
Abstract
The limited availability of native vessels suitable for the application as hemodialysis shunts or bypass material demands new strategies in cardiovascular surgery. Tissue-engineered vascular grafts containing autologous cells are considered ideal vessel replacements due to the low risk of rejection. However, endothelial cells (EC), which are central components of natural blood vessels, are difficult to obtain from elderly patients of poor health. Umbilical cord blood represents a promising alternative source for EC, but their allogeneic origin corresponds with the risk of rejection after allotransplantation. To reduce this risk, the human leukocyte antigen class I (HLA I) complex was stably silenced by lentiviral vector-mediated RNA interference (RNAi) in EC from peripheral blood and umbilical cord blood and vein. EC from all three sources were transduced by 93.1% ± 4.8% and effectively, HLA I-silenced by up to 67% compared to nontransduced (NT) cells or transduced with a nonspecific short hairpin RNA, respectively. Silenced EC remained capable to express characteristic endothelial surface markers such as CD31 and vascular endothelial cadherin important for constructing a tight barrier, as well as von Willebrand factor and endothelial nitric oxide synthase important for blood coagulation and vessel tone regulation. Moreover, HLA I-silenced EC were still able to align under unidirectional flow, to take up acetylated low-density lipoprotein, and to form capillary-like tube structures in three-dimensional fibrin gels similar to NT cells. In particular, addition of adipose tissue-derived mesenchymal stem cells significantly improved tube formation capability of HLA I-silenced EC toward long and widely branched vascular networks necessary for prevascularizing vascular grafts. Thus, silencing HLA I by RNAi represents a promising technique to reduce the immunogenic potential of EC from three different sources without interfering with EC-specific morphological and functional properties required for vascular tissue engineering. This extends the spectrum of available cell sources from autologous to allogeneic sources, thereby accelerating the generation of tissue-engineered vascular grafts in acute clinical cases.
Introduction
C
In the past, much effort has been undertaken to generate tissue-engineered vascular grafts composed of autologous cells of the vascular system such as endothelial cells (EC) and mural cells (pericytes, smooth muscle cells, and fibroblasts), which were integrated in appropriate scaffold materials.5,6 While the differentiation of mesenchymal stem cells from several tissue types such as bone marrow, 7 adipose tissue, 8 and umbilical cord 9 into mural cells like smooth muscle cells is largely established, the isolation of autologous EC is restricted to peripheral blood, which is associated with a number of hurdles. First, the number of endothelial progenitor cells in peripheral blood is with 0.01% extraordinarily small 10 demanding for large blood volumes (≥500 mL) which is incompatible for patients with a fragile state of health. Second, the isolation and expansion of sufficient cell numbers are labor intense and time consuming, preventing their off-the-shelf application.
Moreover, in contrast to these more practical considerations, there is a general restriction for the extraction of endothelial progenitor cells from peripheral blood. We 11 and others 12 showed that successful isolation and expansion of peripheral blood outgrowth endothelial cells (PB-OEC) were restricted to young and healthy donors, whereas the blood-derived EC of elderly patients with health impairments were not expandable. Recipients of vascular prostheses often belong to this group; in consequence, peripheral blood is an unsuitable source for the isolation of autologous EC for vascular tissue engineering purposes. In contrast, the isolation of EC from umbilical cord blood 13 and the umbilical cord vein 14 has been described to be easier, quicker, and more reproducible,15,16 even when compared to peripheral blood from young and healthy donors.
The major obstacle of these allogeneic cells, however, is the expression of human leukocyte antigen class I (HLA I) on their cell surface, which ultimately leads to graft rejection by an antibody-mediated immune response due to its high variability. 17
Recently, RNA interference (RNAi) has been used as a tool to stably silence the expression of HLA I proteins in hematopoietic progenitor cells,18,19 induced pluripotent stem cells, 20 and umbilical cord blood-derived EC. 21 Lentiviral transduction of the interfering RNA construct leads to a stable integration into the respective target cell genome and thus to a permanent knockdown of HLA I expression. Downregulation instead of a complete knockout is mandatory to maintain a residual expression of HLA I of at least 10% 21 since both the presence and complete absence of HLA I evoke immunological responses. Silencing HLA I expression was demonstrated to reduce cellular and humoral allogeneic immune responses by in vitro tests and prevent rejection after allotransplantation.20–23
Vector-mediated transfer of the RNAi cassette, however, may compromise the functionality of HLA I-silenced cells by integration in relevant genomic regions for cell viability and functionality. In particular, EC for vascular tissue engineering purposes require an array of properties to exert their homeostatic, vessel tone-regulating, and capillary-forming functions. Thus, to ensure that HLA I-silenced EC remain functional for vascular tissue engineering, these properties should be evaluated thoroughly.
We here silenced HLA I expression in EC from three different sources and characterized their specific morphological and functional properties with respect to expression of specific cell surface antigens and factors, uptake of acetylated low density lipoprotein (ac-LDL), and alignment under laminar flow and capillary formation capability, which is of pivotal importance for graft revascularization.
Materials and Methods
Primary cells
Human umbilical vein endothelial cells (HUVEC) used for HLA I silencing were purchased from Pelobiotec, Planegg/Martinsried, Germany.
Peripheral blood outgrowth endothelial cells
Mononuclear cells were isolated from healthy volunteers (26.5 ± 0.7 years, male) after informed consent. Buffy coats were diluted 1:2 with phosphate-buffered saline (PBS) and density gradient centrifugation in 1.077 g/mL Biocoll solution (Biochrom, Berlin, Germany) was performed. CD34-positive cells were isolated by magnet-activated cell sorting according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany) using magnet bead-coupled anti-CD34 antibodies and cultivated at a density of 0.5 × 106 cells per well in 24-well plates (Sarstedt, Nümbrecht, Germany) coated with 50 μg/mL rat tail collagen type I (Life Technologies, Darmstadt, Germany) in complete EGM-2 Bulletkit (Lonza, Basel, Switzerland) supplemented with 5% fetal bovine serum (FBS; Pan Biotech, Aidenbach, Germany) and 1% penicillin/streptomycin (P/S; Biochrom). 24 The medium was changed thrice a week. PB-OEC appeared after 10–14 days of cultivation and were expanded to confluence on cell culture plates coated with 10 μg/mL human recombinant fibronectin (Biochrom).
