
Abstract
The intervertebral disc (IVD) is composed of nucleus pulposus (NP) surrounded by multilamellated annulus fibrosus (AF), and is located between the vertebral bodies. Current treatments for chronic neck or low back pain do not completely restore the functionality of degenerated IVDs. Thus, developing biological disc replacements is an approach of great interest. Given the complex structure of the IVD, tissue engineering of the individual IVD components and then combining them together may be the only way to achieve this. The engineered disc must then be able to integrate into the host spine to ensure mechanical stability. The goal of this study was to generate an integrated model of an IVD in vitro. Multilamellated AF tissues were generated in vitro using aligned nanofibrous polycarbonate urethane scaffolds and AF cells. After 3 weeks in culture, it was placed around NP tissue formed on and integrated with a porous bone substitute material (calcium polyphosphate). The two tissues were cocultured to fabricate the IVD model. The AF tissue composed of six lamellae containing type I collagen-rich extracellular matrix (ECM) and the NP tissue had type II collagen- and aggrecan-rich ECM. Immunofluorescence studies showed both type I and II collagen at the AF-NP interface. There was evidence of integration of the tissues. The peel test for AF lamellae showed an interlamellar shear stress of 0.03 N/mm. The AF and NP were integrated as the pushout test demonstrated that the AF-NP interface had significantly increased mechanical stability by 2 weeks of coculture. To evaluate if these tissues remained integrated, allogeneic IVD model constructs were implanted into defects freshly made in the NP-inner AF and bone of the bovine coccygeal spine. One month postimplantation, the interfaces between the AF lamellae remained intact and there was integration with the host AF tissue. No inflammatory reaction was noted at this time period. In summary, an engineered IVD implant with mechanically stable integration between AF lamellae and AF-NP can be generated in vitro. Further study is required to scale up the size of this construct and evaluate its ability to serve as a biological disc replacement.
Introduction
T
Previous studies showed that NP tissues with compressive strength can be formed scaffold free and integrated to the top surface of a porous bone substitute material such as calcium polyphosphate (CPP).
13
The bone substitute will help anchor the implanted tissue as bone ingrowth will fix it into the bone.14,15 In addition, another advantage of this approach is that, by forming tissue on a bone substitute, it allows surgical resection of the sclerotic calcified endplate as the porous bone substitute can fill the defect. As the material degrades, it will facilitate formation of a healthy endplate. AF tissue has been generated using biodegradable electrospun-aligned nanofibrous polycarbonate urethane (PU). This scaffold has the tensile strength of a native AF lamella and fiber diameters similar to the native collagen fibrils, allowing seeded AF cells to accumulate collagen aligned parallel to the scaffold.3,16–19 However, the successful integration of these in vitro generated tissues is crucial for it to be mechanically functional in vivo and for the longevity of the engineered
Thus, the goal of engineering of an
Materials and Methods
Synthesis of PU scaffolds
PU was synthesized as previously described. 16 Briefly, the base PU was formed by the reaction of poly (1,6-hexyl 1,2-ethyl carbonate)diol, 1,6-hexane diisocyanate and 1,4-butanediol in N,N-dimethylacetamide (DMAC) at 60–70°C. Anionic dihydroxyl oligomer (ADO) was synthesized by reacting polytetramethylene oxide, hydroxyethylmethacrylate (HEMA), and lysine diisocyanate in DMAC overnight at 50–60°C. 17 ADO was added to PU at a concentration of 0.15% by weight and dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol, followed by electrospinning onto a rotating mandrel at 1150 rpm and an applied potential difference of 18 kV into nanofibrous scaffolds. The scaffolds were dried in an Isotemp Vacuum Oven (Model 280A; Fisher Scientific, Pittsburgh, PA) overnight at 45°C and sterilized by gamma irradiation (2.5 MRad). 3 The scaffolds were cut into strips (0.5 × 23 cm) and coated with fibronectin (20 μg/mL F0895; Sigma-Aldrich, St. Louis, MO) dissolved in sterile phosphate-buffered saline solution without Ca2+ and Mg2+ (phosphate-buffered saline [PBS]−/−) overnight at 4°C. 17 PU scaffolds were rolled around perforated Teflon® PFA tubing (9.5 mm inner diameter and 11 mm outer diameter; McMaster-Carr, Aurora, OH) into circular multilamellated PU constructs composed of six scaffold layers before cell seeding.
