
Abstract
Lacrimal gland (LG) insufficiency is a main cause for severe dry eye leading to pain, visual impairment, and eventually loss of sight. Engineering of transplantable LG tissue with secretory capacity is a desirable goal. In this study, a three-dimensional decellularized LG (DC-LG) scaffold with preserved LG morphology was generated by treatment with 1% sodium deoxycholate and DNase solution using porcine LG tissue. To address clinical applicability, the primary in vitro culture of secretory active LG cells from a small tissue biopsy of 1.5 mm diameter was introduced and compared with an established isolation method by enzymatic digestion. Cells from both isolation methods depicted an epithelial phenotype, maintained their secretory capacity for up to 30 days, and exhibited progenitor cell capacity as measured by aldehyde dehydrogenase-1 activity, side population assay, and colony-forming units. Cells from passage 0 were reseeded into the DC-LG and secretory active cells migrated into the tissue. The cells resembled an LG-like morphology and the constructs showed secretory activity. These results demonstrate the possibility of engineering a secretory competent, three-dimensional LG construct using LG cells expanded from a small tissue biopsy and DC-LG as a matrix that provides the native structure and physiological niche for these cells.
Introduction
D
Culture of LG cells has proven to be challenging, but previous studies established secretory active cell cultures from mouse, rat, rabbit, monkey, and human LGs using several enzymatic digestion and filtration steps (suspension culture [SC]).7,8 These techniques require relatively large tissue pieces, which diminish their clinical applicability. Therefore, we investigated the expansion of secretory active LG cells from a small LG biopsy (explant culture method [EC]).
So far, LG epithelial cells have been cultured on collagen, Matrigel, 3T3 feeder cells, amniotic membrane, and in a microgravity bioreactor environment maintaining the secretory function for about 28 days.9,10 Ideally, the reconstruction of transplantable LG tissue demands a scaffold, which not only maintains the secretory function but also possesses three dimensionality, biocompatibility, and surgical manageability. The domestic pig (sus scrofa) provides an easily accessible mammalian model and tissue source to study human diseases. 11 Decellularized xenogeneic organs feature a tissue-specific ultrastructure and molecular composition, they lack cellular immunogenicity, and have shown to enhance cell adhesion, proliferation, and differentiation. 12 The aim of this study was to engineer a three-dimensional LG construct on the basis of decellularized LG (DC-LG) tissue for recellularization with secretory competent cells from a small LG biopsy to engineer functional LG tissue for clinical applications.
Materials and Methods
LG extraction
Fresh LGs were obtained from 8-month-old domestic pigs. All experiments were conducted in accordance with the Association for Research and Vision in Ophthalmology (ARVO) statement for the use of animals in ophthalmic research.
Cell isolation and culture of LG epithelial cells
The cells were either isolated by an SC technique from a 100 mm3 LG piece (10 mm length × 5 mm width × 2 mm height) or by an EC technique from a 3.4 mm3 LG piece (1.5 mm length × 1.5 mm width × 1.5 mm height).
Cell SC technique
The LG was chopped into pieces and incubated with an enzyme cocktail at 37°C (collagenase [350 U/mL], hyaluronidase [300 U/mL], and DNase [40 U/mL]; Sigma-Aldrich, Schnelldorf, Germany) twice (for 20 and 30 min, respectively), filtered through a 70-μm mesh (Roth, Karlsruhe, Germany), centrifuged (Heraeus Megafuge 16; Thermo Scientific, Schwerte, Germany), and filtered through a Ficoll gradient (Sigma-Aldrich). The cell pellet was resuspended with culture medium (Dulbecco's modified Eagle's medium with glutamax [DMEM/F12]) containing 10% (v/v) fetal bovine serum (FBS; Life Technologies, Carlsbad, CA) supplemented with 0.4 μg/mL hydrocortisone, 0.1 nM cholera toxin, 0.18 mM adenine, 5 μg/mL transferrin, 5 μg/mL insulin (all Sigma-Aldrich), 10 ng/mL epidermal growth factor, and antibiotics (Life Technologies) and plated at a density of 4.0 × 104 cells per cm2 on a 3T3/J2 mouse fibroblast feeder layer (a gift from Julie Daniels, Moorfields Eye Hospital).
EC technique
For EC, the tissue piece was chopped into small bits with a mean diameter of 0.1 mm; 15 of these were distributed onto a 3T3/J2 feeder layer and the culture medium was carefully added.
Assembly of the 3T3/J2 feeder layer
3T3/J2 mouse fibroblasts were cultured in DMEM containing 10% (v/v) FBS. For coculture applications, 3T3/J2 cells were inactivated with 10−5 M mitomycin C.
Flow cytometry
For cytokeratin staining, the cells were fixed with Cytofix/Cytoperm™ buffer supplemented with 0.2% (v/v) Triton X-100 for 30 min at 4°C (BD Bioscience, Heidelberg, Germany) and washed with appropriate washing buffers. One million cells were incubated with 2 μL of Alexa Fluor® 488-conjugated isotype control antibody (mouse IgG1 kappa) or with Alexa Fluor 488-conjugated anti-pan-cytokeratin (AE1/AE3) antibody (both eBioscience, Affymetrix, San Diego, CA) for 30 min at room temperature (RT).
For detection of aldehyde dehydrogenase-1 (ALDH-1) activity, an ALDEFLOUR™ kit (StemCell Technologies, Köln, Germany) was used. One million cells were incubated with 1 μM ALDH substrate-containing buffer or with 50 mM of the ALDH-1-specific inhibitor, diethylaminobenzaldehyde (negative control), for 1 h at 37°C in the dark.
For side population analysis, the Hoechst 33342 dye exclusion technique was used. One million cells were incubated in DMEM containing 2% (v/v) fetal calf serum either with 5 μg/mL Hoechst 33342 or 50 μM verapamil (Sigma-Aldrich) for 90 min at 37°C. Two micrograms per milliliter propidium iodide was added for nuclear staining. Analysis was performed with a flow cytometer (CyFlow Space; Partec, Münster, Germany).
Cell proliferation analysis
Cell population doubling time (PDT) was calculated according to the formula PDT [d] = H*ln2/ln(c2/c1), where H = duration of passage (days), c2 = cell count at the end of the passage, and c1 = cell count at the start of the passage. To determine the colony-forming units, 1000 cells per well were plated on a 3T3/J2 feeder layer, fixed after 8–12 days, and stained with 1% (w/v) Rhodamine red (Sigma-Aldrich). Cell aggregates >50 cells were regarded as a colony-forming unit.
Decellularization of the porcine LG matrix
The LGs were cut into pieces of about 3 mm diameter, incubated overnight (o/n) in cold phosphate-buffered saline solution (PBS; Sigma-Aldrich) containing 5% penicillin/streptomycin, then incubated thrice for 12 h in three changes of 1% (w/v) sodium deoxycholate monohydrate solution (Sigma-Aldrich), thoroughly washed, transferred into DNase solution (200 U/mL; Roche, Penzberg, Germany) for 24 h, and washed again. All incubation steps were performed at 4°C under continuous agitation. The decellularized scaffolds were γ-irradiated with 25 kGy (BBF Sterilisation-Service GmbH, Rommelshausen, Germany).
Histological characterization and verification of DNA absence
For the Feulgen reaction, a DNA staining kit was used according to the manufacturer's protocol (Merck, Darmstadt, Germany). For DNA quantification, homogenized tissue was mixed with 2 M sodium acetate, water-saturated phenol was added, incubated with chloroform/isoamyl alcohol (49:1 solution), and centrifuged for 20 min at 10,000 rpm at 41°C. The interphase and the lower organic phase were used to precipitate DNA. One hundred percent ethanol was added, incubated for 5 min at RT, and centrifuged again. The DNA pellet was washed with 0.1 M sodium citrate and resuspended in 75% (v/v) ethanol. The supernatant was centrifuged and decanted. DNA was quantified at 280 nm and loaded on a gel for illustration (GeneRuler 1 kb DNA Ladder, 250–10,000 bp; Thermo Scientific).
Determination of collagen content and basement membrane components
For collagen I-V determination, a dye-binding colorimetric assay was used (Sircol™ collagen assay; Biocolor, Newtownabbey, United Kingdom). The tissue was incubated at a pepsin concentration of 0.1 mg/mL in 0.5 M acetic acid at 4°C o/n and processed according to the manufacturer's protocol. Collagen samples delivered by the manufacturer were used as standards. Samples were measured at 550 nm and the collagen amount was normalized to the tissue dry weight (VICTOR™ X multilabel plate reader; PerkinElmer, Waltham, MA).
