
Abstract
Liver transplant is the only treatment option for patients with end-stage liver failure, however, there are too few donor livers available for transplant. Whole organ tissue engineering presents a potential solution to the problem of rapidly escalating donor liver shortages worldwide. A major challenge for liver tissue engineers is the creation of a hepatocyte microenvironment; a niche in which liver cells can survive and function optimally. While polymers and decellularized tissues pose an attractive option for scaffold manufacturing, neither alone has thus far proved sufficient. This study exploited cell's native extracellular matrix (ECM) producing capabilities using two different histone deacetylase inhibitors, and combined these with the customizability and reproducibility of electrospun polymer scaffolds to produce a “best of both worlds” niche microenvironment for hepatocytes. The resulting hybrid poly-capro-lactone (PCL)-ECM scaffolds were validated using HepG2 hepatocytes. The hybrid PCL-ECM scaffolds maintained hepatocyte growth and function, as evidenced by metabolic activity and DNA quantitation. Mechanical testing revealed little significant difference between scaffolds, indicating that cells were responding to a biochemical and topographical profile rather than mechanical changes. Immunohistochemistry showed that the biochemical profile of the drug-derived and nondrug-derived ECMs differed in ratio of Collagen I, Laminin, and Fibronectin. Furthermore, the hybrid PCL-ECM scaffolds influence the gene expression profile of the HepG2s drastically; with expression of Albumin, Cytochrome P450 Family 1 Subfamily A Polypeptide 1, Cytochrome P450 Family 1 Subfamily A Polypeptide 2, Cytochrome P450 Family 3 Subfamily A Polypeptide 4, Fibronectin, Collagen I, and Collagen IV undergoing significant changes. Our results demonstrate that drug-induced hybrid PCL-ECM scaffolds provide a viable, translatable platform for creating a niche microenvironment for hepatocytes, supporting in vivo phenotype and function. These scaffolds offer great potential for tissue engineering and regenerative medicine strategies for whole organ tissue engineering.
Introduction
A
While scientists can maintain liver organoids within a laboratory environment, decades worth of scientific research has failed to translate this into an organ that could be used for patient transplant.5,6 This in part is due to the complexities of recapitulating a vital part of the in vivo multicellular environment, the extracellular matrix (ECM). 18
Multiple methods have been employed in an attempt to provide hepatocytes with an optimal environment; in particular decellularization of whole livers and novel three-dimensional (3D) tissue-engineered scaffolds. Decellularization provides a whole organ ECM-based bioscaffold with the potential to maintain the 3D site-specific architecture and highly conserved sinusoidal ECM gradient required for hepatocyte function upon their repopulation of the organ. 18 Several studies have repopulated decellularized organs with hepatocytes and endothelial cells, which subsequently survive and exhibit some level of function.19–23 However, this approach still requires precious donor organs, sourced from potentially immunogenic human or animal donors, and methods used are not without their drawbacks.
Detergents such as sodium dodecyl sulfate, Triton-X100, and sodium deoxycholate are employed to strip the ECM of cells. These detergents disrupt native tissue ultrastructure, decrease glycosaminoglycan content, and reduce collagen integrity26,27 and disrupt lipid–lipid, lipid–protein, and protein–protein interactions. 28 Scaffold manufacture employs engineering technologies to create a synthetically derived structure, which mimics the characteristics of the native ECM. There are several different methods of creating a scaffold, and they can be made from a myriad of substances; both natural and synthetic. 29
Hydrogels have been of particular interest to liver tissue engineers, gels biofuctionalized with collagen I enhance P450 (Cyp450) activity, cell adhesion markers, and innate hepatocyte fibronectin production. 30 Gels biofunctionalized with galactose increased albumin (Alb) production and promoted the proliferation of hepatocytes. 31 Viability and hepatic functions of primary hepatocytes are improved by culturing them in hydrogels made with liver ECM, 32 and when encapsulated in collagen-alginate composite hydrogels. 33 Polyethylene glycol hydrogels have been shown to augment cell–cell interactions of bipotential mouse embryonic liver cells, subsequently improving their survival and function, 34 however, incorporating ECM components into scaffolds at the manufacturing stage has proved challenging, with the harsh solvents require to solubilize major components of ECM alter their microstructure and functionality.35,36 Ongoing research demonstrates great promise, however to date no scaffold has been created which allows hepatocytes to function as well as they do in vivo.11,37
Both ECM components and scaffolds have shown great promise in tissue engineering.9,10,15 With these considerations in mind, we combined decellularization and scaffold engineering to create a novel environment in which hepatocytes thrive (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea). By manipulating ECM production using pharmaceuticals and exploiting polymer characteristics we created a novel hybrid polymer-ECM platform for liver tissue engineering. To validate the platform we used cells representative of the liver (HepG2s).
Materials and Methods
Electrospinning
A 12% wt/vol solution of poly-capro-lactone (PCL; Sigma-Aldrich) and hexafluoroisopropanol (Manchester Organics) was dissolved overnight at room temperature with agitation. Solutions were placed into a 10 mL syringe and pumped using syringe pump EP-H11 (Harvard Apparatus) into the EC-DIG electrospinning system (IME Technologies) via a 27G bore needle under the following parameters Table 1.
Conditions under which the electrospun poly-capro-lactone scaffold was fabricated.
The mandrel was coated in nonstick aluminium foil for collecting the electrospun fibers. The sheets of electrospun fibers were allowed to dry overnight in a fume hood when the electrospinning session was completed. The average fiber size was 1.14 μm as calculated by ImageJ plugin “Diameter J” (Fig. 1). 38

Characterization of electrospun scaffolds. The scaffolds were assessed for consistency and fiber size via SEM and subsequent image analysis. Fibers consistently displayed a mean diameter of 1.14 μm as determined by DiameterJ, 34 n = 4. 200 × magnification. SEM, scanning electron microscopy.