Umbilical cord blood-derived outgrowth endothelial cells
Mononuclear cells were isolated from 50 to 80 mL cord blood of healthy volunteers after informed consent. Buffy coats were diluted 1:2 with PBS and density gradient centrifugation in 1.077 g/mL Biocoll solution (Biochrom) was performed. Mononuclear cells were cultivated in cell culture flasks (Thermo Fisher Scientific, Braunschweig, Germany) in EGM-2 Bulletkit (Lonza) without gentamycin and amphotericin B (GA), but supplemented with 1% P/S (Biochrom). Umbilical cord blood-derived outgrowth endothelial cells (UC-OEC) appeared after 5–8 days of cultivation and were expanded to confluence on Nunc cell culture flasks (Thermo Fisher Scientific).
Human adipose tissue-derived mesenchymal stem cells
Adipose tissue-derived mesenchymal stem cells (ASC) were isolated from patients (46.0 ± 21.2 years, female) scheduled for visceral surgery after informed consent. Briefly, adipose tissue was washed with PBS, minced, and digested in serum-free Dulbecco's modified Eagle medium (DMEM) (Lonza) containing 37.5 μg collagenase (Liberase) and 25 U DNase (both Roche, Penzberg, Germany) per gram adipose tissue for 45 min at 37°C under shaking conditions. The cell suspension was washed with PBS and filtered through a 100 and 40 μm cell strainer (Corning, Kaiserslautern, Germany). Residual erythrocytes were lysed using distilled water and the cellular fraction was plated on tissue culture flasks (Sarstedt) in DMEM containing 10% FBS, 1% P/S, and 1% GA (all from Biochrom) and 10 ng/mL basic fibroblast growth factor (b-FGF) (Peprotech, Hamburg, Germany). The medium was changed thrice a week.
Generation of HLA-silenced EC
HLA I expression was silenced on EC using a lentiviral vector encoding for a short hairpin RNA (shRNA) targeting a specific β2-microgobulin sequence [shRNA construct: 5′-GAATGGAGAGAGAATTGAA-(loop: TTCAAGAGA)-TTCAATTCTCTCTCCATTCTT-3′)] and the enhanced green fluorescent protein (GFP) as reporter gene, as previously described. 18 Cells transduced with the same vector backbone, but encoding for a nonspecific shRNA, were used as a control. Cells were grown to 70% confluency and transduced for 8 h in the presence of 8 μg/mL protamine sulfate (Sigma-Aldrich, Steinheim, Germany). The vector-containing medium was exchanged for fresh medium after incubation. Transduction efficiency was assessed by flow cytometric detection of GFP expression frequencies and only EC populations showing more than 75% GFP expression were used for further experiments.
Determination of the HLA I silencing effect
The silencing efficiency of transduced EC was analyzed directly after 2 days and up to five passages after lentiviral transduction.
Flow cytometric analysis
For evaluation of the HLA I silencing effect, nontransduced (NT), nonspecifically transduced (NST, expressing shNS), and HLA I-silenced EC from donors were stained with anti-HLA I (w6/32) antibodies conjugated with Alexa fluor 647 (AF647) (AbDSerotec, Düsseldorf, Germany) and analyzed by flow cytometry. The percentage of GFP-expressing cells indicated the transduction efficiency and the mean fluorescence intensity (MFI) detected by the AF647-conjugated w6/32 antibody showed the level of HLA I expression on the cell surface.
Real-time polymerase chain reaction
β2 microglobulin (β2m) mRNA levels of EC from donors were analyzed as described before. 18 Briefly, the cells were harvested and total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The high-capacity cDNA reverse transcription kit (Applied Biosystems, Darmstadt, Germany) was used to reverse transcribe the RNA into cDNA. β2m transcript levels were analyzed by real-time PCR using specific gene expression assays (Hs00984230_m1; Thermo Fisher). Expression values were calculated as ratio to glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Hs02758991_g1; Thermo Fisher), which was used as internal control (relative quantification [RQ]).
Characterization of NT, NST, and HLA I-silenced EC
The characterization of NT, NST, and HLA I-silenced EC was performed after one to five passages after lentiviral transduction.
Immunohistochemistry
EC were fixed with 4% paraformaldehyde, permeabilized using 0.1% Triton-X-100, and blocked with PBS containing 10% FBS, 0.1% bovine serum albumin, and 0.05% Tween-20. Cells were stained for endothelial markers using the following antibodies: polyclonal rabbit anti-human CD31 (Santa Cruz Biotechnology, Heidelberg, Germany), dilution 1:750; monoclonal mouse anti-human VE-cadherin/CD144 (BD Biosciences, Heidelberg, Germany), dilution 1:100; polyclonal rabbit anti-human von Willebrand factor (vWF) (Dako, Jena, Germany), dilution 1:8000; and monoclonal mouse anti-human endothelial nitric oxide synthase (eNOS) (BD Biosciences), dilution 1:100, and the corresponding secondary antibody (Alexa fluor 555 anti-mouse 1:250 or anti-rabbit 1:100; Life technologies). After incubation, cells were washed with PBS and mounted with a medium containing 4′,6-diamidino-2-phenylindole (Roth, Karlsruhe, Germany) to counterstain cell nuclei. Staining was assessed by fluorescence microscopy (Nikon Eclipse TE300, Düsseldorf, Germany).