Western blot analysis of basement membrane proteins
Denaturated protein samples were separated by 8% (v/v) Bis-Tris gels and blotted to polyvinylidene fluoride membranes (Roche, Mannheim, Germany). The membranes were blocked with 5% (w/v) skim milk powder in Tris-buffered saline with 0.05% (v/v) Tween 20 for 1 h at RT and incubated o/n at 4°C with anticollagen IV (rabbit polyclonal, dilution 1:250), antilaminin (rabbit polyclonal, dilution 1:500), or antifibronectin (mouse monoclonal, dilution 1:200) (all Abcam, Cambridge, England). As secondary antibodies, the peroxidase-conjugated goat anti-rabbit (dilution 1:10,000; Sigma-Aldrich), horse anti-mouse (dilution 1:4000; Cell Signaling, Danvers, MA), and goat anti-mouse (dilution 1:10,000; Sigma-Aldrich) antibodies were used for 1 h at RT. Mouse anti-β-actin (dilution 1:10,000; Sigma-Aldrich) served as the housekeeping protein.
Evaluation of cytotoxicity by cell viability assay
The DC-LGs were extracted in DMEM containing 10% (v/v) FBS for 48 h at 37°C. 3T3/J2 feeder cells were cultured in 96-well plates at a density of 50,000 cells per well with 100 μL of the leaching liquor or DMEM with 10% (v/v) FBS (control). After 1 and 24 h, the luminescence as a linear measure of the viable cells was determined (CellTiter-Glo® Luminescent Cell Viability Assay, Madison, WI). Briefly, 100 μL of the provided reagent was added to each well, the content was mixed for 2 min on an orbital shaker and incubated for 10 min at RT. The luminescence was recorded with a plate reader (VICTOR X, PerkinElmer).
Reseeding of the decellularized LG matrix
DC-LG pieces were dab-dried and put into 96-well plates. One million SC or EC cells in 150 μL media were added and allowed to adhere for 3 h before another 100 μL medium was added. The culture medium was carefully changed daily. Before analysis of the secretory capacity, the recellularized pieces were washed with serum-free medium thrice. Before fixation for immunochemistry or electron microscopy, the pieces were washed with PBS. All analyses were performed after 7 days of cultivation time.
To investigate the degree of recellularization after a longer cultivation time, EC cells were seeded on the DC-LG for 28 days and analyzed histologically using hematoxylin and eosin (H&E) staining.
Transmission electron microscopy
Tissue pieces or the cell pellets were fixed in 2.5% (v/v) glutaraldehyde with 4% (v/v) paraformaldehyde (PFA) in 0.1 M cacodylate buffer, embedded in Spurr media (Modified SPURR Embedding Kit; Serva, Heidelberg, Germany), cut into 60–80-nm-thin sections, and analyzed with a Hitachi H600 transmission electron microscope (Krefeld, Germany). Images were taken with a Gatan BioScan camera (München, Germany).
Assessment of the secretory capacity by β-hexosaminidase assay
The cells were seeded at a density of 5 × 105 cells per well in 24-well plates, allowed to adhere o/n, washed with serum-free DMEM, and incubated with 500 μL serum-free DMEM for 2 h (baseline value). Carbachol was added for 15 min at a final concentration of 100 μM, and a stimulated sample was removed. For measurement of β-hexosaminidase activity, 4-methylumbelliferyl N-acetyl-β-D-glucosaminide (Sigma-Aldrich) was used as a substrate. The fluorescence intensity was determined at 360 nm excitation and 450 nm emission (FLUOstar Omega; BMG LABTECH, Ortenberg, Germany).
The recellularized DC-LGs were washed and incubated with 1000 μL serum-free DMEM for 2 h (baseline value). Carbachol was added and β-hexosaminidase activity was measured as described above.
Histology and immunochemistry
For H&E staining, sections were deparaffinized, rehydrated, and incubated in H&E solution for 5 and 7 min, rinsed in running tap water, dehydrated, and embedded (Roti-Histokit 2; Roth). For periodic acid-Schiff (PAS)–alcian blue reaction (Merck), sections were deparaffinized, rehydrated, and incubated in alcian blue solution for 5 min, washed with running tap water for 3 min, rinsed with distilled water, and incubated with Schiff's reagent for 15 min. Nuclear staining was performed with hematoxylin solution.
For immunostaining, anti-pan-cytokeratin (mouse monoclonal, ab6401, 1:400; Abcam), anti-smooth muscle actin (mouse monoclonal, M0851, 1:400; Dako, Glostrup, Denmark), anti-vimentin (rabbit monoclonal, D21H3, 1:500; Cell Signaling), anti-Rab3D-C-terminal (rabbit polyclonal, ab170057, 1:200; Abcam), and the primary antibodies utilized for the western blot analysis were used. Slices were deparaffinized, washed with PBS, and boiled for 5 min in 0.01 M citric acid buffer at pH 6 containing 0.05% (v/v) Tween 20 to induce antigen retrieval. Cryosections were washed with PBS for 10 min, fixed methanol for 10 min, and permeabilized with 0.1% (v/v) Triton X-100 in PBS. Cells were fixed with ice-cold methanol or 10% (v/v) PFA. The slices or cells were blocked for 1 h at RT with 5% (v/v) normal goat serum (VWR, Radnor, PA), incubated o/n at 4°C with the primary antibody, washed, and incubated with the appropriate fluorochrome-conjugated secondary antibody (Alexa flour) for 1 h at RT. As a control, the samples were incubated with the secondary antibody only. Microscopic analysis and visualization were performed with an optical microscope (Leica DM4000 B and camera system Leica DFC450 C, Wetzlar, Germany or with BIOREVO BZ9000; Keyence, Montabaur, Germany).
All experiments were performed with at least three biological replicates and statistical analysis was done with SPSS 21.1 (IBM, Ehningen, Germany). The paired or independent Student's t-test was applied to analyze the significance of difference between related or unrelated samples with p ≤ 0.05 as the level of significance.
Results
Characterization of the native porcine LG and comparison of SC and EC
The main cell types in the porcine LG were acinar and ductal epithelial cells (Fig. 1). Alpha-smooth muscle actin (alpha-SMA)-positive myoepithelial cells encircled the LG acini, but not the ductal structures. The stroma contained vimentin-positive mesenchymal cells. The PAS-alcian blue reaction, which stains neutral mucins purple and acid mucins light blue, showed that the native porcine LG is a mucoserous gland with mainly neutral mucous secretions. Ductal and serous acinar cells did not stain. The transmission electron microscopy (TEM) analysis illustrated serous and mucous secretory vesicles in the acinar cells.