Plasma coating
Ten millimeter discs of scaffold were cut from the dry fiber sheet. The scaffolds were soaked in 70% ethanol for 30 min, rinsed three times in phosphate-buffered saline (PBS) for 15 min each, and allowed to dry completely at room temperature. We then freeze dried the scaffolds overnight in a FreeZone® 4.5 freeze-drier (Labconco®) before plasma coating (Harrick Plasma) at 10.2 W for 30 s. Scaffolds were removed from the plasma chamber and placed into an antibiotic/antimycotic treatment solution of Eagles Minimal Essential Media supplemented with 10% fetal bovine serum (FBS), 2 mM
Initial layer cell seeding and culture
Scaffolds were removed from the antibiotic/antimycotic treatment solution and rinsed three times for 15 min each in complete media; Eagles Minimal Essential Media supplemented with 10% FBS, 2 mM
5637 human urinary bladder epithelials (ATCC) were trypsinized using standard methods from tissue culture flasks and counted using the trypan blue exclusion method. About 1 × 104 cells at passage 23 were suspended in 100 μL of complete media and seeded directly on to the scaffolds. The cells were allowed to incubate in this small volume on the scaffolds for 2 h, before an additional 400 μL of complete media was added.
Media was changed after 24 h to either 750 μM valproic acid (VA) or 750 μM sodium butyrate (NaB; Sigma-Aldrich) in complete media and changed every 48 h. Controls were scaffold only, that is, not seeded with an initial cell layer and no drug treatment that is, the initial layer cultured in drug-free complete media only. Drug concentrations and initial layer cells were chosen following results of a drug response curve for each histone deacetylase inhibitor (iHDAC) (data not shown). VA and NaB are used as epigenetic control mechanisms of gene transcription. They function by inhibiting HDACs (as iHDACs) to modulate chromatin structure, creating an open, transcriptionally active euchromatin configuration at gene coding and regulatory regions of the chromosome. This renders the chromatin accessible to transcription factors, and facilitates gene transcription. 39
VA and NaB are both commonly used in industry, particularly antibody production due to their ability to upregulate protein production in such a manner.40,41 This initial layer of cells was cultured for 7 days at 37°C and 5% CO2 in a humidified incubator.
Decellularization
Decellularization was performed using methods adjusted from Lu et al., 42 under sterile conditions at room temperature (19–22°C) and agitation (Fig. 2). Scaffolds were washed in PBS for 15 min and then rinsed in 10 mM Tris-buffered saline (TBS) for 15 min.
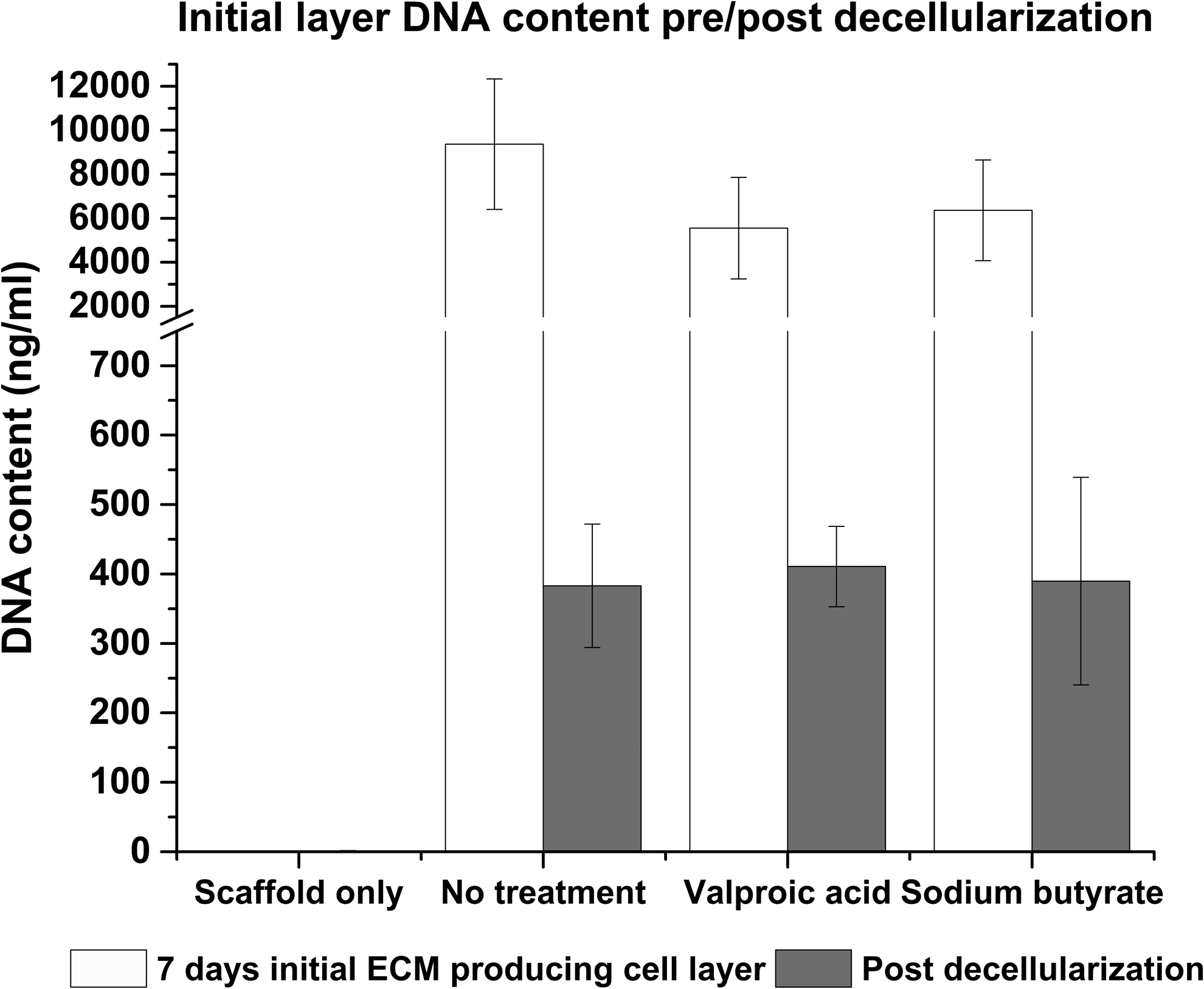
Confirmation of decellularization. Decellularization was confirmed using the Quant-IT™ Picogreen® dsDNA assay and SEM (not shown). Additionally, CellTiter-Blue® cell viability assay was performed on the decellularized scaffolds (data not shown).