Synthesis of bone substitute by solid free-form fabrication
CPP porous bone substitute was synthesized as previously described. 22 Calcined CPP amorphous particles (size ranging from 75 to 150 μm) blended with the polymeric binding agent polyvinyl alcohol (PVA) (Alfa Aesar, WardHill, MA) (10% weight) were injected with aqueous solvent Zb™58 (Z Corporation, Burlington, MA). 22 In a layer-by-layer manner, cylinders were 3D printed by a Zprinter® 310plus (Z Corporation, Burlington, MA) using a rotating roller that spread and compacted CPP particles delivered at a thickness of 150 μm per cycle until diameters of 10 mm and heights of 6 mm were obtained. 22 PVA was subsequently burnt off by treatment at 400°C for 1 h to anneal amorphous CPP particles. The cylinders were sintered at 950°C to achieve the desired crystalline structure of 30% volume porosity and interconnected pores in the 50–150 μm range. 14 The CPP substrates were placed within clear 3M™ Heat Shrink Thin-Wall Tubing (3M, St. Paul, MN), heated briefly to form a well-like structure, and then sterilized by gamma irradiation (2.5 MRad).
Fabrication of in vitro generated IVDs
IVDs were harvested aseptically from bovine caudal spines and placed in serum-free Dulbecco's modified Eagle's medium (DMEM). The discs were dissected, separating outer AF and NP tissues. Each tissue type was further diced into fine pieces under sterile conditions and digested separately with 0.3% protease for 90 min followed by collagenase overnight (0.1% for NP tissue and 0.2% for AF tissues). NP and AF cells were individually filtered using a sterile mesh and washed thrice with DMEM supplemented with 5% fetal bovine serum (FBS) (Wisent Bioproducts, St-Bruno, Quebec, Canada). 2 × 106 NP cells were seeded statically onto the top surface of the CPP substrate within the tubing and cultured in DMEM supplemented with 20% FBS for 4 weeks as previously described. 13 This time was selected as it was shown to be optimal for NP tissue formation. 30 × 106 AF cells in DMEM supplemented with 20% FBS were placed in a spinner flask (Bellco Glass, Vineland, NJ) to dynamically seed four circularized multilamellated PU scaffolds securely affixed to the impeller of a spinner flask rotating at 35 rpm. The nonadherent cells were removed 1 day after seeding by a complete media change. Ascorbic acid (Sigma-Aldrich, St. Louis, MO) was added on the third day to both cultures (100 μg/mL final concentration). Seventy-five percent of media was changed every other day.
In vitro generated

Schematic diagram of the method used to generate the intervertebral disc (IVD) in vitro. AF cell-seeded multilayered PU scaffold was grown in spinner culture for 3 weeks. NP cells were grown on a bone substitute for 4 weeks. The two tissues are combined and grown in coculture for up to 3 weeks. AF, annulus fibrosus; NP, nucleus pulposus; BS, bone substitute; PU, polycarbonate urethane. Color images available online at www.liebertpub.com/tea
Evaluation of AF and NP ECM accumulation and DNA content by the in vitro generated IVDs
After 2 weeks of coculture, the tissues of the constructs were separated using a scalpel blade and digested with papain (40 μL/mL; in digestion buffer containing 20 mM ammonium acetate, 1 mM EDTA, and 2 mM DTT at pH 6.2; Sigma-Aldrich, St. Louis, MO) for 48 h at 65°C. DNA content was determined using Hoechst 33258 dye (Polysciences, Warrington, PA) binding assay and fluorometry (excitation wavelength 365 nm and emission wavelength 458 nm) as described previously. 3 Proteoglycan content was determined by quantifying the amount of sulfated glycosaminoglycans (GAG) using the dimethylmethylene blue binding assay (Polysciences, Washington, PA) and spectrophotometry (525 nm) as described previously. 23 Collagen content was determined by quantifying the amount of hydroxyproline content in acid-hydrolyzed digests with a chloramine-T/Erlich's reagent assay and spectrophotometry (560 nm). 23 OH-Pro was converted to collagen based on the assumption that OH-Pro constituted 10% of the weight of collagen. 24
Evaluation of the interlamellar shear strength between adjacent layers of in vitro generated AF tissue
Multilamellated AF tissue (after 3 weeks of culture) was harvested and the mechanical strength between adjacent lamellar tissues was assessed by the peel test. Briefly, the outer layer of the AF was visualized and clamped on a crosshead. Using an Instron® model 8501 (Instron, Norwood, MA), which pulled the lamella at 10 mm/min, the interlamellar stress-to-failure was determined. 27 The peak stress (normalized to the width of the AF tissue) was the point at which the adjacent lamellae separated.