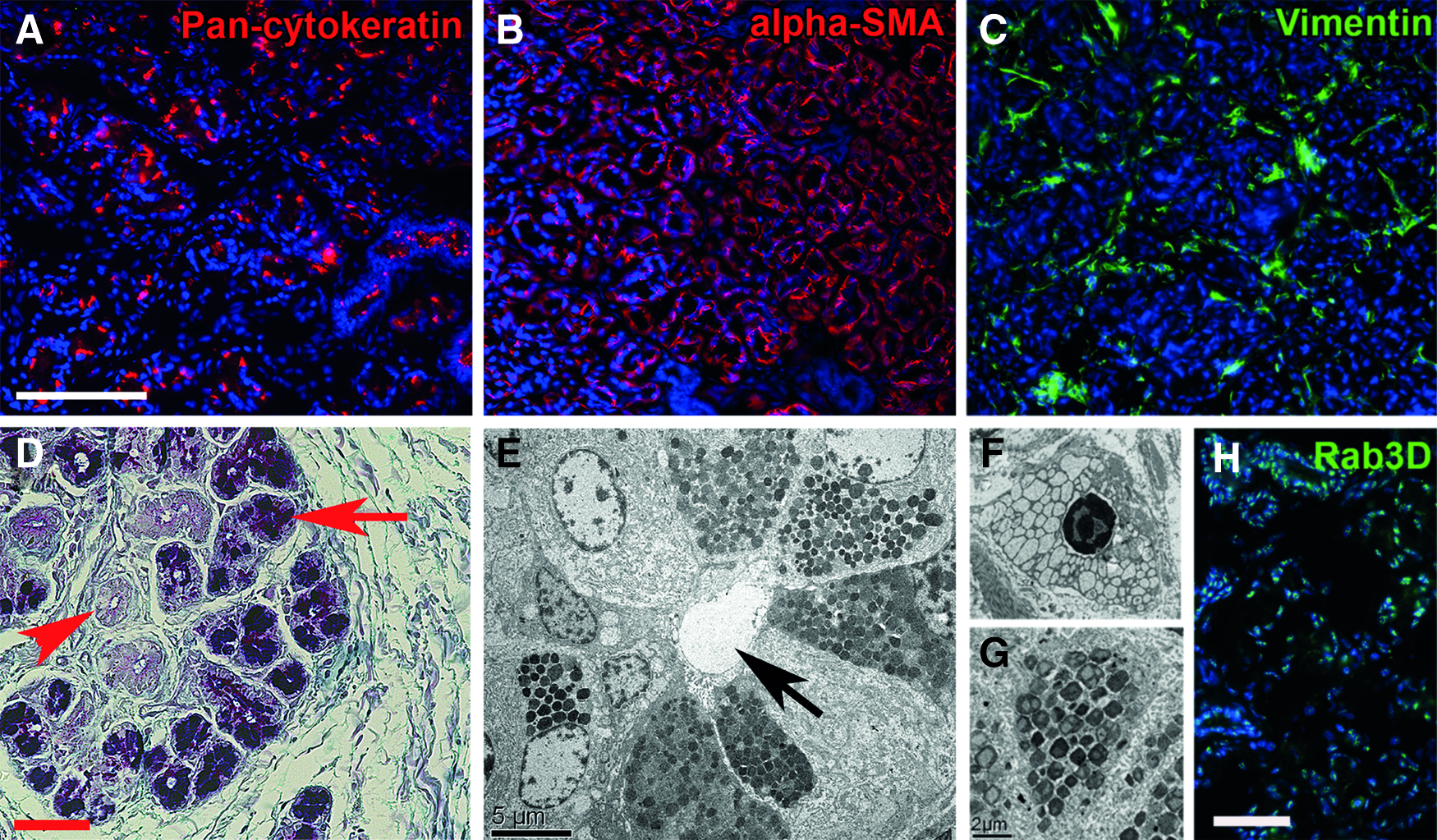
Histological characterization of fresh porcine lacrimal gland (LG). The acinar and ductal epithelial cells stained for pan-cytokeratin
Using the SC isolation method, a 100 mm3 LG piece generated ∼2.8 ± 1.1 million viable cells. 13 A single-cell suspension containing almost exclusively epithelial cells was obtained as described earlier and colonies formed within 3–5 days, reaching subconfluency after 9 ± 1 days. 9
Applying the EC method, cells grew out from the explants after about 7 days, reaching subconfluency after 16 ± 2 days. A 3.4 mm3 LG piece generated 2.7 ± 0.6 million viable cells, which in relation to the tissue size was about 30 times more compared with isolation by SC. The epithelial phenotype of SC and EC cells was verified by immunostaining and flow cytometry, exhibiting a 14.7 and 12.6-fold mean increase in fluorescence intensity compared with the isotype control, respectively (Fig. 2). Very few cells showed immunoreactivity for alpha-SMA or vimentin.
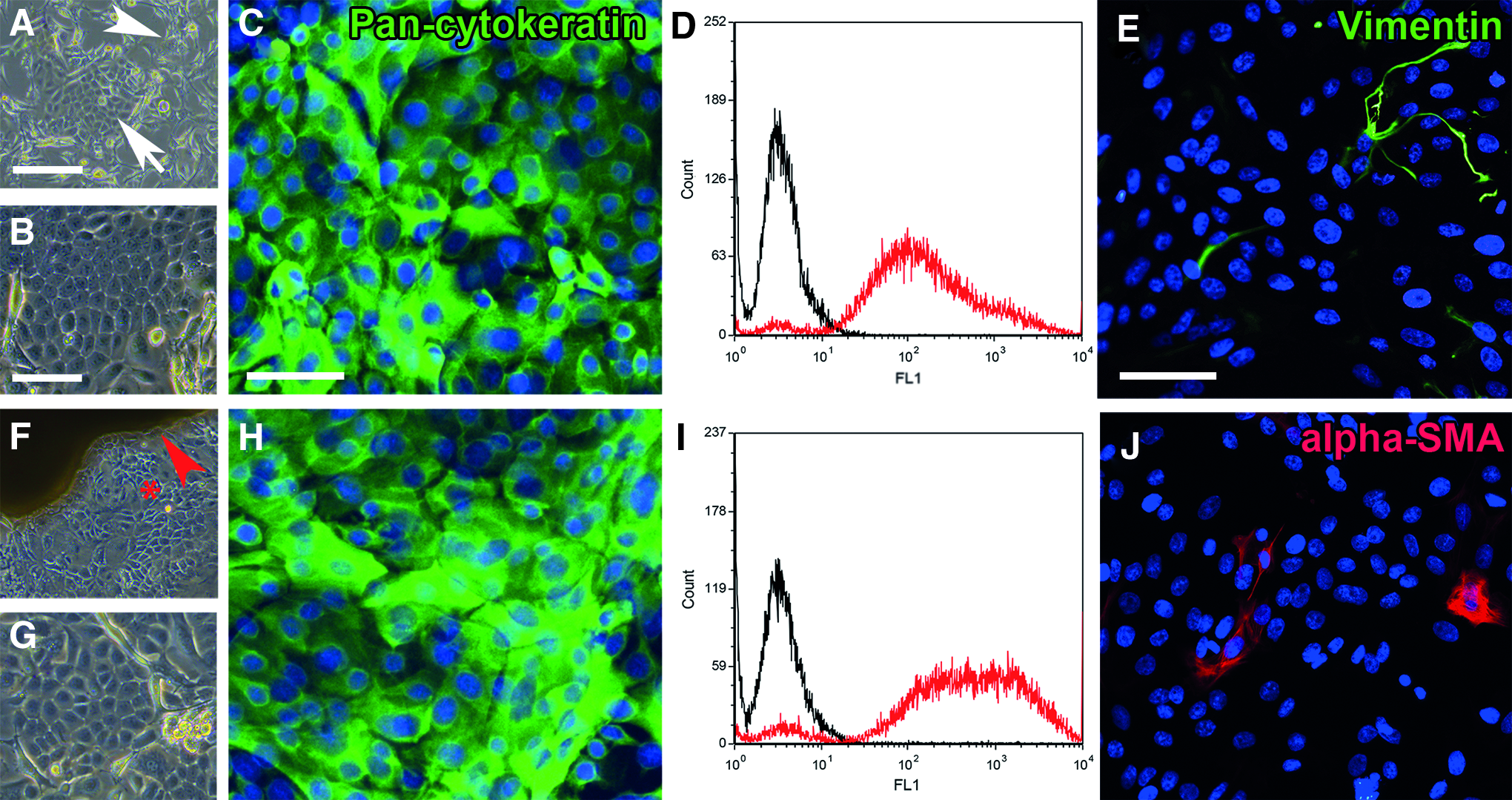
Characterization of the LG epithelial cells. The images in the upper row show the suspension culture (SC) cells, while the explant culture (EC) cells are illustrated below. The SC cells build colonies around day 3
Proliferation potential of primary cells in vitro
Cells expanded by SC and EC revealed similar PDTs until passage 3 (SC: 1.3 ± 0.20 days, EC: 1.3 ± 0.15 days, p = 0.10) and cells from passage 0 comprised a similar colony-forming efficacy (colony-forming units SC/EC: 43.3 ± 19.2/51.8 ± 14.0, p = 0.19, Fig. 3). The amount of ALDH-1-high and side population cells from passage 0 was measured to quantify the cells with potential stem cell capacity. SC and EC cells contained a similar amount of ALDH-1-high and side population cells (SC/EC cells: ALDH-1-high cells = 13.4% ± 1.2%/11.8% ± 1.2%, p = 0.19; side population = 5.4% ± 2.2%/4.7% ± 2.9%, p = 0.75, Fig. 3).