The scaffolds were submerged in a 0.1% vol/vol Triton X-100 (Sigma-Aldrich), 1.5 M potassium chloride (Acros Organics), and 50 mM TBS for 4 h. They were rinsed for 15 min in 10 mM TBS before being submerged in fresh 10 mM TBS overnight.
Scaffolds were given a final rinse in 10 mM TBS for 15 min before being incubated in complete media for 15 min and then transferred to new 48-well plates for seeding.
Functional layer cell seeding and culture
HepG2 cells were trypsinized using standard methods from tissue culture flasks and counted using the trypan blue exclusion method. About 3 × 105 cells at passage 17 were suspended in 100 μL of complete media and seeded directly on to the scaffolds. The cells were allowed to incubate in this small volume on the scaffolds for 2 h, before an additional 400 μL of complete media was added.
Media was changed after 24 h and changed every 48 h after the initial 24 h adherence and recovery period. This functional layer (FL) of cells was cultured using standard methods for either 3 or 5 days at 37°C and 5% CO2 in a humidified incubator.
Live/Dead® viability/cytotoxicity assay
To determine cellular viability, cell/scaffold constructs were incubated with 10 μm calcein and 2 μm ethidium ho-modimer-1 (Ethd-1) for 30 min as part of the two color live/dead assay (Molecular Probes). Calcein is actively converted to calcein-AM in living cells, which then appear green when excited during fluorescence microscopy. Ethd-1 only accumulates in dead cells, which subsequently appear red. The method allows differentiation between dead and viable cells. The scaffolds were rinsed three times in CaCl2/MgCl2-free PBS to remove excess dye and placed onto a standard microscope slide with a 25 mm glass coverslip (VWR). All images were captured using a Zeiss Axio Imager fluorescent microscope (COIL; University of Edinburgh) at 40 × magnification and postprocessed using ImageJ.
CellTiter-Blue® Cell viability assay
The assay was performed according to manufacturer's instruction (Promega). For each condition group, n = 3. Importantly, cell/scaffolds constructs were moved into fresh 48-well plates to prevent reading activity from tissue culture plastic bound cells. Measurements were read in a Modulus™ II microplate reader at an excitation wavelength of 525 nm and emission wavelength of 580–640 nm and reported as fluorescence.
Picogreen DNA quantification
The Quant-IT™ Picogreen® dsDNA assay kit (Life Technologies™) was employed to establish the efficiency of the decellularization method in removing cellular material and to estimate cell number on the cell/scaffold constructs.
The assay was performed according to manufacturer instructions. In brief, constructs (n = 6) were digested in a solution of CaCl2 and MgCl2-free PBS (Sigma), containing 2.5 U/mL papain extract (Sigma), 5 mM cysteine-HCl (Sigma), and 5 mM ethylenediaminetetraaceticacid (Sigma) and incubated overnight at 60°C. Picogreen solution was added to the digests and fluorescent intensity measurements read in a Modulus II microplate reader at an excitation wavelength of 480 nm and emission wavelength of 510–570 nm. A standard λ dsDNA curve of graded known concentrations was used to calibrate fluorescence intensity versus dsDNA concentration.
Sectioning and staining
The samples were rinsed three times in PBS (Gibco) for 15 min each, then fixed in 4% v/v formalin buffered in saline for 15 min at room temperature. After rinsing with fresh PBS, constructs were embedded in low melting temperature polyester wax (Electron Microscopy Supplies) using methods adapted from Steele et al. 9 In brief, samples are dehydrated through 70–100% ethanol, then incubated in 50:50 ethanol:wax overnight at 45°C overnight with agitation. The samples were moved into 100% wax for 3 h at 45°C and then fresh 100% wax for 1 h at 45°C. Samples were embedded and allowed 72 h to fully cure before sectioning.
Immunohistochemical staining was undertaken using antibodies for Collagen I (Stratech), Laminin (Stratech), and Fibronectin (Sigma). All images were captured using a Coherent Anti-Stokes Raman Scattering system (Bioimaging Facility, University of Edinburgh) at 10 × and 100 × magnification and postprocessed using ImageJ.
Scanning electron microscopy
Scanning electron microscopy (SEM) was used to characterize the scaffold architecture. Samples were rinsed three times in PBS for 15 min each, then fixed in 2.5% v/v glutaraldehyde (Fisher Scientific) in 0.1 M phosphate buffer (PB) (pH 7.4) at 4°C overnight. They were then rinsed three times in 0.1 M PB before being postfixed in 1% v/v osmium tetroxide (Electron Microscopy Supplies) buffered with 0.1 M PB. Samples were again rinsed three times in 0.1 M PB and dehydrated through an ethanol gradient (30–100%). They were dried by placing them in hexamethyldisilazane (Sigma), which was allowed to evaporate off at room temperature overnight. We mounted the samples onto SEM chucks using double-sided carbon tape and coated them with a thin layer of gold and palladium alloy (Polaron Sputtercoater).
All images were captured at 5 kV using a Hitach S-4700 SEM (BioSEM; University of Edinburgh).
Mechanical testing
Nanoindentation experiments were undertaken to establish the dynamic properties of scaffolds and decellularized ECM/scaffold constructs using the Keysight/Agilent 5200 nano indenter testing system.