Evaluation of the interfacial shear strength between AF and NP tissues in the in vitro generated IVDs
In vitro combined AF-NP biphasic constructs were cocultured for various time points (between 1 and 3 weeks) and the mechanical strength of the interface formed between the AF and NP tissues was assessed by the pushout test. 26 The construct was placed in a specially designed jig attached to an Instron 4301 (Instron Norwood, MA) and loaded at a rate of 0.5 mm/min on the CPP until the NP-CPP tissue was displaced from the surrounding AF tissue (Fig. 5A). The interface area where AF and NP tissues were integrated was determined by measuring the circumference of the NP tissue and the height of the interface between the two tissues. The interfacial stress-to-failure was the peak shear stress (normalized to the interface area) at which the two tissues separated.
Evaluation of the compressive mechanical properties of in vitro generated IVDs
The compressive mechanical properties of the in vitro generated
Histological processing
In vitro generated
Immunostaining
Before immunostaining for type I and type II collagen, antigen retrieval was performed by sequential digestion of rehydrated sections with pepsin (2.5 mg/mL TBS buffer pH 2.0; Sigma-Aldrich, St. Louis, MO) at room temperature for 10 min, followed by trypsin (2.5 mg/mL TBS buffer pH 7.3; Sigma-Aldrich, St. Louis, MO) at room temperature for 30 min and then hyaluronidase (25 mg/mL PBS; Sigma-Aldrich, St. Louis, MO) at 37°C for 30 min. Sections immunostained for aggrecan were first incubated with chondroitinase ABC (0.25 U/mL in 100 mM Tris-HCl, 100 mM sodium acetate, pH 7.4; Sigma-Aldrich, St. Louis, MO) at 37°C for 30 min. After blocking with 20% goat serum diluted in PBS containing 0.1% Triton X-100, the sections were incubated with rabbit polyclonal anti-collagen type I antibody (1:100, CL50111AP-1; Cedarlane, Burlington, Ontario, Canada), mouse monoclonal anti-collagen type II antibody (1:250, MAB8887; Millipore, Etobicoke, Ontario, Canada), or mouse monoclonal anti-aggrecan antibody (1:250, AHP0022; Life Technologies, Burlington, Ontario, Canada) overnight. Following washes in PBS, the sections were incubated with secondary AlexaFluor®-labeled antibodies (1:1000; Life Technologies, Burlington, Ontario, Canada): AlexaFluor 594-labeled goat anti-rabbit IgG for type I collagen, AlexaFluor488-labeled goat anti-mouse IgG for type II collagen, and AlexaFluor594-labeled goat anti-mouse IgG for aggrecan. Nuclei were stained with 4′6-diamidino-2-phenylindole (DAPI). For negative controls, control mouse or rabbit IgG was used to replace the primary antibody followed by incubation with respective fluorophore-matched goat anti-mouse or anti-rabbit IgG and DAPI.