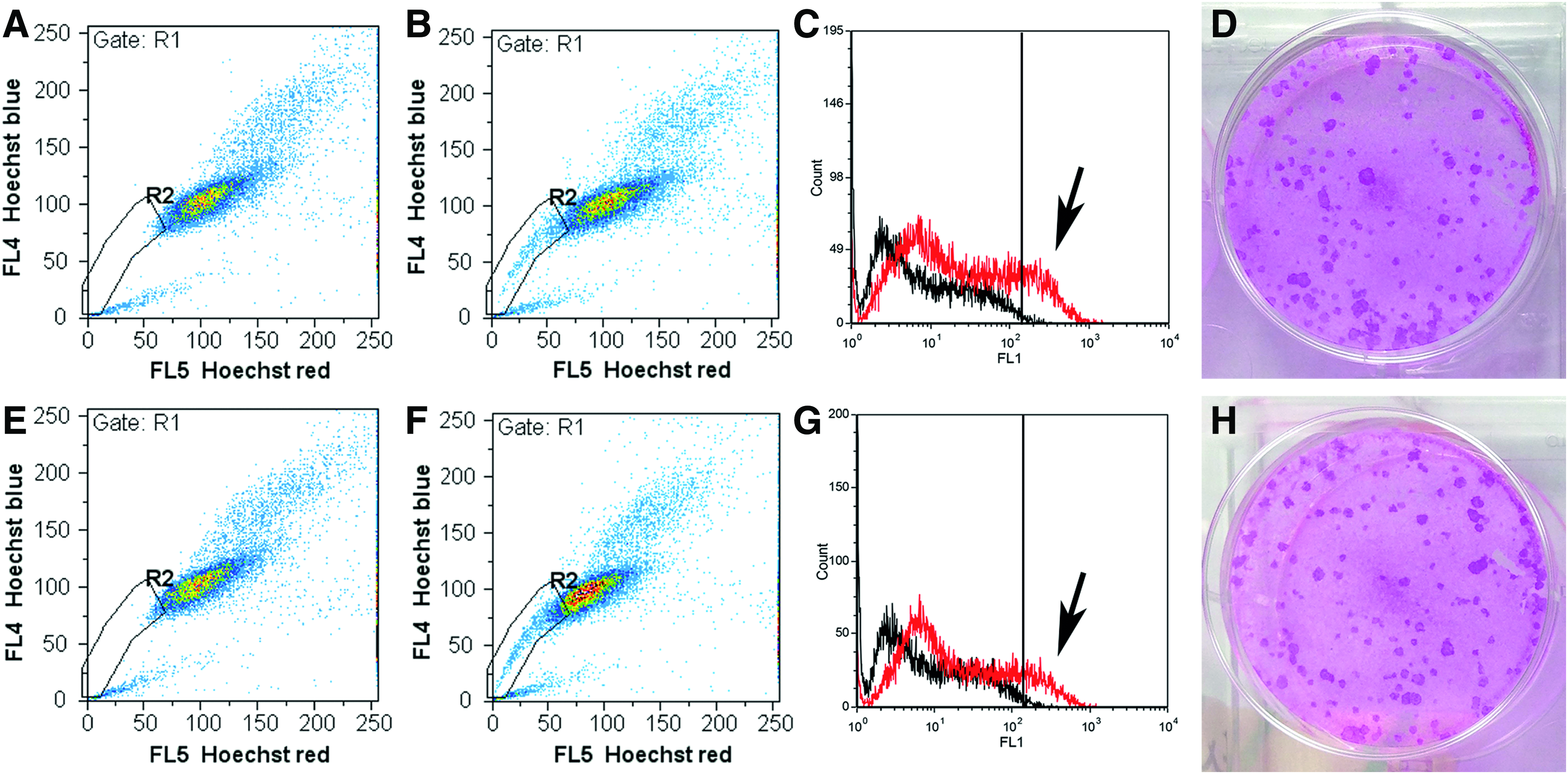
Proliferative capacity of the LG epithelial cells. The images in the upper row show the SC cells, while the EC cells are illustrated below. Cells from both isolation methods contained side population cells (SC: 5.4% ± 2.2% and EC: 4.7% ± 2.9%, p = 0.75), which are illustrated in the R2 gate
Assessment of the secretory function
Both, SC and EC cells exhibited histological and immunocytochemical features typical for cells with secretory function. Similar to the native gland, the PAS-alcian blue reaction revealed a purple-colored cytoplasmic staining, typical for neutral mucous secretions. The TEM analysis illustrated secretory vesicles in SC and EC cells and the immunocytochemistry showed strong granular staining for Rab3D, a marker for mature secretory vesicles (Fig. 4). The secretory capacity measured by β-hexosaminidase assay revealed a significant increase in secretory activity after parasympathetic stimulation up to day 30 (SC and EC cells: days 10–15: p = 0.004 and p = 0.01, day 23: p = 0.03 and p = 0.04, day 30: p = 0.03 and p = 0.01). The secretory response of the SC and EC cells declined over time and was not significantly different at the individual time points (days 1–15: p = 0.10, day 23: p = 0.10, day 30: p = 0.85) (Fig. 5).
Secretory capacity of the LG epithelial cells. The images in the upper row show the SC cells, while the EC cells are illustrated below. After expansion in passage 0, cells from both isolation methods contained multiple secretory granules, as illustrated by a PAS reaction
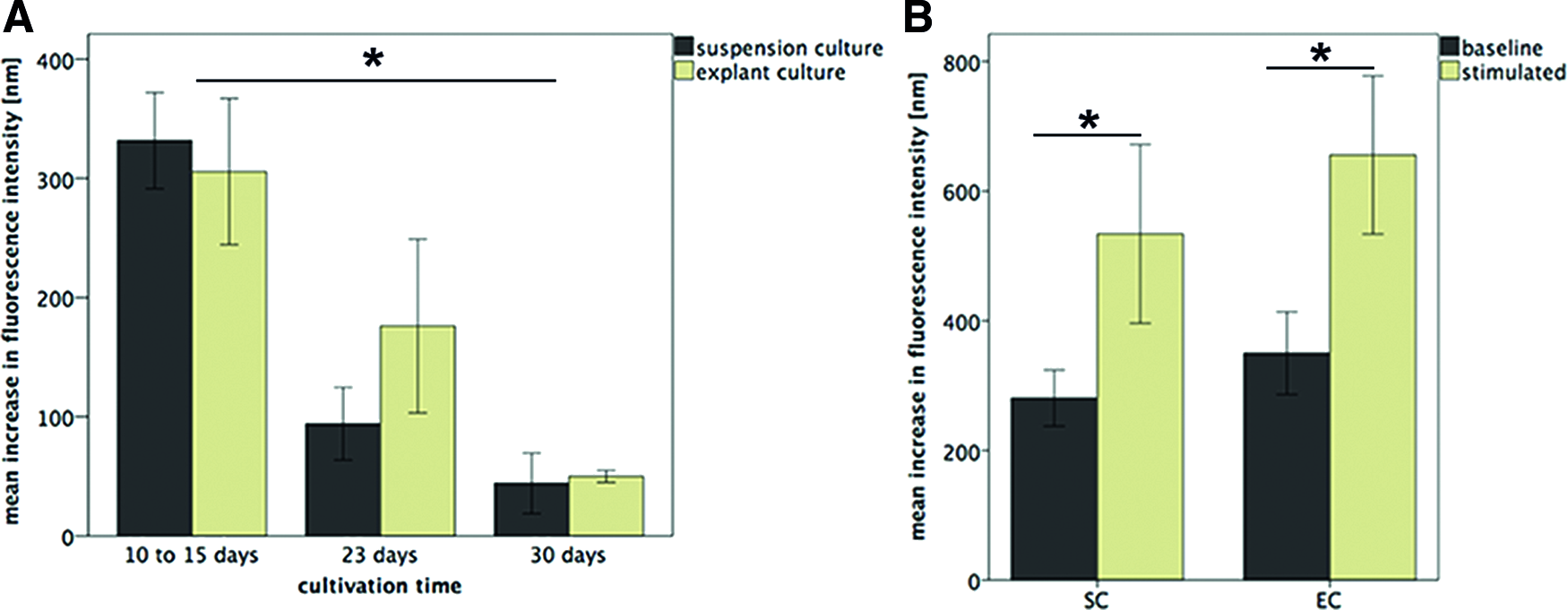
Proof of the secretory capacity in the two- and three-dimensional culture. When cultured on plastic, SC and EC cells revealed secretory capacity until day 30. The secretory capacity declined over time (*p = 0.02) and was not significantly different between both isolation methods
Characterization of the decellularized LG tissue
The DC-LG had a whitish color and a tensile sponge-like structure. Histologically, it depicted mainly intact acinar structures with some interruptions of the extracellular matrix integrity (Fig. 6). The TEM analysis showed an intact collagen network without cellular debris. The mean collagen I-V content was 1.3 times decreased, from 20.7% ± 4.1% in the native LG to 13.7% ± 2.6% in the DC-LG, but this effect was not statistically significant (p = 0.08) (Fig. 7). The native LG showed a linear expression of collagen IV and laminin at the basement membrane of the acini and ducts (Fig. 8). This staining was mainly preserved after decellularization. Fibronectin revealed a more diffuse staining of the extracellular matrix, which was not limited to the basement membrane and maintained after decellularization. The immunoblotting verified the preservation of laminin and fibronectin, while collagen IV was reduced, but still clearly evident after decellularization (Fig. 7). The Feulgen staining indicated the absence of DNA, which was illustrated by DNA gel electrophoresis (Figs. 6 and 7) and DNA quantification (decrease from 2565.1 ± 893.5 ng/mg before to 15.2 ± 26.4 ng/mg after decellularization). The cytotoxicity assay confirmed no toxic effects of the DC-LG on cultured cells (Fig. 7).