Scaffolds and constructs were subject to indentation by a DCM II actuator flat-ended cylindrical punch (D = 100 μm) using a max load of 1 gf. All nanoindentation experiments were carried out on fresh, hydrated, unfixed samples that were adhered to the chuck using double-sided tape. A total of 36 indentations were carried out on each sample, 50 nm apart. Indent sites were selected using the high precision X–Y stage within the testing system (Agilent). Poissons ratio was assumed to be 0.5 for each sample.43,44 Allowable drift rate was 0.1 nm/s. A NanoSuite (Keysight Technologies) test method “G-Series DCM CSM Flat Punch Complex Modulus” was used for all testing.45,46
Gene expression analysis
RNA was extracted from constructs using standard TRIzol (Fisher Scientific) methods and purified using Qiagen's RNeasy spin column system. cDNA was synthesized using the Promega's ImProm-II™ Reverse Transcription System.
Quantitative real-time polymerase chain reaction was performed using the LightCycler® 480 Instrument II (Roche Life Science) and Sensifast™ SYBR® High-ROX (Bioline) system. Results were normalized to HepG2s of the same passage number grown on tissue culture plastic and compared to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase. Analysis was performed using the 2−ΔΔCt method, 47 n = 4. Alb, Cytochrome P450 Family 1 Subfamily A Polypeptide 1 (Cyp1A1), Cytochrome P450 Family 1 Subfamily A Polypeptide 2 (Cyp1A2), Cytochrome P450 Family 3 Subfamily A Polypeptide 4 (Cyp3A4), Collagen Type I alpha 1, Collagen Type 4 alpha 1, and Fibronectin Type 1 were investigated, forward and reverse primers (Sigma) are detailed in Supplementary Table S1.
Statistical analysis
One-way analysis of variance and Tukey post hoc testing were performed and graphs generated using Origin software (OriginLab). A minimum of n = 3 and max of n = 6 was used for all analysis. Study groups were as follows; Scaffold only, No drug treatment, treatment with VA, and treatment with NaB. HepG2 cells were grown on each scaffold for 3 and 5 day time points.
Results
Cell attachment and survival on scaffolds
When compared to normal ECM (N-ECM), a lower number of HepG2s adhered to each of the drug-induced scaffold-ECM constructs, and to the scaffold alone (Fig. 3); demonstrating that “normal” ECM is efficient for cell seeding purposes. Scaffold only (SO), valproic acid ECM (VA-ECM) and sodium butyrate ECM (NaB-ECM) conditions demonstrate longer term maintenance or the HepG2s, however, (Fig. 3) and results are confirmed by both CellTiter-Blue viability and Picogreen DNA quantitation. Live/dead viability/cytotoxicity images (Fig. 4) further demonstrates the metabolic viability of the functional HepG2 cell layer (FL), although it should be noted that imaging reveals the FL was not yet confluent at 5 days culture, as further confirmed by SEM images (Fig. 5).

Seeding efficiency/viability on scaffolds. Cell adherence was assessed by Quant-IT Picogreen dsDNA assay
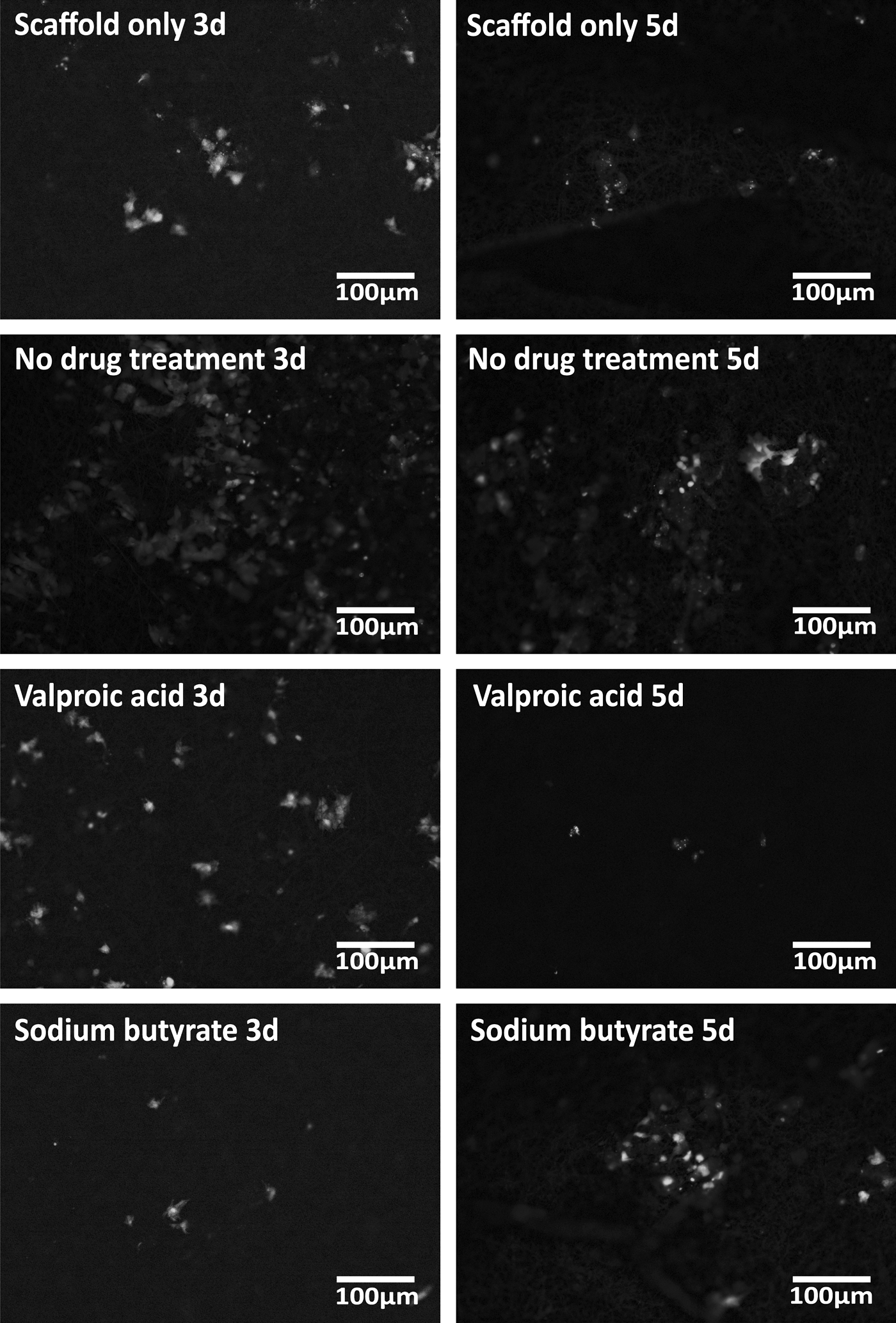
Live/Dead® viability/cytotoxicity assay. Viability of the FL was assessed by live/dead viability/cytotoxicity staining assay. Results demonstrate the FL is metabolically viable at all assessed time points. 40 × magnification. FL, functional layer.