In vivo evaluation of interfaces
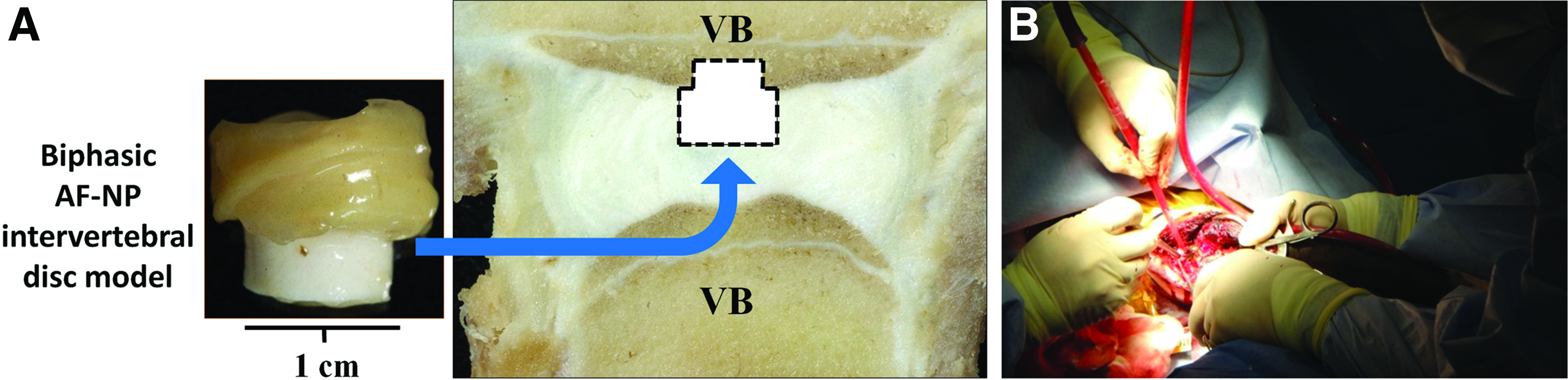
In vitro generated model discs were prepared as described above using cells obtained from allogeneic bovine caudal spine discs. A bovine caudal disc acute defect model was utilized. The protocol was approved by the Research Ethics Board of Ontario Veterinary College, University of Guelph. Calves were induced with diazepam (1 mg/kg, Sandoz, Holzkirchan, Germany) and ketamine (5 mg/kg, Telesta Therapeautics, Inc., Saint-Laurent, Quebec), and then intubated in sternal recumbency. The animal was anesthetized with Isoflurane (1.5–2.0%, IsoFlo; Abbott, Montreal, Quebec, Canada). Penicillin G (20,000 IU IV; Pharmaceutical Partners of Canada, Inc., Richmond Hill, Ontario, Canada), gentamicin (Gentocin, 2.2 mg/kg; Merck, Kirkland, Quebec, Canada), and flunixin meglumine (2.2 mg/kg IV Wyeth, Guelph, Ontario, Canada) were administered intraoperatively. The tail, sacrum, and perineum were scrubbed with chlorhexidine soap followed by 70% alcohol and then draped in an adhesive barrier drape (Ioban; 3 M Healthcare, London, Ontario, Canada). The spine was exposed by a paramedial incision. An annular flap was created and the caudal disc, either Cy 2 or 4, was visualized. The NP and inner AF were removed, leaving the outer AF intact except for the entry site flap. A defect ∼1 cm wide and 1 cm deep was created in the underlying bone with an osteotome in one VB. The implant was placed in the defect with the bone substitute placed into the site prepared in the VB. The AF flap was sutured closed with 3-0 caprosyn. The muscle and soft tissue were closed with 2-0 biosyn. After surgery, the calves were fully ambulated and the tail was allowed unrestricted movement. Two implants per animal were inserted (three animals) resulting in a total of six implants. The spinal unit containing the implant was harvested 1 month postimplantation and imaged by X-Ray. The segment containing the VBs, IVD, and the implant was excised as a block, fixed in 10% neutral buffered formalin, and embedded in plastic. One hundred fifty micrometer sections were cut and ground to 30 μm before staining with toluidine blue and light green, and visualized by light microscopy.
Statistical analysis
All conditions were done in triplicate and at least three independent experiments were performed unless otherwise stated. Results are expressed as mean ± standard deviation. Student's t-test was used to determine significance between two groups. One-way analysis of variance with the Tukey's post-hoc test was used to evaluate significance if more than two groups were compared. Significance was assigned at p < 0.05.
Results
Coculture of combined AF and NP tissues formed a model IVD
Combining NP and AF tissues generated a construct that resembled a model of IVD and could be handled (Fig. 2). The AF cell-seeded PU scaffolds formed multilamellated AF-like tissue. The AF cells were spindled in shape and accumulated ECM between the PU layers. The AF lamellae were adherent to each other and at its most inner layer, this tissue was integrated to the outer aspect of the NP. The AF tissues accumulated significantly more collagen and significantly less proteoglycans than NP tissues (Fig. 3). The NP tissues contained NP and notochordal cells in abundant ECM rich in type II collagen and aggrecan, while the AF tissue contained type I collagen only as determined by immunostaining (Fig. 4). These features are similar to that seen in native discs. Immunohistochemical studies demonstrated both type I and type II collagen and small amounts of aggrecan at the interface between the AF and NP tissues.