Population doubling rate
To determine the population doubling rate (n) of NT, NST, and HLA I-silenced cells per day, EC were cultivated over several passages and the population doubling rate was calculated as follows:
Phenotypic analysis
EC subpopulations were distinguished according to their phenotype. Therefore, the cells were stained using anti-CD14-PerCP, anti-CD31-APC/Cy7, anti-CD144-APC, CD309-PE/Cy7 (all Biolegend, Koblenz, Germany), and CD34-PE (BD Bioscience) for 15 min. After a washing step, the cells were analyzed by flow cytometry. The expression of the surface markers was considered individually or in combination for the identification of specific EC subpopulations as suggested by Steurer et al. 25 Hence, EC populations were separated into circulating EC (CEC; CD14−CD31+CD34+CD144+), circulating endothelial progenitors (CEP; CD14−CD31+CD34+CD144high), EC (CD14−CD31+CD34lowCD144+), and myeloid EC (mEC; CD14+CD31+CD34lowCD144−) according to the expression levels of CD14, CD34, CD31, and CD144.
ac-LDL uptake assay
EC were incubated for 4 h with a medium containing 10 μg/mL 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate-labeled-ac-LDL (Dil-ac-LDL; Alfa Aeser, Karlsruhe, Germany). Afterward, Dil-ac-LDL-containing medium was replaced by fresh medium. The uptake of labeled Dil-ac-LDL was analyzed by fluorescence microscopy using an Olympus IX81 microscope and the Xcellence Pro image Software (Olympus, Hamburg, Germany). To perform a quantitative analysis, EC were detached as described above, washed, and evaluated by flow cytometry.
Laminar flow assay
Laminar flow assays were performed with a perfusion system (Ibidi, Planegg/Martinsried, Germany). Briefly, 1.2 × 105 EC were seeded on a flow chamber (μ-slide I 0.4 Luer). After 24 h, the chamber was perfused for 48 h by continuous unidirectional flow resulting in 25 and 44 dyn/cm2 shear stress. Adaptation to flow was visualized by F-actin filament-staining by phalloidin-tetramethylrhodamine B isothiocyanate (50 μg/mL; Sigma-Aldrich).
Isolation of fibrinogen from human fresh frozen plasma by cryo-precipitation
Fibrinogen was isolated from 50 mL of fresh frozen plasma obtained from Hannover Medical School blood bank. After thawing the plasma at room temperature, precipitated fibrinogen was collected by centrifugation for 5 min at 1600 rpm and 2°C, and dissolved in 5 mL supernatant at 37°C. Protein content of the fibrinogen plasma solution was determined using the bicinchoninic assay method (Thermo Fisher Scientific) according to the manufacturer's instructions. The fibrinogen concentrations were measured using the Clauss method 26 based on the clotting time (Hematology, Hannover Medical School).
Tube formation assay
Tube formation assays were performed with NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC in monocultures or in co-culture with ASC as described previously. 11 Briefly, 375,000 EC or 375,000 EC plus 187,500 celltracker orange CMRA-labeled ASC for a ratio of 1:0.5 were suspended in 340 μL fibrinogen plasma solution corresponding to 0.29 ± 0.08 mg fibrinogen. A fibrinogen-cell suspension of 50 μL/well was polymerized in 96-well plates by addition of 50 μL human thrombin-calcium chloride solution (20 U/mL; Vascular Solutions, Minneapolis). Three-dimensional (3D) capillary networks formed within 72 h in the presence of 40 ng/mL vascular endothelial growth factor (VEGF)165, 40 ng/mL b-FGF (both Peprotech), and 1000 U/mL aprotinin (Bayer, Leverkusen, Germany). Tube formation was visualized by fluorescence microscopy in 12 (monocultures) and 6 (co-cultures) replicates. Tube area, tube length, number of junctions, and number of tubes were quantified on two photos per replicate using the Angiotool software. 27
For detection of endothelial lumen formation, co-cultures of NT HUVEC and ASC cultivated for 72 h in fibrin gels were fixed in 10% formalin, embedded in Technovit® 9100 (Heraeus Kulzer, Hanau, Germany), and 5 μm sections were stained by hematoxylin/eosin (Merck KGaA, Darmstadt, Germany).
Statistics
Statistical analyses were performed using Graphpad Prism 5 or 6 (Graphpad Software, San Diego, CA). Normal distribution of data was tested using the d'Agostino & Pearson omnibus normality test. Comparisons between two groups were performed by Student's t-test for normally distributed data and by Mann–Whitney U-test for nonparametric data. Comparisons between three or more groups were performed by one-way ANOVA plus Tukey's post-test for parametric data and by Kruskal–Wallis Test plus Dunn's post-test for nonparametric data. Differences were considered significant at p < 0.05. Significance levels were given as follows: *p < 0.05; **p < 0.01; ***p < 0.001; and #p < 0.0001.
Results
Determination of HLA I silencing
To reduce the immunogenicity of EC for vascular tissue engineering, EC from three different sources were transduced by lentiviral vectors encoding for a shRNA specific for β2m. Flow cytometric analysis for the detection of GFP expression revealed similar transduction efficiencies in the three EC types for up to five passages. NST cells were transduced by 92.0% ± 6.3% (PB-OEC), 90.4% ± 4.0% (HUVEC), and 86.3% ± 1.7% (UC-OEC) and HLA I-silenced cells were transduced by 89.2% ± 7.3% (PB-OEC), 94.8% ± 0.4% (HUVEC), and 95.4% ± 2.3% (UC-OEC). This corresponds to a mean transduction efficiency of 89.5% ± 4.0% and 93.1% ± 4.8% for NST and HLA I-silenced cells, respectively. In terms of reduction of the HLA I-expression, for all cell types, already one passage after transduction, a silencing effect was detectable by β2 m-specific flow cytometry and persisted for more than five passages (data not shown). Figure 1A shows a histogram of a representative experiment. NT HUVEC showed an MFI of 14,037 ± 10,021, the NST HUVEC 10,964 ± 7306, and the HLA I-silenced HUVEC 7468 ± 3314. This represented a decrease of 47% on HLA I expression compared to NT (p = 0.0080). Also, HLA I-silenced PB-OEC showed a significantly reduced MFI value of 7674 ± 5034, representing a silencing effect of 67% in comparison to the MFI of NT cells (23,475 ± 12,480, p = 0.0017). NST cells expressed a reduced value of 13,743 ± 5306. HLA I-silenced UC-OEC showed an MFI of 6857 ± 2709 representing a significant reduction of 52% in HLA I expression compared to NT cells (MFI 14,204 ± 6565, p = 0.0005).