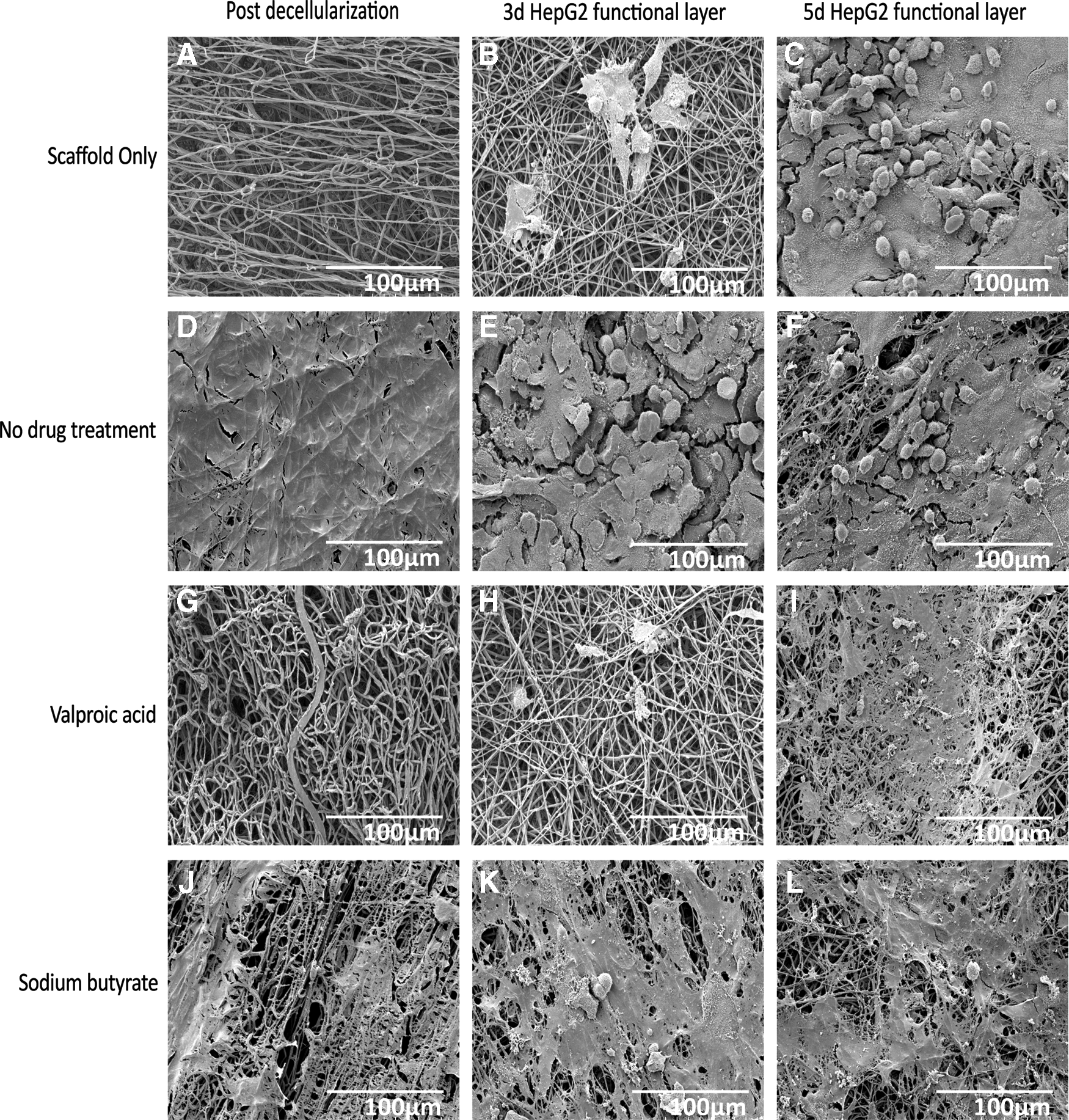
SEM of scaffold-ECM constructs and functional cell layers. SEM images of the decellularized scaffold-ECM constructs
Mechanical characterization of scaffolds
Interestingly, mechanical differences between the scaffold construct were negligible (Fig. 6). No significant differences in storage (Fig. 6A) or loss (Fig. 6B) modulus between the four conditions (Supplementary Tables S2 and S3).