Evaluation of AF and NP tissues after 2 weeks of coculture. The tissue components were separated and quantified for

Multilamellated AF-like tissue showed lamellar adhesion with time
AF/PU multilamellar tissues (six lamellae) were formed in vitro. Early (less than 1 week) manual manipulation of the in vitro formed AF tissues resulted in the separation of the layers (data not shown). At 2 weeks of culture, the AF tissues were able to maintain their shape and not delaminate by handling, but the mechanical strength of the AF lamellar interfaces was not successfully evaluated as it was below the limit of detection. Therefore, the test was performed after 3 weeks of culture. The interlamellar shear stress was 0.03 ± 0.005 N/mm (n = 12) (Table 1), significantly lower compared to the native bovine AF tissues (0.6 ± 0.07 N/mm, n = 4).
Values were expressed as mean ± standard deviation.
At 5 mm/min.
p < 0.01 compared to values measured for native intervertebral disc.
At 0.5 mm/min.
At 0.1 Hz.
At 10% strain.
AF, annulus fibrosus; NP, nucleus pulposus.
IVD models generated in vitro had mechanically stable tissue integration
Pushout tests were done to evaluate the interfacial shear strength between the AF and NP cocultured over time (Fig. 5). There was a significant increase in strength between 1 and 2 weeks of coculture and no further increase was seen at 3 weeks. The interfacial shear strength at 2 weeks was 96 ± 16 kPa (n = 6), which was a magnitude lower compared to the native disc (487 ± 14 kPa, n = 6). These data suggested that 2 weeks of coculture was the minimum time required for the integration of the AF and NP tissue to form an interface with some mechanical strength. Thus, this culture time was chosen for further studies.
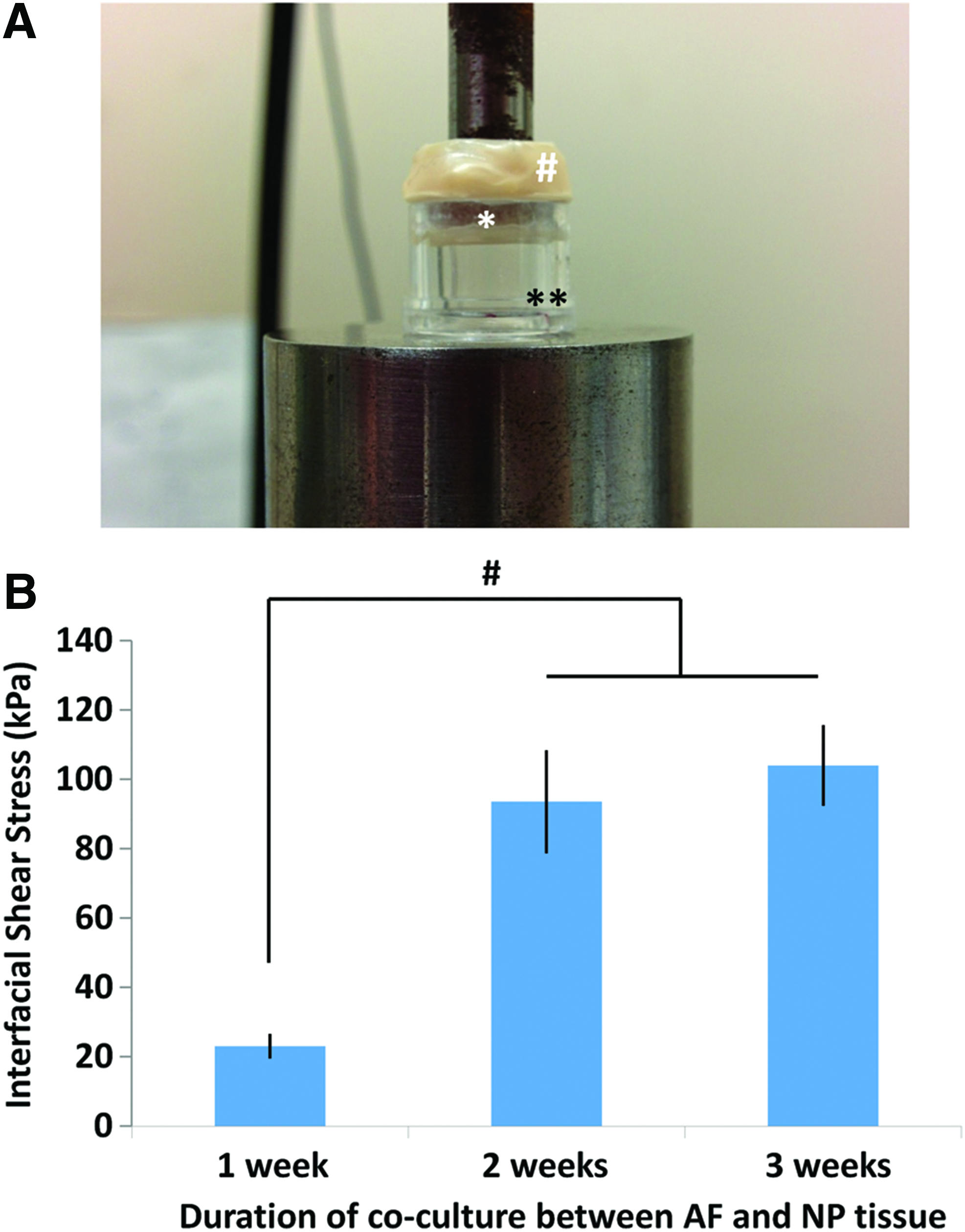
The strength of the interface formed between AF and NP tissues was determined using a pushout test after different times of coculture as an IVD model.
In vitro generated IVD showed compression-bearing mechanical function
The in vitro generated IVDs had compressive properties as determined after 2 weeks of coculture (Table 1). The model disc had a hysteresis of 64.3% ± 0.007 and a compressive modulus of 17 ± 0.007 kPa (n = 4), in comparison to the bovine native
Histological assessment of the IVD construct implanted in a bovine disc defect model
After 2 weeks of coculture, the model IVD with the bone substitute was implanted into a defect that had been created in the native disc and the adjacent bone. The bone substitute was placed in the defect created in the VB with the disc tissue located in the defect created in the native disc. The residual native AF surrounded the implant and the AF flap generated during the surgery was sutured closed to hold the implant in place (Fig. 6). The tail was allowed to move freely and no limited movement was appreciated during the 1-month follow-up. One month after implantation, the implants were harvested and X-Rays showed that the implants were present. All implants had