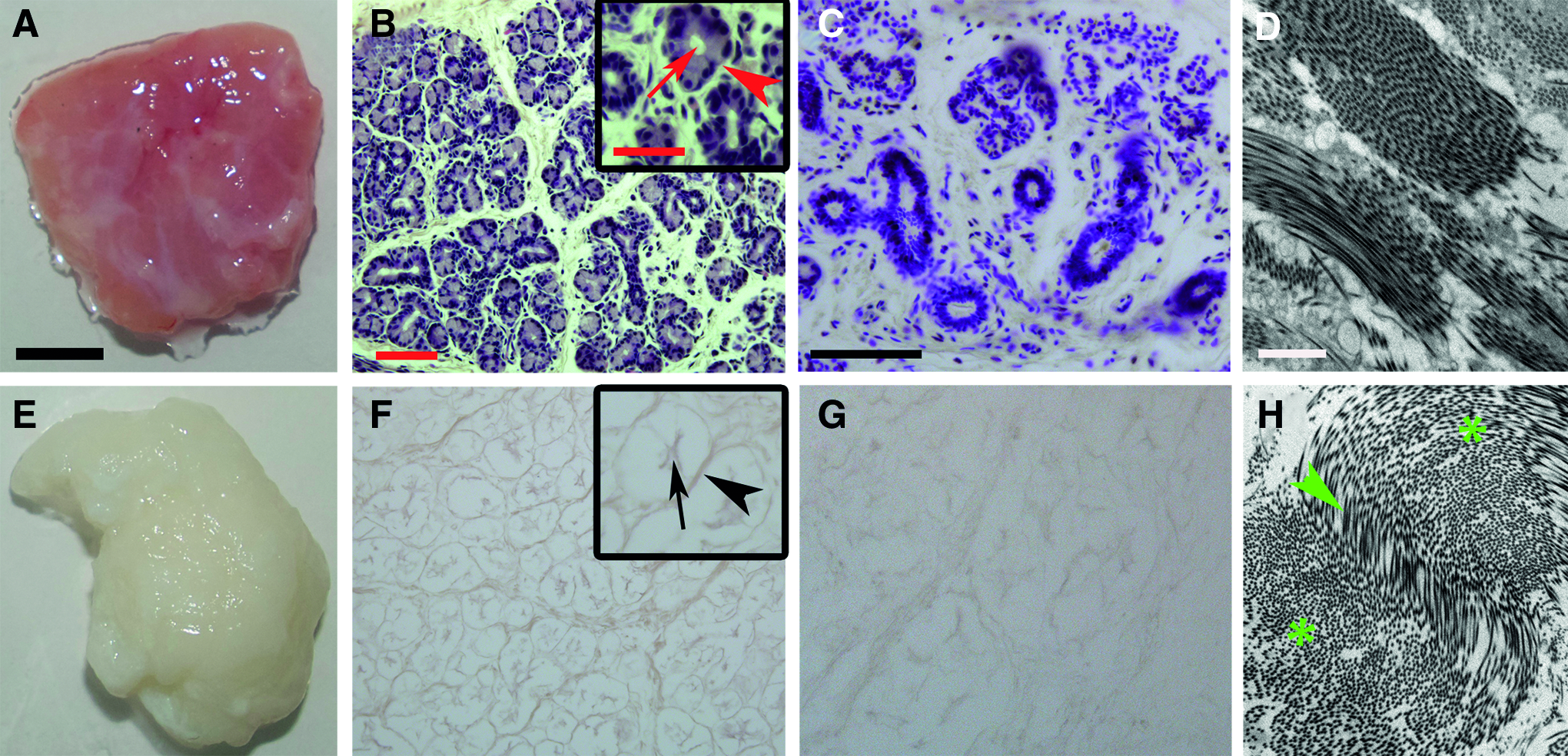
Histological and ultrastructural characterization of the native and decellularized LG. The images in the upper row show the native LG, while the DC-LG is illustrated below. Compared with the untreated LG
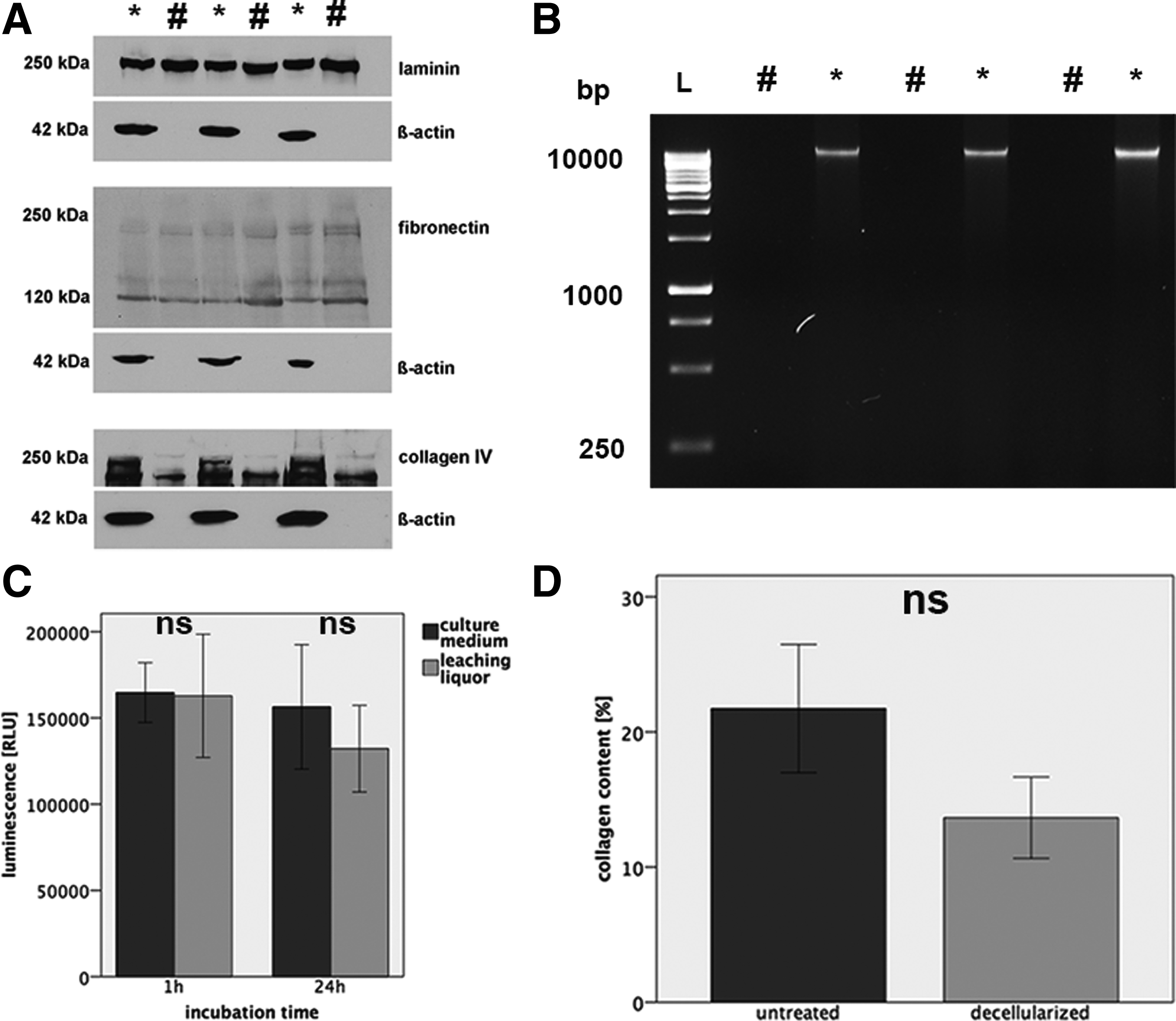
Protein content and toxicity testing of the decellularized LG tissue.