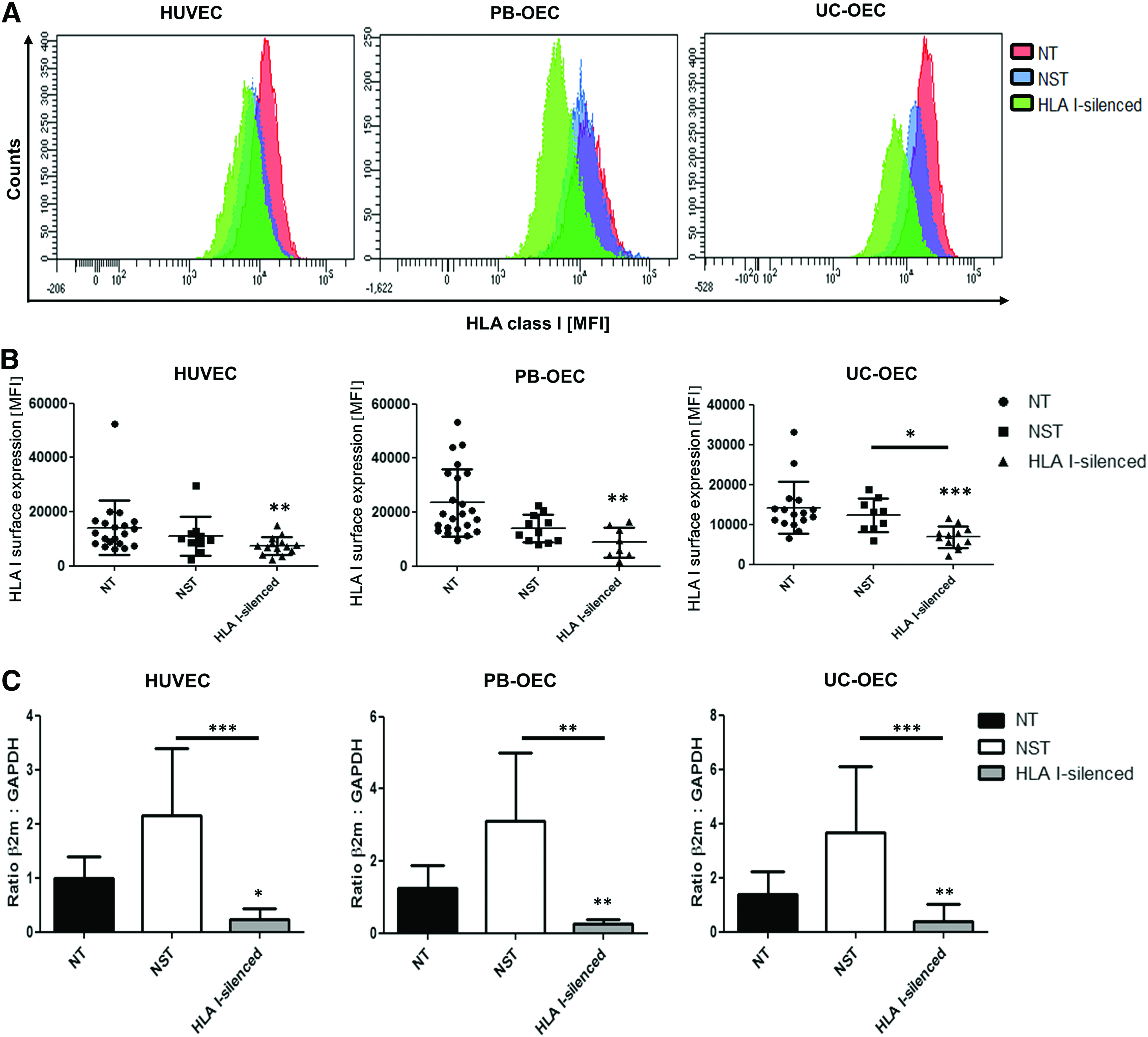
Determination of the HLA I-silencing efficacy by flow cytometry and real-time polymerase chain reaction of PB-OEC, HUVEC, and UC-OEC. The MFI detected by anti-HLA I-staining showed the level of HLA I-expression on the cell surface. Exemplary histogram overlays are shown for NT, NST, and HLA I-silenced EC of all three cell types
In comparison to NST cells (12,328 ± 4121), HLA I-silenced cells showed a significant reduction in HLA I expression of 47% (p = 0.0111) (Fig. 1B).
Transcript levels of β2 m of HLA-silenced HUVEC (RQ: 0.22 ± 0.21) were significantly decreased in comparison to NT (RQ: 0.99 ± 0.41, p = 0.0324) and NST cells (RQ: 2.16 ± 1.23, p = 0.0003).
A similar effect was shown for PB-OEC and UC-OEC. Levels of β2 m mRNA levels were significantly decreased in HLA I-silenced PB-OEC (RQ: 0.26 ± 0.11) in comparison to NT cells (RQ: 1.24 ± 0.63, p = 0.0092) and NST cells (RQ: 2.58 ± 1.88, p = 0.0027). Also, decreased β2m levels were measured in HLA I-silenced UC-OEC (RQ: 0.4 ± 0.61) in comparison to NT (RQ: 1.38 ± 0.86, p = 0.0014) and NST cells (RQ: 3.66 ± 2.47, p = 0.0003) (Fig. 1C).
Morphological, phenotypic, and functional characterization of HLA I-silenced EC
In a next step, the effect of HLA I silencing on EC was morphologically and functionally assessed. In a first set of experiments, NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC were immunohistochemically stained for EC-specific markers. Staining revealed a similar expression of CD31 (Fig. 2) and VE-cadherin/CD144 (Fig. 3) in NT, NST, and HLA I-silenced EC from all three sources, indicating intact cell–cell junctions, which are necessary for EC layer that functions as a selective barrier. Moreover, NT, NST, and HLA I-silenced EC were shown to synthesize vWF (Fig. 4) and eNOS (Fig. 5) in similar amounts, which are indispensable for blood coagulation and vessel tone regulation.
