling by directly binding the BMP receptor, 34 the produced sclerostin has no direct effect on BMP signaling involving Smad phosphorylation and Smad-driven transcriptional reporter activation. 35 Instead, sclerostin inhibits the Wnt signaling that is required for BMP-stimulated osteoblastic differentiation, which explains the high bone mass in diseases associated with SOST mutations, such as sclerosteosis and van Buchem disease.36,37 The induction of SOST by BMPs is a controversial component of BMP action. It is reported that this catabolic activity of BMP is modulated by several steroids at the transcriptional level. 38 DEX has been shown to be an antagonist of SOST in BMP4-treated cells; in contrast, retinoic acid or 1,25(OH)2D3 enhances the levels of SOST in vitro. 16 A recent study reported that estrogen inhibits BMP2 signaling, including the expression of ALP and osteocalcin, in the murine stem cell line, C2C12. 39 This tendency was also observed in the current studies using human MSCs and osteoblasts. Estrogen showed an inhibitory action on the upregulation of the catabolic marker, SOST, as well as anabolic markers (ALP and IGF1) by BMP2. Although there was no direct effect of estrogen alone on SOST expression in vitro, these results imply a molecular link between estrogen signaling and SOST expression with indirect modulation through BMP2 signaling.
We found that the Wnt/ERα/β-catenin signaling pathway participates in estrogen signaling for SOST suppression. Consistent with previous reports in cell lines derived from rodents, our findings showed that estrogen activated the Wnt/ERα/β-catenin signaling pathway in hMSCs.40,41 E2 alone increased transcriptional activity of β-catenin and the level of active β-catenin, whereas BMP2 failed to do so. β-Catenin expression with combined treatment of E2 and BMP2 was higher than that induced by BMP2 treatment alone, but lower than that induced by E2 treatment alone. The role of ERα was critical in estrogen-mediated β-catenin activation and SOST suppression. The suppressive effect of E2 on SOST induction by BMP2 was abolished both in ERα-silenced cells and by treatment with the Wnt inhibitor, ICI 182,780, which causes ERα degradation, 42 whereas BMP2 signaling in SOST induction was not affected under the same conditions. These results suggest that even in the presence of BMP2 signals, which suppress the Wnt pathway, estrogen/ERα exerts positive control in the activation of β-catenin. The current findings support the initial suggestion that estrogen and BMP signaling converge on SOST expression and Wnt pathways in bone cells.4,10,25,26 Sutherland et al. suggested participation of steroid hormone receptor complexes created with DEX, which may interfere with the binding of SMADs or create an inactive complex, resulting in a decrease in the BMP-induced transcription of SOST, thus tuning BMP2 activity toward favoring bone formation when BMPs upregulate antagonists such as SOST. 16 Current findings suggest that estrogen is such a modulating factor that downregulates SOST levels induced by BMP2 in human bone-forming cells by activating the Wnt/β-catenin pathway through ERα.
This study also investigated the role of the Smad pathway in estrogen signaling. It is reported that BMP2 upregulates SOST expression through the Smad pathway, ultimately blocking the Wnt pathway.9,11 The current finding that E2 inhibited BMP2-mediated SOST expression suggests that there is a connection between ERα/β-catenin and the Smad pathway. Our experiments targeting Smad4 expression indicated that the Smad pathway plays a critical role in BMP2-mediated SOST induction, as seen in a previous report. 9 However, estrogen did not exert any influence on Smad4 activity. Because of lack of evidence from the current study, modulation of the Smad pathway by activated β-catenin needs to be explored further. This question might be explained by previous studies.31,39 It was reported that in the presence of E2, ERα suppresses the prodifferentiation actions of BMP2 by inhibiting BMP2-induced activation of Smads and Smad-mediated transcription. 31 In addition, it is also speculated that the inhibitory effect of E2 on BMP2 signaling is mediated by phosphorylation of ERKs in the Smad1 linker region, which in turn increases the proteasomal degradation of Smad1 and ultimately contributes to the suppressive effect of estrogens on osteoblastogenesis. 39 Taken together, these results imply that estrogen signaling is probably not linked directly to the Smad-related signaling pathway, but instead indirectly inhibits the Smad pathway in the presence of BMP2.
The current results indicate that the effect of E2 on BMP2 signaling changes according to the stage of osteoblast development. E2 had an inhibitory effect on the BMP2-responsive genes, ALP, IGF1, and SOST, in undifferentiated hMSCs independent of the donor. However, in human osteoblasts, the effect of E2 showed individual differences. Specifically, E2-mediated inhibition of SOST induction by BMP2 was observed in two of the tested osteoblast lines, whereas E2 did not inhibit ALP or IGF1 expression in any of the osteoblast lineages tested, in contrast to the observations for hMSCs. To confirm the cell specificity of these stimuli, we examined the effects of estrogen and BMP2 in other types of human cells, such as skin fibroblasts and liver cells (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea). There was no estrogen-mediated suppression of SOST induction by BMP2 in either cell line, although the fibroblasts showed SOST induction by BMP2. These results indicate that estrogen-mediated suppression of BMP signaling is specific to bone cells and might be more apparent in the undifferentiated state.