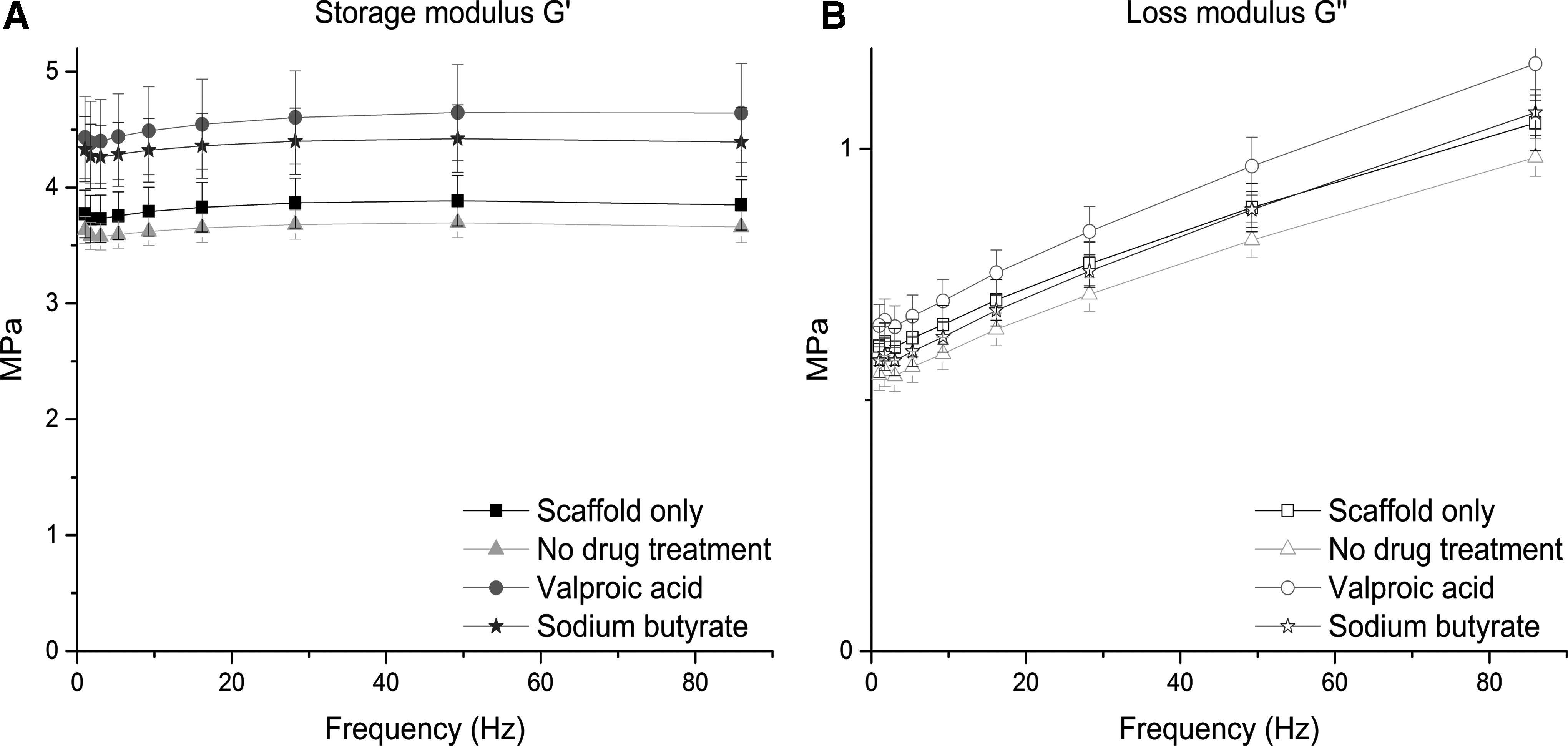
Mechanical characterization of decellularized scaffolds. Decellularized ECM/scaffold constructs were subject to nanoindentation experiments to assess their dynamic properties. Results demonstrate no significant differences in storage
Testing was performed at frequencies experienced by the human liver in vivo. 48 Storage modulus (G′) ranged from 3.58 ± 0.12 to 4.64 ± 0.43 MPa and loss modulus (G′′) from 0.55 ± 0.03 to 1.17 ± 0.07 at the frequencies detailed in Supplementary Tables S2 and S3. This reassures us that differences in cell attachment, viability, and function are due to differences in the biochemical and topographical profile of the hybrid scaffolds, as opposed to potential dynamic differences to which cells are known to be so sensitive.49–52
Biochemical characterization of the hybrid polymer-ECM scaffolds
Differences in the biochemical profile of the different ECMs were demonstrated by immunohistochemistry performed on the hybrid scaffold sections (Fig. 7). Hepatic phenotype has long been known to be influenced by ECM proteins; particularly Collagen I, Laminin, and Fibronectin,15,53,54 all of which are present on the scaffolds to varying degrees.
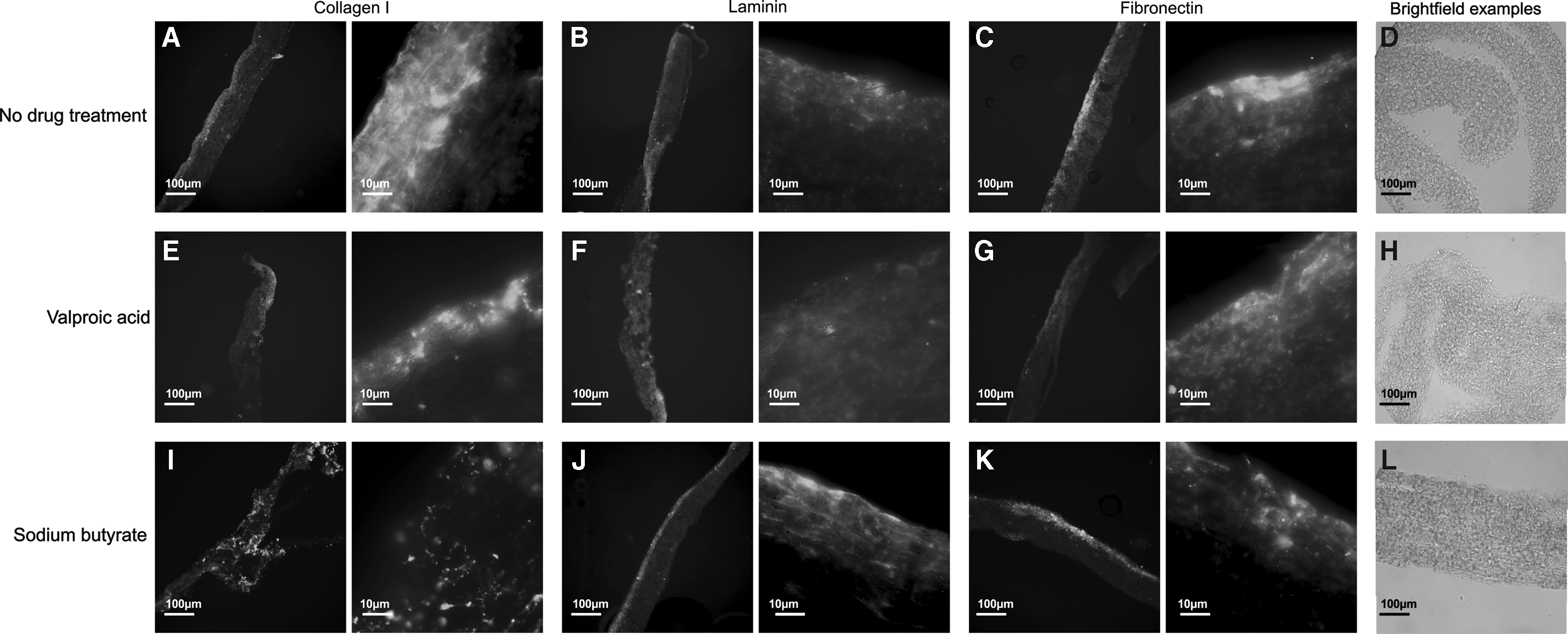
Immunohistochemical investigation. Immunohistochemistry staining of the decellularized ECM/scaffold constructs revealed significant differences in ECM components. Stains were performed for Collagen I
Laminin is of particular importance in the regenerating liver and for cell adhesion, and it is increased in injured or developing states.55,56 Laminin is present in each construct, but it appears to be most prevalent in NaB-ECM scaffolds (Fig. 7J). These constructs demonstrate higher “maintenance” of the HepG2s; that is cell number and viability increases between 3 and 5 days (Fig. 3).