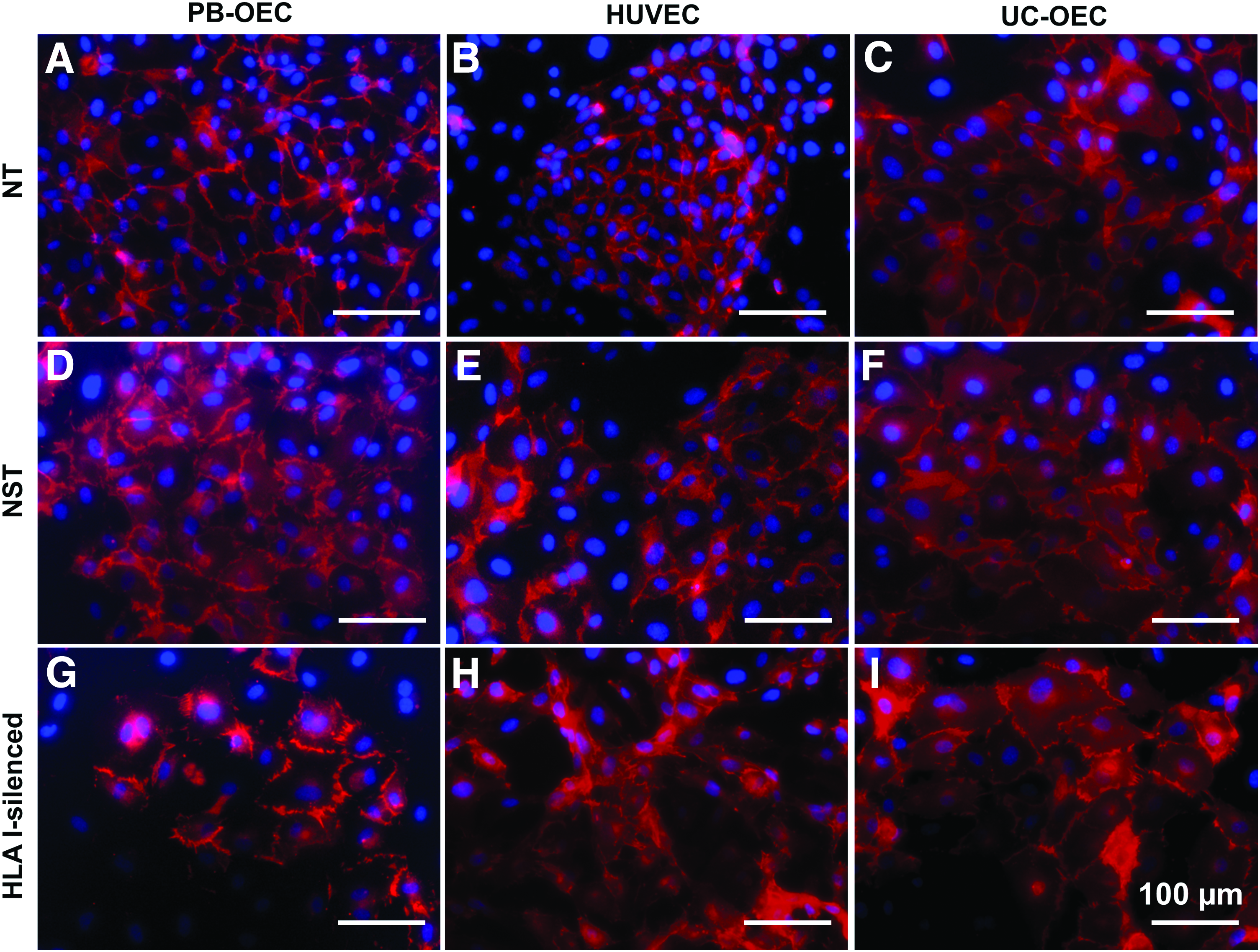
Expression of CD31 in NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC. EC from three different sources were transduced with lentiviral vectors encoding a β2-microglobulin-specific shRNA to downregulate HLA I expression. NT and NST cells transduced with a nonsense shRNA served as controls. Red immunofluorescence staining for CD31 revealed a similar expression in NT cells
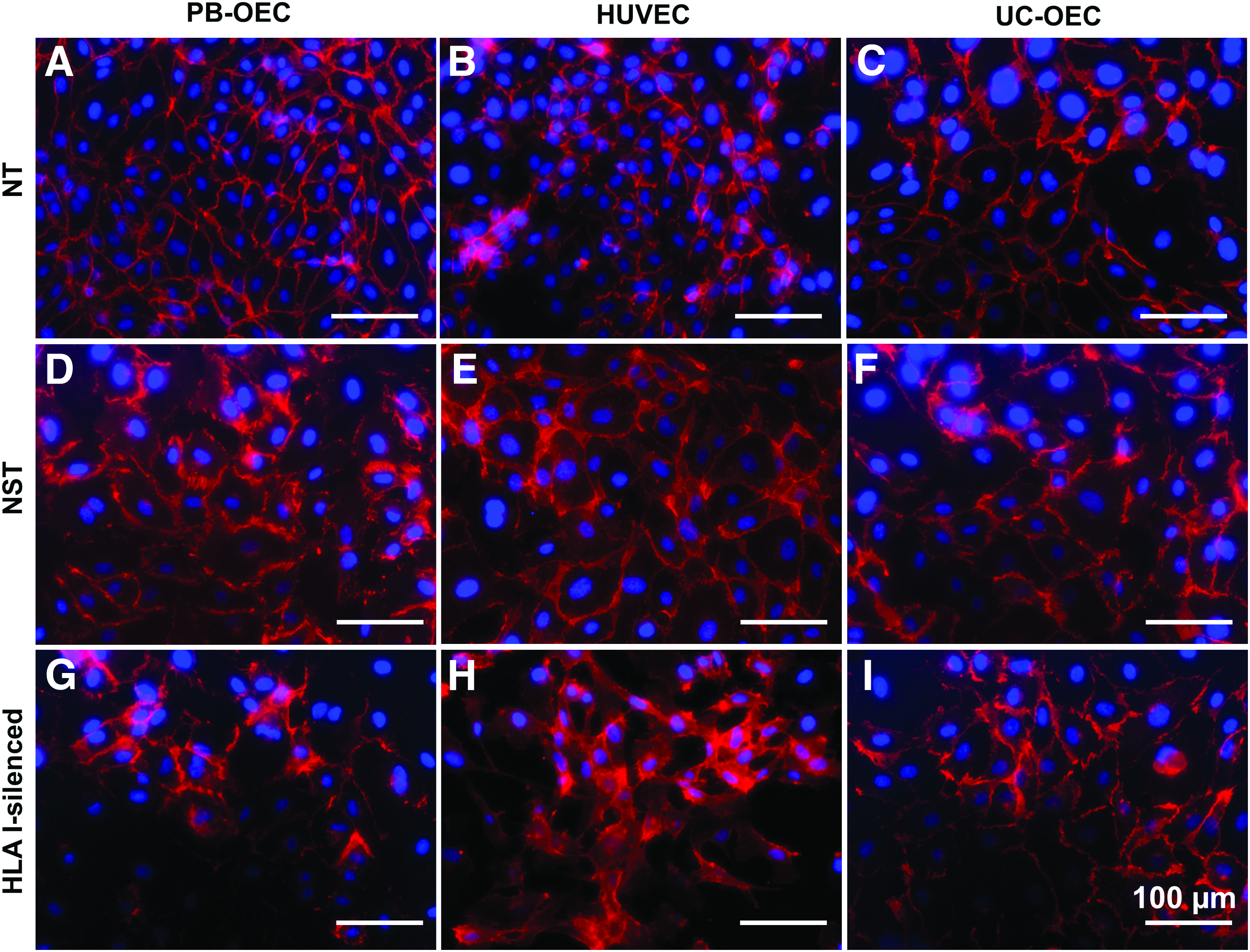
Expression of VE-cadherin/CD144 in NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC. EC from three different sources were transduced with lentiviral vectors encoding a β2-microglobulin-specific shRNA to downregulate HLA I expression. NT and NST cells transduced with a nonsense shRNA served as controls. Red immunofluorescence staining for VE-cadherin revealed a similar expression in NT cells
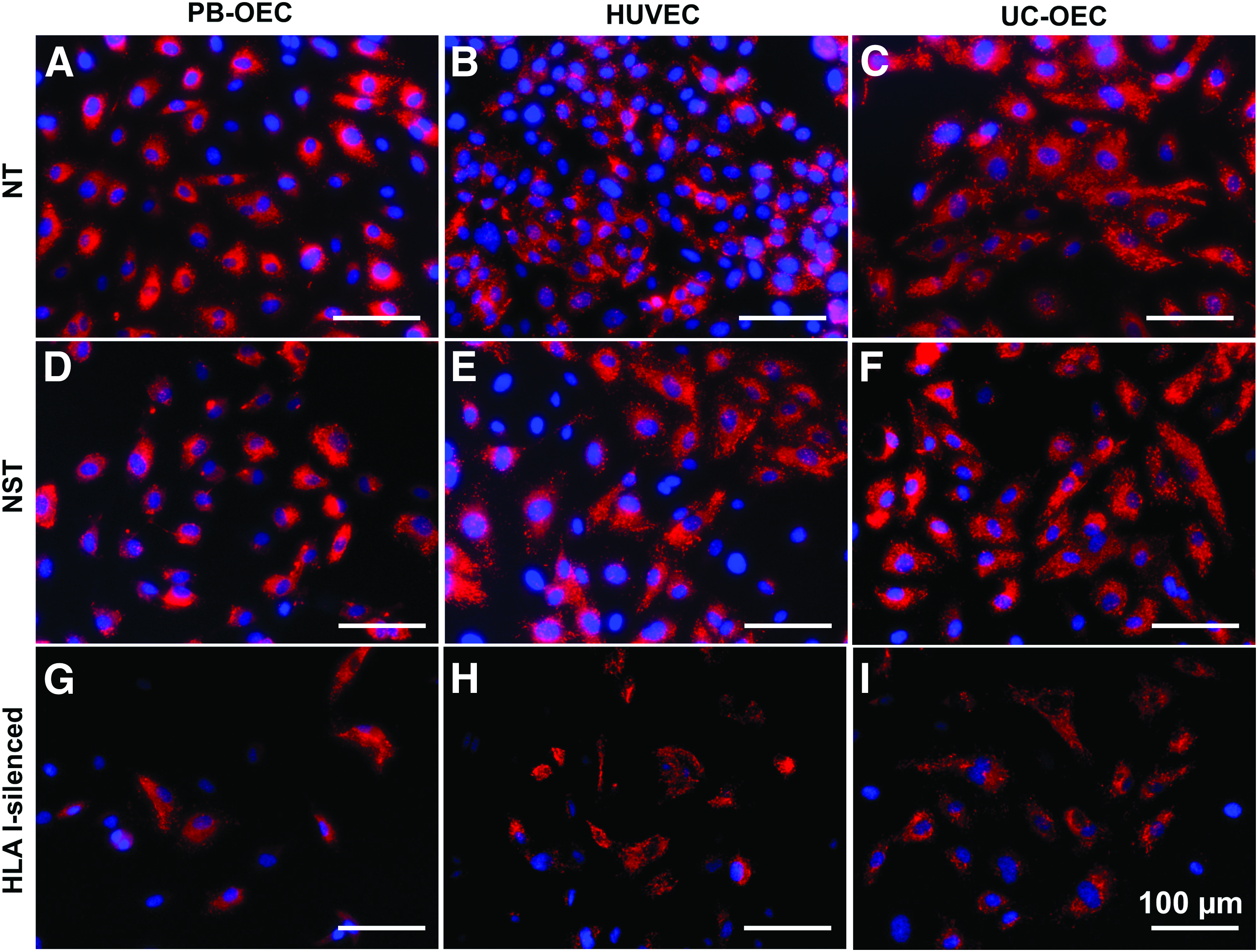
Expression of vWF in NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC. EC from three different sources were transduced with lentiviral vectors encoding a β2-microglobulin-specific shRNA to downregulate HLA I expression. NT and NST cells transduced with a nonsense shRNA served as controls. Red immunofluorescence staining for vWF revealed a slightly higher expression in NT cells
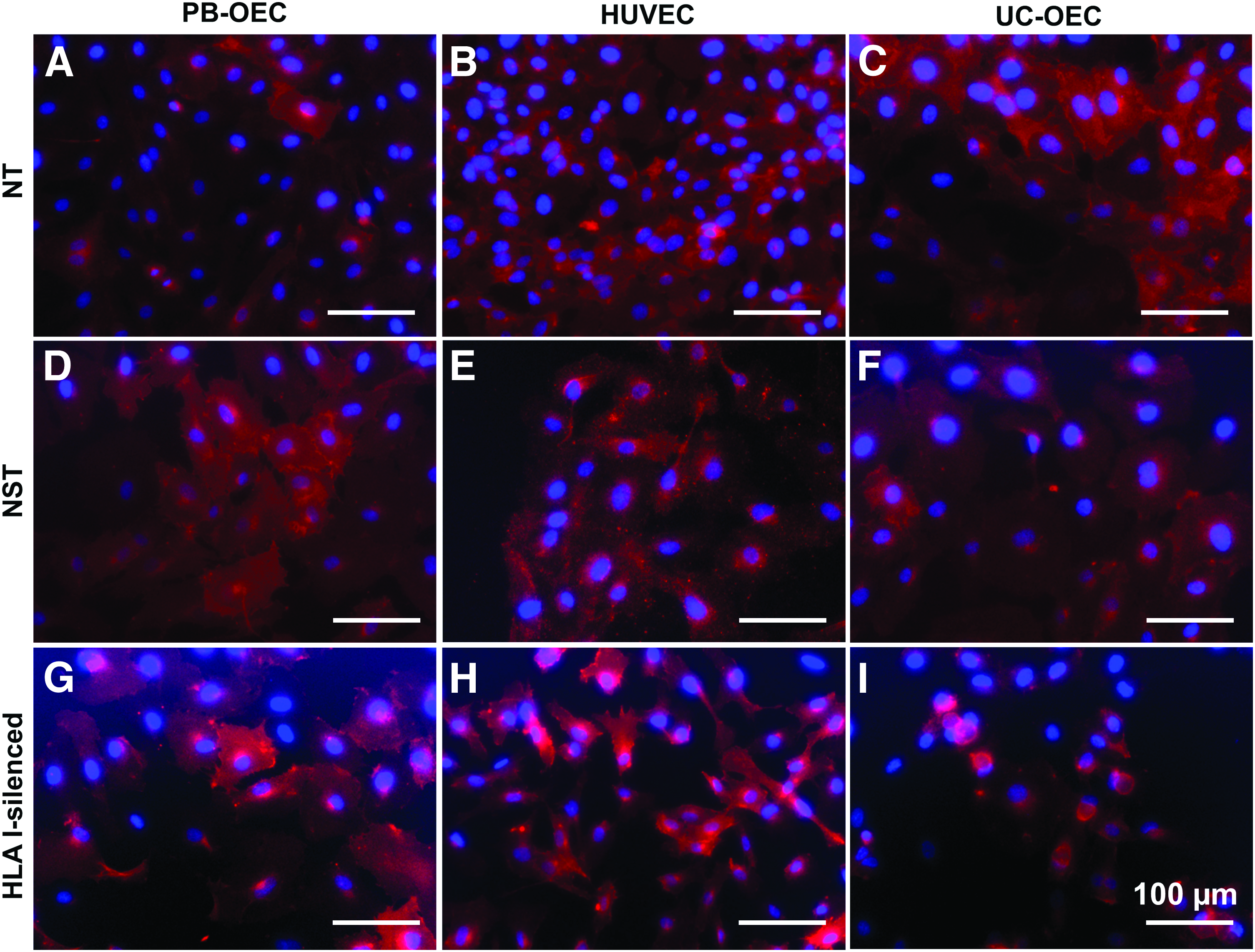