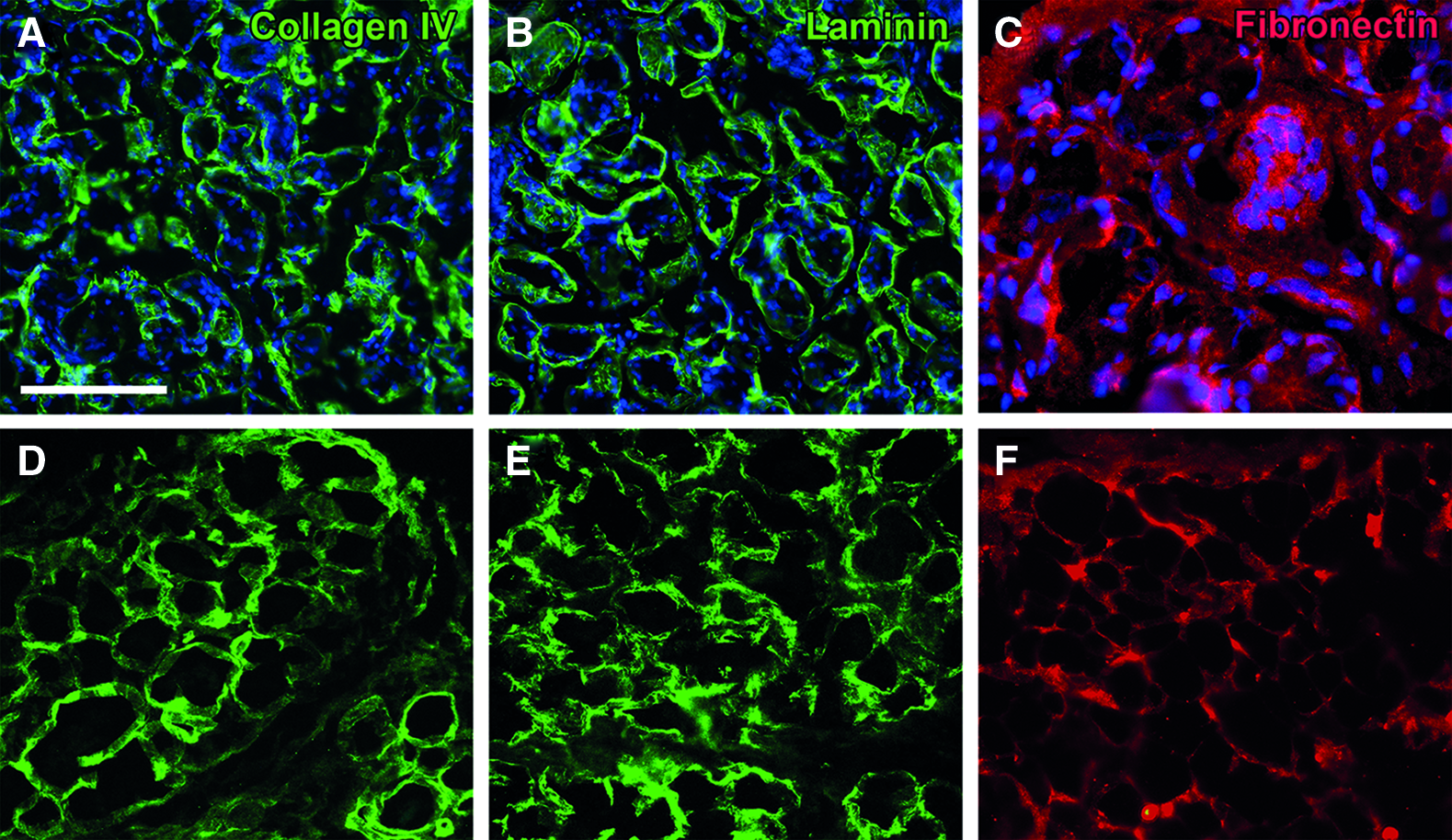
Structural integrity of the basement membrane after decellularization. The images in the upper row show the native LG, while the DC-LG is illustrated below. In the untreated LG, the staining for collagen IV
LG recellularization
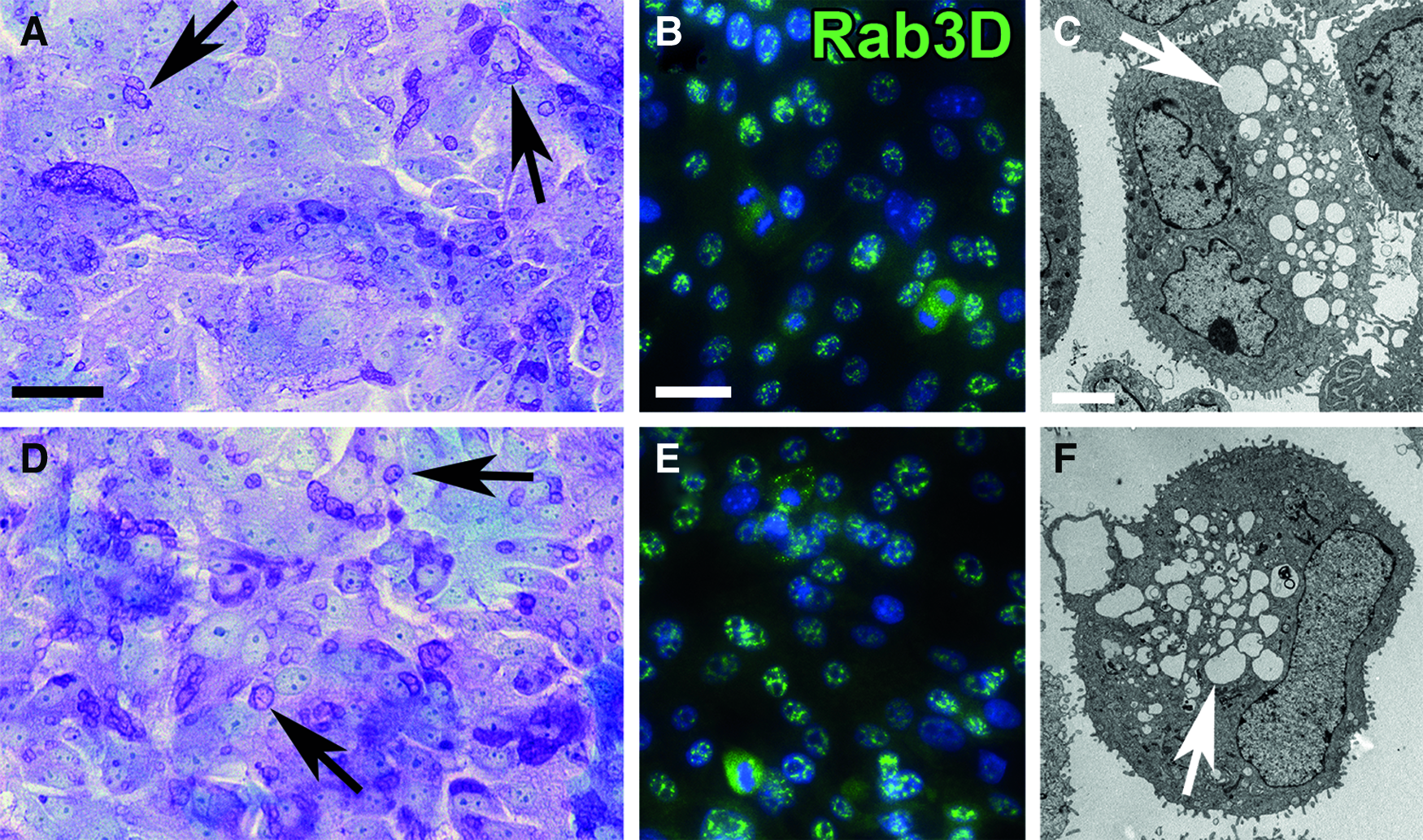
After 7 days of culture on the DC-LG, SC and EC cells formed a multilayer, maintaining an epithelial phenotype (Fig. 9A–D). The cells started to migrate into the tissue and partly build duct- and sphere-like structures. The PAS-alcian blue reaction identified mucous in the cytoplasm of the seeded cells and the Rab3D staining revealed evidence of secretory vesicles, as verified by TEM (Fig. 10).