A unique point is that estrogen does not inhibit BMP2-mediated bone-forming activity in cells that have differentiated into osteoblasts, but still suppresses the antibone-forming activity of BMP2 signaling in an individual-dependent manner. All of the osteoblast cell lines used in this study failed to show a significant increase in ALP activity after treatment with BMP2, as seen in hMSCs. 29 Moreover, the development of in vitro mineralization exhibited little difference after treatment with E2, BMP2, E2/BMP2, or DEX from the nontreated control group, although all experimental groups, including the control group, were positively stained. These data imply that all of the osteoblast cell lines might be at a later stage of osteogenic differentiation at the time of isolation. Nevertheless, SOST expression was different and highly dependent on the individual donor. Taken together, our data imply that estrogen might be a modulator of BMP2 activity that antagonizes SOST activity at all stages of osteoblast development in adult hMSCs and human osteoblasts. However, this leaves the question of whether this action of estrogen coupled with BMP signaling differs according to the differentiation state of bone cells or has individual variations. The estrogen response implies that estrogen plays a dynamic role in enhancing bone formation at the level of osteoblast cells.
SOST activity is linked to BMP-BMPR1A signaling in osteoblasts and triggers RANKL-OPG pathway-induced osteoclastogenesis in vivo. 39 We aimed to identify a downstream marker of BMP2-mediated SOST induction in hMSCs or human osteoblasts. The current results showed that OPG was significantly downregulated when hMSCs were treated with BMP2, whereas there was no basal expression of RANKL. In contrast to the absence of basal RANKL expression in hMSCs, there was a distinct relationship between SOST expression and the ratio of RANKL to OPG in human osteoblasts. A previous animal study using Bmpr1a-deficient mice showed that the effect of BMP was mediated through Bmpr1A and that Bmp increased SOST expression, as well as the ratio of RANKL to OPG expression. 39 In contrast to this animal study, clinical studies showed that treatment of postmenopausal women with estrogen did not induce any changes in Opg levels, whereas estrogen significantly reduced the levels of serum sclerostin and other markers of bone resorption. 4 Current data using human samples showed that OPG expression was downregulated by BMP2, but the addition of E2 did not influence the BMP2-mediated inhibition of OPG expression in either hMSCs or osteoblasts.
The effects of BMP2 and E2 on RANKL expression differed depending on the individual. Above all, the ratio of RANKL to OPG expression was consistent with that of SOST expression, which was increased by BMP2 and decreased by cotreatment with E2 and BMP2 in differentiated osteoblasts, but not in hMSCs. Notably, the inhibitory effect of estrogen on the ratio of RANKL to OPG expression was similarly independent of the E2 dose, meaning that E2 is active at low doses. This result is similar to data reported by a study of Bmpr1a-modified mice, in which the ratio of RANKL to OPG expression was a determining factor when Bmpr1a was deficient or constitutively expressed. 39 Consistent with these data, we also demonstrated in a recent publication that the ratio of RANKL/OPG s correlated with in vivo bone-resorbing activity of rhBMP-2 during the early healing period. 43 Taken together, these findings suggest that the target of SOST inhibition by E2 in bone metabolism might be the ratio of RANKL to OPG rather than the concentration of OPG or RANKL alone at the osteoblast level. However, further study is required to elucidate which factor is linked to individual differences in the effect of estrogen on SOST expression.
In conclusion, we investigated the molecular mechanism by which estrogen regulates SOST expression in association with BMP2 signaling. We demonstrated that the mechanism of E2 action on SOST expression involved activation of the Wnt pathway involving ERα and β-catenin. In differentiated osteoblasts, the effect of E2 on SOST expression differed among individuals. It is intriguing that the relative ratio of RANKL to OPG expression after BMP2 or E2 treatment was a downstream marker of SOST expression in differentiated osteoblasts, but was indeterminate in hMSCs because of the extremely low-level expression of RANKL. The strength of this study is the demonstration of cross talk between estrogen and BMP2 signaling in SOST regulation, which is probably one of the mechanisms involved in bone homeostasis by estrogen at the level of human osteoblast cells. Therefore, this study provides a logical basis for recent therapies targeting sclerostin in osteoporotic patients. However, further studies are needed to more precisely define why estrogen signaling requires interaction with BMP signaling to regulate SOST expression and whether the effect of estrogen on BMP2 signaling differs depending on the differentiated state or among individuals.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education (NRF-2011-0017104), and by a grant from the Korea Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (A120850). The authors thank T. Cho and S. Ryu for assistance in the preparation of the manuscript and Novosis®-Dent, BioAlpha, Inc., for providing the rhBMP2.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.