subsided into the bone. However, a portion of the disc could be seen in the disc space in most animals (Fig. 7A). Histological examination showed that the implanted CPP bone substitute was present in the VB (Fig. 7B). The in vitro engineered AF tissues were present and still adherent to each other and to the bone substitute material. There was fibrous tissue between the native and implanted AF, but no inflammatory reaction was noted. The implanted NP could not be definitively identified as the tissue appeared to be chondroid-like (Fig. 7D).

Discussion
This study shows that it is possible to form a model of the IVD in vitro by combining preformed AF and NP tissues. These tissues integrate and have mechanical stability as indicated by the pushout test. The strength of this integration increased between 1 and 2 weeks of coculture. In addition, the layers of the multilamellated AF are adherent to each other. Although the peel test indicated that the strength between the layers was an order of magnitude less than native AF, it may be sufficient for weight bearing as the AF remains intact after implantation into the bovine caudal disc. As it was only in place for 1 month, it could be argued that it was not long enough to assess this. However, the fact that the implanted AF/PU tissue remains intact during the postoperative period and its reparative response is an important finding.
The approach of seeding fibronectin-coated nanofibrous scaffolds with AF cells in spinner culture allowed for formation of multilamellated AF/PU tissues. The cells adhered to the scaffold and accumulated type I collagen-rich tissues that served as the biological “glue” preventing the delamination of the engineered AF tissue. It is possible that culture in spinner flasks was critical as attempts to form this tissue under static conditions were not successful (data not shown). There are several possible explanations for this. First, the stirring motion could provide convective flow for nutrient exchange and mass transport, which could enhance tissue formation.27,28 Second, the application of flow perfusion has been reported to increase the density and uniformity of cell adhesion, the infiltration into fibrous scaffold matrices, and the accumulation of matrix molecules such as collagen and elastin in other cell types.27,29 Third, in a previous study, we demonstrated that the presence of fibronectin is critical for proper cellular and collagen alignment in vitro, a phenomenon also observed in the fetal developing disc.18,32 Shear flow has been shown to enhance fibronectin synthesis and organization by cultured cells and so may be acting in this manner in our culture system. 31 Fourth, the AF cells may benefit from the application of the shear forces during tissue formation as preconditioning enhances matrix accumulation as shown for cartilage and vascular tissue engineering.32,33 Further study is required to determine which factor(s) are important for the formation of multilamellated AF/PU tissue.