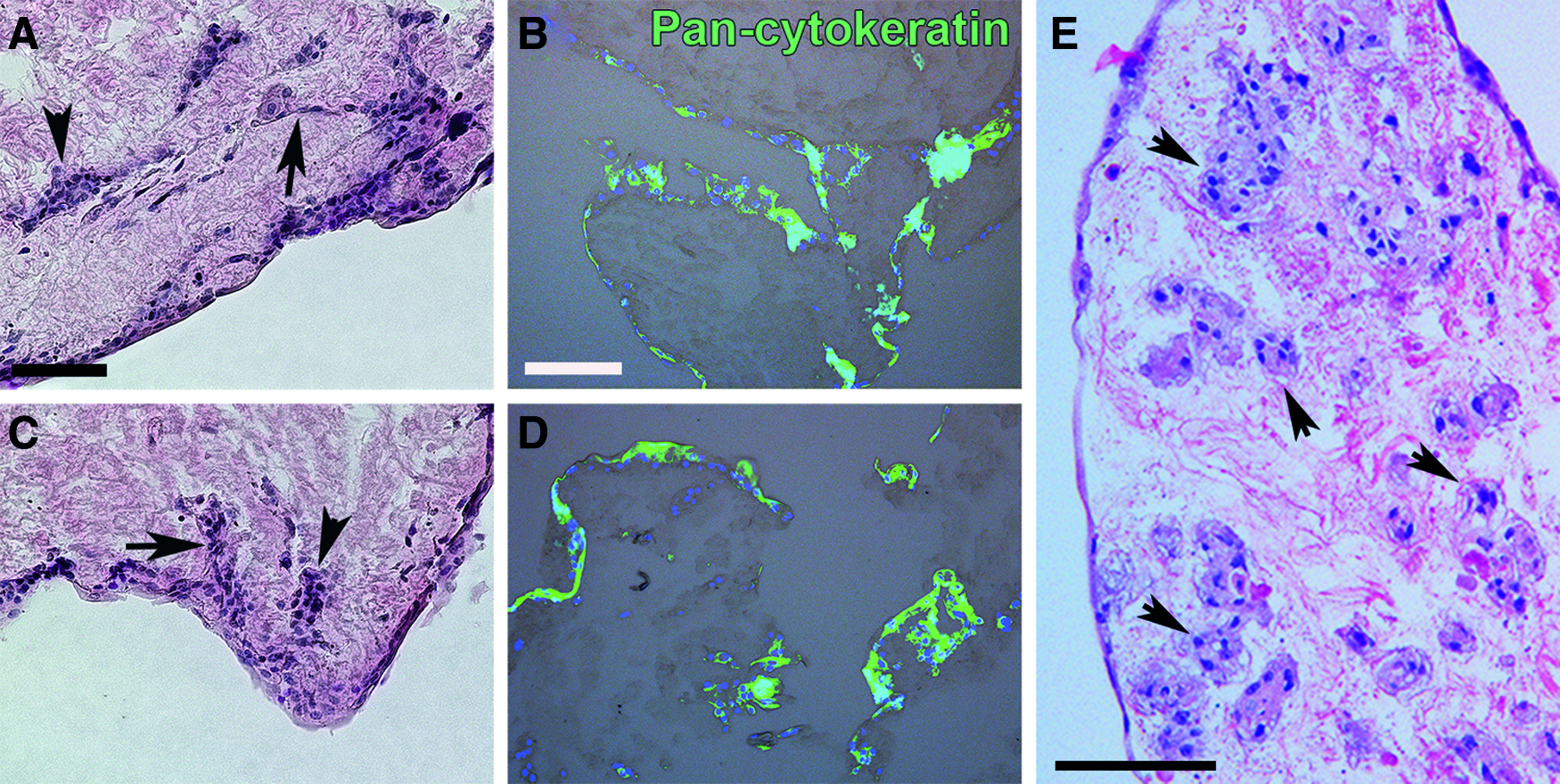
Histological analysis of the recellularized DC-LG.

Secretory phenotype of the recellularized DC-LG. The images in the upper row show the SC cells seeded onto the DC-LG, while the EC cells are illustrated below. The PAS-alcian blue reaction illustrated that the cells contained many secretory granules
Both, SC and EC cells showed a significant secretory response after parasympathetic stimulation (SC: p = 0.007, EC: p = 0.001). The secretory capacity was not significantly different between cells from each isolation method (p = 0.88) (Fig. 5). Normalized to the cell count in the two-dimensional culture, the secretory response of the cells remained stable after seeding into the DC-LG (SC and EC cells: p = 0.22 and p = 0.90). A detailed TEM analysis showed that the cells adopted a polarized phenotype with microvilli on the surface of the apical cell membrane, secretory vesicles in the cytoplasm, and basal nuclei (Fig. 11). The cells exhibited cell–cell contacts and those, which had grown deeper into the DC-LG, partly showed a more undifferentiated phenotype as they were smaller and had less secretory vesicles and a larger nucleus-to-cytoplasm ratio. After a 4-week culture period, the cells migrated deeper into the DC-LG and organized themselves into acinar-like structures between the extracellular matrix (Fig. 9E).
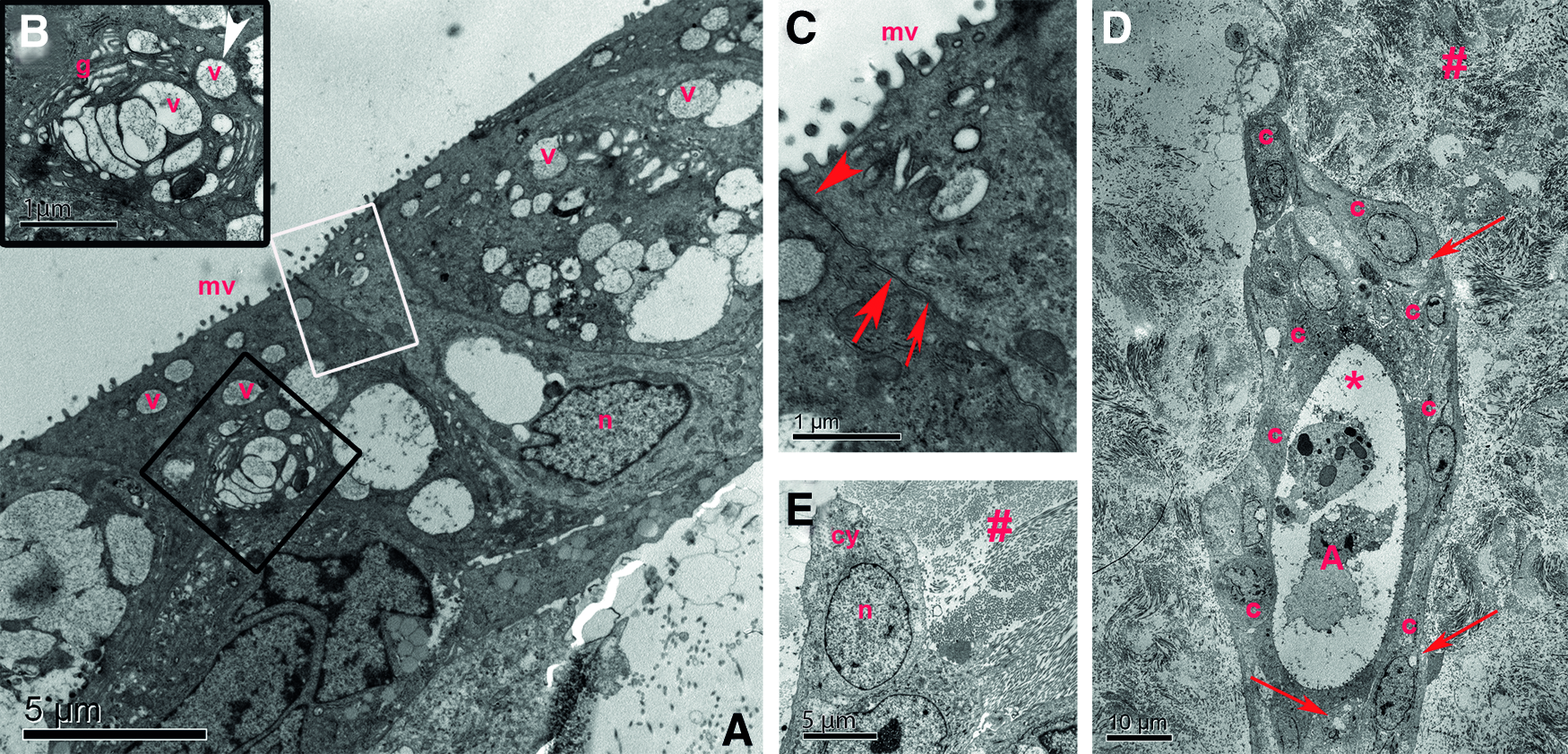
Ultrastructural analysis of the recellularized DC-LG. The TEM images showed that the cells seeded into the DC-LG depicted a polarized phenotype with apical microvilli (mv), secretory vesicles (v) in the apical cytoplasm, and basal nuclei (n)
Discussion
Severe dry eye due to LG deficiency is an incurable disease causing visual impairment with painful destruction of the ocular surface and eventually loss of sight. LG reconstruction is a desirable goal, but requires functionally competent cells and a suitable, three-dimensional transplantable matrix. To date, few studies investigated regenerative LG models in vitro using rodent tissue.9,14,15 In a study by Hirayama et al., a bioengineered LG organ germ from embryonic mice was developed, which achieved tear production after orthotopic transplantation into tear-deficient adult mice. 16 So far, this model lacks clinical feasibility because the embryonic cell source implies ethical concerns and the allogeneic character of the tissue raises issues of transplant rejection. Moreover, the anatomy and size of rodent LG tissue differ considerably from human tissues. 17 The domestic pig (sus scrofa) provides an easily accessible mammalian animal model and tissue source to study human diseases. 11 The porcine LG has a similar secretory system and arterial perfusion compared with human LG, thus providing a promising model to study lacrimal function in vitro and in vivo. 18
Secretory competent LG cells have been successfully isolated previously by various enzymatic digestion, filtration, and centrifugation steps, but this approach is time-consuming and the number of viable cells is considerably reduced during tissue processing.19,20 The strength of the extracellular matrix increases with age, which leads to a further decrease in cell gain when using adult compared with embryonic or juvenile tissue. 21 With regard to clinical applications, where a small biopsy from an adult patient would be available, one aim of this study was to investigate the possibility to obtain and expand functional LG epithelial cells from a small biopsy.