Expression of eNOS in NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC. EC from three different sources were transduced with lentiviral vectors encoding a β2-microglobulin-specific shRNA to downregulate HLA I expression. NT and NST cells transduced with a nonsense shRNA served as controls. Red immunofluorescence staining for eNOS revealed a comparable expression in NT cells
It was striking that the total number of stained cells was highly reduced in HLA I-silenced cells compared to NT and NST cells. As shown by the population doubling rate (Table 1), the proliferation capacity of NST and HLA I-silenced cells was considerably decreased compared to NT cells. This might explain the relatively low cell number of HLA I-silenced cells, although the cell number of NST cells was comparable to NT cells. Independent of the number of cells, the ratio of stained to unstained cells seemed to be similar in all three groups.
EC were cultivated over several passages before the population doubling rate was calculated and normalized to a culture period of 24 h.
EC, endothelial cells; HLA I, human leukocyte antigen class I complex; HUVEC, human umbilical cord vein endothelial cells; NST, nonspecifically transduced; NT, nontransduced; PB-OEC, peripheral blood-derived outgrowth endothelial cells; UC-OEC, umbilical cord blood-derived endothelial outgrowth cells.
The effect of transduction and silencing procedure on the different EC phenotypic markers was also evaluated at a quantitative level (Fig. 6A). Flow cytometric analysis of the frequency of individually expressed CD14 (monocyte marker), CD34 (EC progenitor marker), CD31, CD144 (VE-cadherin), and CD309 (vascular endothelial growth factor receptor-2 [VEGF-R2]) as mature EC markers revealed no significant differences between NT, NST, and HLA I-silenced HUVEC and UC-OEC. In PB-OEC, HLA I silencing slightly reduced the frequency of CD14 and CD34 compared to NST PB-OEC (p = 0.0262 and p = 0.0336, respectively). Also, on the mRNA level, no differences with CD31 and CD144 between NT and HLA I-silenced cells were observed, although the levels in NST cells in all EC types were somewhat increased (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea). According to the definition of EC subpopulations, 25 a gating strategy was established to first distinguish between CD34low and CD34+ cells and then divide the CD34low population into CD14+ mEC and CD14− EC. The CD34+ population was divided into CD144+ CEC and CD144high CEP subpopulations (Fig. 6B). Hence, the transduction procedure and HLA I silencing did not affect the number of circulating, progenitor, mature, and mEC in HUVEC and UC-OEC, whereas in PB-OEC, a significant decrease in the CEC population (p = 0.0350) and an increase in the EC population (p = 0.333) in HLA I-silenced PB-OEC in comparison to NST were observed (Fig. 6C).