Others have formed IVD-like tissues, but they have differed from the method described in this study. For example, Mizuno et al. used poly(lactic-co-glycolic) acid scaffold seeded with passaged sheep AF cells, but the lamellar architecture of the AF was not recreated.34,35 In addition, the NP cells were seeded into alginate hydrogel and tissue formation occurred in vivo rather than in vitro. Several groups have used type I collagen to generate the lamellated structure of the AF.10,36 Bowles et al. gelled the collagen around alginate containing NP cells, 10 whereas the other group wrapped crosslinked collagen around an acellular core of collagen-chondroitin-6-sulphate co-polymer, which served as the NP. 36 Mauck et al. have used electrospun aligned nanofibers for the engineering of multilamellated AF tissues, similar to the current study. 12 However, they used poly(ɛ-caprolactone), a different polymer. 12 In addition, the NP portion was composed of alginate containing mesenchymal stromal cells and not scaffold-free NP tissue as in this study. 12 No disc to date has incorporated a bone substitute component.
This is the first report, to the best of our knowledge, describing integration of in vitro formed AF and NP tissues and evaluation of the interfacial shear strength. The mechanism that led to this integration is unknown. A previous study that examined bioengineered cartilage–cartilage integration suggested that the matrix between the two tissues intermingle as transmission electron microscopy studies showed thin collagen fibers that were produced by the bioengineered cartilage admixed with the mature collagen fibers of the native cartilage across the interface with the host tissue. 26 The presence of both type I and II collagens at the AF-NP interface suggests that this may be occurring in this situation as well. Interestingly, it has been proposed that the integration of distinct tissue types requires an intervening region that serves as a gradual and continuous transition in ECM properties and/or mechanical properties. 37 Other multiphasic tissue types include the ligament–bone interface, in which unmineralized fibrocartilage merges into mineralized fibrocartilage, which is the transitional tissue between the ligament and the bone. Similarly the interface between articular cartilage and the subchondral bone is characterized by a zone of mineralized cartilage. 37 This layer is critical for force distribution and to prevent separation of the two tissues with loading. 38 In the native IVD, the inner AF serves as a transition zone between NP and type I collagen-rich outer AF tissues. It is not clear, given the short time period of time in culture, whether this construct is developing this interface. The tissue interface strength did increase between week 1 and 2, but no further increase was seen between 2 and 3 weeks. It is possible that with weight bearing, in vivo mechanical properties may further increase, as was observed for implanted in vitro formed scaffold-free cartilage. 15
This implant study was performed only to evaluate the stability of disc tissue integration postimplantation; so restoration of disc height and function was not evaluated. Two previous studies have reported the outcome of implanted tissue-engineered
The optimal sources of cells to form AF or NP tissue for use in a biological disc replacement still need to be determined. Cells utilized to date include NP, AF, chondrocytes, mesenchymal stromal cells, and induced pluripotent stem cells (iPSCs).11,12,45,46 Cocultures of NP cells and notochordal cells have also been investigated.47,48 All of these have their issues, such as inadequate tissue formation by stem cells and the potential for tumor formation when using iPSCs.50 Differentiated NP and AF cells would be optimal, but they are limited in numbers. Interestingly, our studies to date suggest that bovine AF cells passaged to provide sufficient cell numbers will retain their phenotype (data not shown); however, this is yet to be shown for human cells. In contrast, NP cells appear to lose their phenotype with time in culture. Further study is required to identify an optimal source of NP cells if this approach is to be translated into clinical use.
In summary, this study demonstrates that it is possible to generate a model of an
Footnotes
Acknowledgments
This research was supported by CIHR (MOP342825) and MI-POP. The authors would like to thank Harry Bojarski and Ryding-Regency Meat Packers for providing bovine tissues, Dr. Meilin Yang for synthesizing PU scaffolds, and Drs. Pilliar and Eugene Wang for their help in producing CPP substrates.
Disclosure Statement
No conflicting financial interests exist.