Our results show that secretory active cells can be expanded from a 1.5-mm diameter biopsy and generate a similar amount of cells as a 30 times bigger LG piece processed by SC. Although the outgrowth from the explants took around 16 days, while the SC cells reached subconfluency after about 9 days, this extended culture period did not affect cell viability and function. This is in agreement with findings from other human tissues such as the placenta as well as delicate ocular tissues such as the anterior lens capsule and aorta, the retina, and the corneal limbus.22–26 EC cells from small limbal biopsies are viable, retain an epithelial phenotype, and are already applied clinically, proving to reconstruct the corneal surface.27,28
In our study, in analogy to the SC culture, EC cells from the LG generated an almost pure epithelial cell population and maintained their secretory capacity for up to 30 days. This is comparable with results from Tiwari et al. who showed that human LG cells isolated by enzymatic digest revealed secretory capacity for about 21 days in vitro. 8 The feasibility of EC for the expansion of functionally competent mammalian LG cells can facilitate the culture of human LG cells, which is an important step toward clinical use.
LG regeneration in vitro and in vivo requires cells with regenerative capacity, but the distinct origin of the LG stem or progenitor cells still remains unclear. Shatos et al. investigated adult rat LG and assumed the myoepithelial cells to be possible LG progenitor cells as they differentiated into multiple lineages in vitro. 29 Voronov et al. found the proliferative capacity of mouse LG epithelial cells, but concluded that it was unclear whether the LG comprises one multipotent or multiple lineage-specific progenitors. 30 Our study showed that acinar epithelial cells from the porcine LG comprise cells with colony-forming capacity, indicating a proliferative potential of this lineage.
For the adult human LG, Tiwari et al. described the presence of ALDH-1-high cells in a freshly isolated population of epithelial, myoepithelial, and mesenchymal cells. 8 ALDH-1 was found to be an important functional enzyme in stem cells from various tissues, such as liver, breast, muscle, bone marrow, neural tissue, and salivary glands, and constitutes a useful marker for stem cell identification and isolation.31,32 The porcine cells contained about 4-fold more ALDH-1-high cells after expansion in passage 0 compared with what has been described for human LG. 8 The 3T3/J2 feeder layer might have supported a proliferative phenotype, as described for other primary epithelial cells. 33 Furthermore, cells isolated by SC or EC contained side population cells, which were found to be stem or progenitor cells in various human tissues and can be identified by flow cytometry due to the ability to exclude Hoechst dye through ATP-binding cassette (ABC) transporter proteins, mainly ABCG2, in the cell membrane.34,35 Mishida et al. isolated side population cells from mouse salivary and LGs. 36 These cells were identified as CD31-positive endothelial cells and induced functional recovery of irradiated glands after transplantation. Our study found that epithelial LG cells comprise side population cells, which is another indicator for the regenerative potential of this lineage. However, further characterization of mesenchymal and myoepithelial cells of the porcine LG, which may potentially include positive effects on the secretory and proliferative capacity of the epithelial cells in coculture, needs to be analyzed in more detail in future.
To mimic a three-dimensional environment, induce sphere formation, and maintain the secretory capacity, LG epithelial cells have previously been cultured on the amniotic membrane, in microgravity bioreactors without additional matrix support, and frequently on Matrigel, a soluble extract from the Engelbreth-Holm-Swarm (EHS) mouse sarcoma.9,15,21,37,38 However, tissue engineering of whole organs requires not only a three-dimensional, biocompatible nonimmunogenic scaffold but also a matrix, which will integrate into the host environment and can be handled surgically. Spheres generated by microgravity culture as well as Matrigel lack the mechanical strength needed for surgical applications. Since Matrigel is derived from mouse sarcoma cells, its clinical application raises biocompatibility and safety concerns. The amniotic membrane is currently used for ocular surface reconstruction, providing a precedent for use in clinical settings, but its availability is limited, there is a risk of disease transmission, and it is degraded quickly by the host's immune system.39,40
In previous studies, decellularized organ scaffolds were proved to largely retain their three-dimensional extracellular matrix structure, the basement membrane, and their tensile strength, thus providing a biocompatible and surgical manageable matrix.40–42 As human tissue is rarely available, decellularized porcine organ scaffolds have been explored in detail for transplantation into humans. They are already used in the clinical setting as they resemble the human organs in terms of size, structure, and matrix composition and also are more readily available.43–45 We explored whether decellularized porcine LG can serve as a three-dimensional native scaffold for cell expansion and potential transplantation.
Decellularization, including sodium deoxycholate and DNase treatment, proved to create nearly DNA-free biocompatible matrices and has been successfully applied in glandular tissues as the lung, gut, or kidney.42,46 47 Although the risk for immunologic reactions or transmission of infectious agents from decellularized xenografts is low, retarded cellular material can induce the major histocompatibility complexes I and II (MHC-I and II), which may lead to a proinflammatory immune response and graft rejection.44,48 The DC-LG retained minimal amounts of DNA, meeting the criteria for the acceptable amount of DNA residues in decellularized tissues (<50 ng/mg DNA/mg dry weight, DNA fragment length <200 bp).49,50 Furthermore, the tissue did not convey cytotoxicity, thus representing a biocompatible matrix for potential recellularization with human LG cells in future.
As the extracellular matrix of each organ is individual, the DC-LG can provide an LG-specific amount, distribution, and composition of the extracellular matrix proteins. 44 The basement membrane, which builds an organ-specific landscape and mainly comprises collagen IV, laminin, and fibronectin, was mainly preserved in the DC-LG.51,52 Collagen IV and fibronectin are the most frequent basement membrane proteins providing cell adhesion domain sequences, which are necessary for recellularization. 53 The amount of these proteins in the DC-LG was sufficient to induce cell adhesion, although collagen IV was slightly diminished after decellularization. However, shorter incubation times with sodium deoxycholate to better preserve the collagen IV content resulted in incomplete cell removal in our experiments.
After 7 days of culture, the LG epithelial cells started migrating into the scaffold and began to form duct- and sphere-like structures in the DC-LG. Lombaert et al. showed that salivary gland epithelial cells developed so-called salispheres after cultivation in three-dimensional collagen gels, but only cell clumps with intact cell–cell contacts and not cells from single-cell suspensions were able to do so. 54 Interestingly, in our study, LG cells from single-cell suspensions were able to resemble cell–cell contacts and arrange themselves around a central lumen as a prerequisite for guided secretion. The cells further maintained their secretory capacity and showed expression of Rab3D, a marker adopted to label mature secretory vesicles and to be involved in regulated exocytosis. 55 An extended culture period of 28 days revealed that the cells were able to migrate deeper into the DC-LG and arrange themselves into acinar-like structures between the extracellular matrix, which underlines the supportive character of the DC-LG.
Conclusion
This study showed the possibility of engineering a secretory active, three-dimensional LG construct from adult mammalian tissue in vitro. LG epithelial cells with secretory function and progenitor cell characteristics were harvested from a small tissue biopsy and expanded on DC-LG as a three-dimensional, supportive, and easily accessible matrix. These preconditions provide a basis for clinical translation and potential future LG regeneration in humans. Functional in vivo studies need to be conducted for further evaluation of this LG construct.
Footnotes
Acknowledgments
The authors thank Ms. Joana Witt, Ms. Sabine Seggewiß, Ms. Mechthild Langkamp-Flock, Ms. Sabine Wilhelm, and Mr. Matthias Schweinlin for their excellent technical assistance. This work was funded by the Research Funding Commission of the University Düsseldorf and supported by the Volkswagen Foundation (Lichtenberg Professorship of Prof. Schrader).
Disclosure Statement
No competing financial interests exist.