Collagen I is one of the major components of normal liver ECM.53,54 Collagen I is most prevalent on NaB-ECM scaffold constructs (Fig. 7I), followed by VA-ECM (Fig. 7E) and N-ECM (Fig. 7A) scaffold constructs. When compared to the N-ECM scaffold constructs, these demonstrate improved maintenance of the HepG2 cell layer (Fig. 3), although not in comparison to the SO conditions.
Fibronectin is also ubiquitous in healthy liver ECM53,57 and is present under each of the conditions, but is most prevalent on NaB-ECM scaffolds (Fig. 7K). SEM of the hybrid scaffolds appears to confirm the fibrillary nature of the Collagen I-enriched NaB (Fig. 5J) and VA-ECMs (Fig. 5G) and the smoother morphology of the Laminin-enriched N-ECM (Fig. 5D).
The ECMs present have penetrated the scaffold in each case, and each protein is present on each scaffold, indicating the robustness and reproducibility of the method (Supplementary Fig. S2 n = 3).
Gene expression of HepG2s in response to hybrid polymer-ECM scaffolds
Multiple genes associated with liver function were assayed for gene expression (Fig. 8). Alb expression is a marker of liver cell differentiation and function and Cytochrome P450 (Cyp) are involved in metabolism of toxic compounds and products of endogenous metabolism such as bilirubin in the liver.6,17

Quantitative polymerase chain reaction analysis of functional cell layer. Quantitative analysis of gene expression was undertaken on the functional cell layer at 3 and 5 days culture, compared to that of HepG2s of the same passage and culture periods grown on tissue culture plastic (Fig. 7). mRNA levels of Albumin
Alb mRNA expression increases between day 3 and 5 as expected, but at day 5 is upregulated in comparison to HepG2s grown on tissue culture plastic in all conditions; with the highest levels observed in SO and N-ECM conditions. Additionally, Alb mRNA expression is downregulated at day 3 in SO and NaB-ECM conditions (Fig. 8A).
Cyp1A1 mRNA expression is consistently upregulated in comparison to tissue culture plastic (Fig. 8B). Cyp1A2 mRNA expression is hugely upregulated in comparison to tissue culture plastic (∼1000–38,000-fold), with the highest levels observed at 3 days on VA-ECM scaffold constructs, and levels at day 5 consistently lower than those at day 3 (Fig. 8C). Cyp3A4 mRNA expression follows a similar pattern, barring the NaB-ECM scaffolds; where expression levels at day 5 are higher than those of day 3. Additionally, the highest levels of expression are observed on SO at day 3 of culture (Fig. 8D).
Additionally, we assayed for three ECM genes, key components of the liver microenvironment; Fibronectin (Fig. 8E), Collagen I (Fig. 8F), and Collagen IV (Fig. 8G). While hepatocytes are not the major producers of ECM proteins in the liver, 53 the expression of such genes are of interest with regards to the cells further modulation of its environment considering the plastic nature of the ECM. 58
Fibronectin mRNA levels are higher in each condition than in that of HepG2s grown on tissue culture plastic, and highest levels are observed at day 3 on VA-ECM scaffold constructs, subsequently dropping to less than a third of day 3 levels by day 5. This may be in response to fibronectin levels observed in Figure 7, where the highest fibronectin levels observed (Fig. 7K) correspond with the lowest mRNA expression in the HepG2s. Collagen I mRNA expression is upregulated in every condition. Levels increase at day 5 in SO and NaB-ECM conditions; N-ECM and VA-ECM conditions follow the opposite trend with lowest levels observed in NaB-ECM conditions at day 3. The highest level of gene expression is observed on the scaffold with correspondingly increased Collagen I levels; NaB-ECM (Fig. 7I). Collagen IV mRNA levels are decreased in comparison to tissue culture plastic.
While the authors refrain from speculation without further analysis, the alterations in mRNA levels indicate that the drug-induced ECMs have a profound effect on liver cellular behavior.
Discussion
Fabrication of a hybrid polymer-ECM scaffold is an important avenue for liver tissue engineering, overcoming the shortage of donor organs and issues regarding animal-sourced biomaterials and creating a platform that can produce consistent, clinically translatable scaffolds for liver cell survival and function. An electrospun fiber approach was taken in the study to mimic the morphology of healthy fibrillary collagen,59,60 which may explain cells favoring environments with more Collagen I. We selected PCL for the fabrication of electrospun scaffolds as it possesses FDA approval for use in medical devices due to its biodegradable nature and elasticity. 9
The ECM is highly plastic, subject to constant modification and varies massively between tissues, 61 with each tissue possessing its own specific ECM “recipe”, composed of water, proteins, and polysaccharides, driven by a dialog between various cellular components and the microenvironment within the tissue.58,62 As the ECM is such a dynamic structure, it stands to reason that its production and maintenance will be influenced by its surrounding environment in 3D culture. 63
We used two different iHDACs to manipulate the cellular environment and alter the ECM production; NaB is widely used in industry to increase yield recombinant protein yields in mammalian cells. VA (ValA) is an FDA-approved anticonvulsant, which also functions as an iHDAC to increase recombinant protein yields. 40 By using iHDACs we can influence gene expression via their role in deacetylation, the process by which DNA renders itself less transcriptionally active. Inhibiting this process in cells results in hyperacetylation of histones and subsequently an increase in transcriptional activity. 39
Our results indicate not only that the iHDACs significantly alter the production and consistency of the ECM, but also that this technique is robust and reproducible and the ECM it produces, when harnessed in combination with 3D scaffolding technologies, creates a biofuctionalized scaffold, which significantly alters the behavior of liver cells.
The ECM produced by a bladder epithelial may be different to that of a liver cell; however, several decellularized ECM products on the market are in clinical use to regenerate tissues from which they are not derived, including ALLOPATCH HD™, MatriStem®, and Tutoplast® Pericardium. 64 Indeed, hepatocytes are often cultured on “ECM” surfaces that are not derived from liver, commonly using Matrigel®, a product derived from murine sarcoma, which is as yet undefined and experiences batch to batch variability.15,65–68
The promising field of whole organ decellularization is hampered by the availability of human livers, and researchers are subsequently investigating alternative organ and cells sources, such as spleen, bone marrow mesenchymal stem cells,69,70 and various animal sources of livers. 37 One of the main advantages of using decellularized native liver ECM is conservation of the highly conserved sinusoidal ECM gradient required to allow hepatocytes to repopulate in their specific niches. The complex ECM of the liver remains a topic of investigation in each of its states, diseased, regenerating, and healthy.53,55,71,72
To assess the performance of the hybrid scaffolds, we investigated the attachment and function of a commonly used liver cell line, HepG2s, at 3 and 5 days post-replating when cultured in vitro on the hybrid scaffolds versus scaffold alone. HepG2s were derived from the hepatocarcinoma of a 15-year-old Caucasian male. They are often used because they are virus-free, possess liver-specific functions such as ammonia metabolism and albumin synthesis, and secrete some growth factors such as insulin and insulin-like growth factor II. 73 We analyzed cell attachment and viability, and gene expression of both liver function genes and ECM genes at both 3 and 5 day time points. Additionally, we validated the decellularization of the ECM producing cell layer and performed immunohistochemical analyses of the hybrid scaffold-ECM constructs upon which the HepG2s were seeded.
While this work is a robust proof of principle regarding manipulation of ECM production, and has produced a novel hybrid polymer-ECM scaffold with great potential for liver tissue engineering, further work is required to analyze results and increase translatability. While HepG2s are a highly valuable research resource, they are derived from a carcinoma and as such criticism of their in vitro relevance abounds within the scientific community. It is important to undertake future work using primary or stem cell-derived hepatocytes and incorporate other stimuli such as fluid flow to combat such criticism. Furthermore, while hepatocytes, the major parenchymal cell of the liver, make up more than 70% of the cellular mass, they do not exist in isolation and the nonparenchymal cells play an essential role in the in vivo liver71,74; future studies should look to include these cells.
In addition, recognizing the value of proteomic and functional assays (such as enzyme-linked immunosorbent assays) in analyzing the function of the primary/stem cell-derived hepatocytes will be important for future validation of the scaffolds, however, at this time these were deemed unnecessary considering the critique of the HepG2 cell line. Additionally, care should be taken to ensure decellularization agents are completely removed from the scaffolds, due to their deleterious effect on both cells and ECM. 24 While such considerations are of importance, this study clearly demonstrates the potential of these hybrid polymer-ECM scaffolds for tissue engineering and provides a robust initial platform for further research.
Conclusion
This study developed a new method of creating hybrid polymer-ECM scaffolds by manipulating cells using electrospun scaffold technologies, clinically relevant iHDACs and methods easily modified to fulfill good manufacturing practice regulation. To do so, a sacrificial, ECM-producing cell layer was seeded onto a novel electrospun scaffold and then treated with VA or NaB to biofuctionalize the scaffold with ECM components. Scaffolds with untreated cells and no initial cell layer at all were used as controls. The initial cell layer was removed with a detergent-based decellularization method, and the resulting hybrid polymer-ECM scaffolds seeded with a liver cell line for validation. The work was validated using robust methods such as Q-PCR, mechanical quantification, and SEM. Drug-induced hybrid polymer-ECM scaffolds had a significant positive influence on the gene expression profile, attachment, and survival of liver cells.
Our data demonstrates promise as a unique method of inducing and altering the production of ECM and that the hybrid scaffolds exert influence upon cells in vitro, as well as future potential as an implantable treatment platform for liver disease patients.
These scaffolds show great potential not only for the future of liver tissue engineering and patient treatment, but are easily adaptable for other organs and tissues. Additionally, they are a useful tool for the development of 3D liver cell platforms, which can be used for in vivo cell analysis and novel pharmaceutical research.
Footnotes
Acknowledgments
The authors would like to thank Dr. Vlastimil Srnsen for technical help, Prof. Alistair Elfick for use of lab facilities (IBioE, University of Edinburgh) and Prof. Stuart Forbes for use of equipment (SCRM, University of Edinburgh). We would also like to thank Steve Mitchell (BioSEM), Dr. David Kelly (COIL), and Dr. Tony Corcoran (Bioimaging Facility) for their imaging expertise (University of Edinburgh). This work is funded by an Engineering & Physical Sciences Research Council [EPSRC] doctoral training partnership studentship, UK Regenerative Medicine Platform II [RMPII] grant MR/L022974/1 and MRC computational and chemical biology of the stem cell niche grant (CCBN) MR/L012766/1.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.