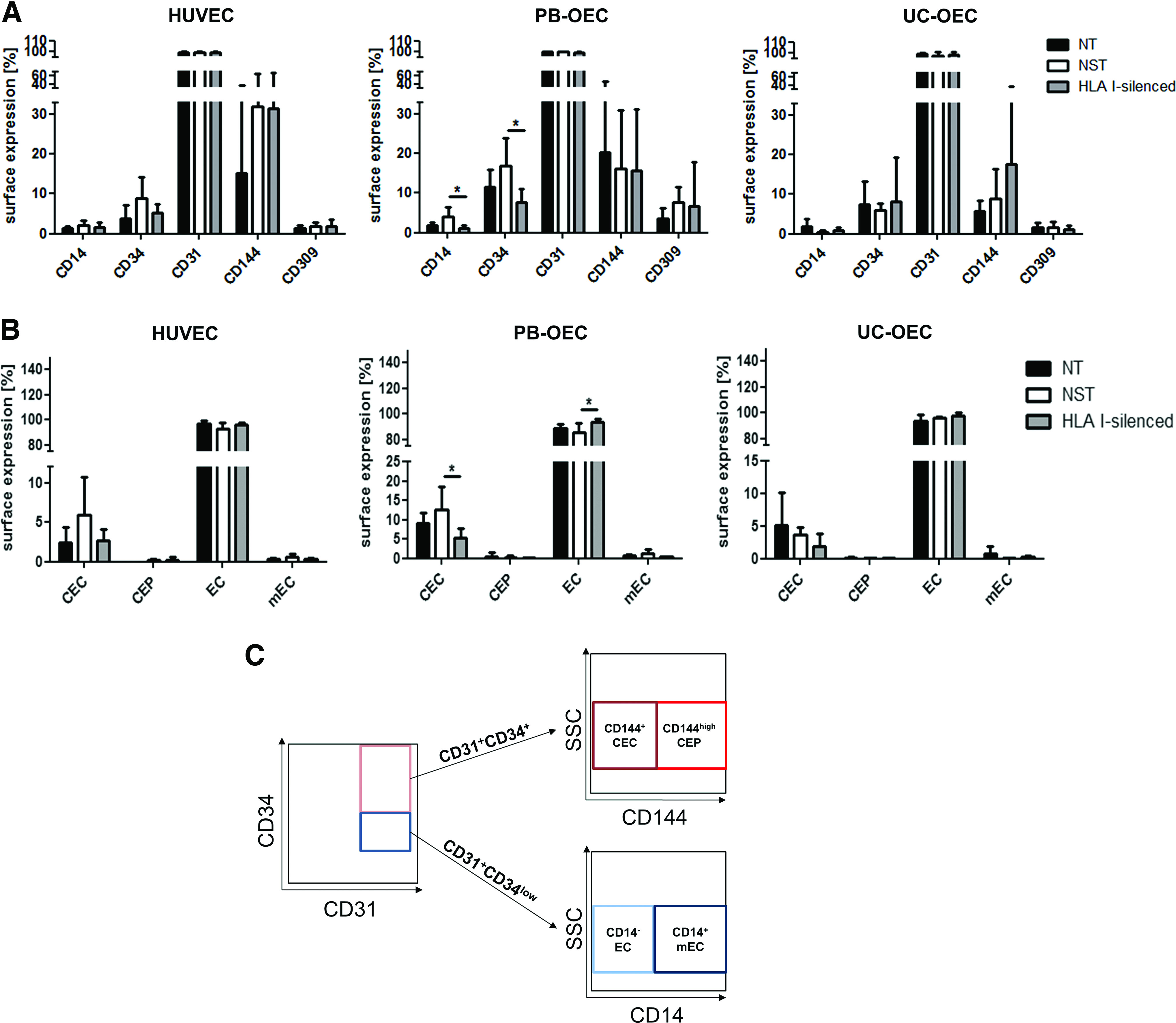
Impact of transduction and HLA I silencing on endothelial phenotype. HUVEC, PB-OEC, and UC-OEC were analysed for the expression of the surface markers CD14, CD34, CD31, CD144, and CD309. The expression of these markers was considered individually
In a second set of experiments, the effect of HLA I silencing on EC functionality like alignment under laminar flow or uptake of ac-LDL was evaluated. The effective uptake of Dil-ac-LDL by NT, NST, and HLA I-silenced EC was shown by bright fluorescence signals for all three cell sources (Fig. 7A). Flow cytometric analysis showed a highly significant increase of fluorescently labeled cells upon incubation with Dil-ac-LDL in HUVEC by up to 99.6% in NT (p = 0.0006), 99.7% in NST (p = 0.0006), and 99.8% in HLA I-silenced cells (p = 0.0012). Also NT, NST, and HLA I-silenced PB-OEC showed a comparably high increase in the incorporation of Dil-ac-LDL by up to 99.6% (p = 0.0022), 98.7%, and 98.7% (p = 0.0286), respectively. The capacity of NT, NST, and HLA I-silenced UC-OEC to take up Dil-ac-LDL was 99.4% (p < 0.0001), 99.8% (p = 0.0079), and 99.6% (p = 0.0079), respectively (Fig. 7B).

Uptake of ac-LDL by HLA I-silenced HUVEC, PB-OEC, and UC-OEC. After 4 h of incubation with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate-labeled ac-LDL (Dil-ac-LDL), NT, NST, and HLA I-silenced cells were analyzed for the frequency of cells that uptake fluorescent- labeled ac-LDL by fluorescence microscopy
The correct alignment of EC is important for a number of functions of intact blood vessels such as an increased expression of eNOS, which prevents thrombogenicity. 28 Laminar flow assays revealed the ability of NT, NST, and HLA I-silenced EC from all three sources to align in the direction of flow under both low (25 dyn/cm2) and moderate (44 dyn/cm2) physiological shear stress. This can be observed by the phalloidin staining of actin filaments indicating long, elongated EC arranged parallel to each other in comparison to EC cultivated under static conditions (Fig. 8).
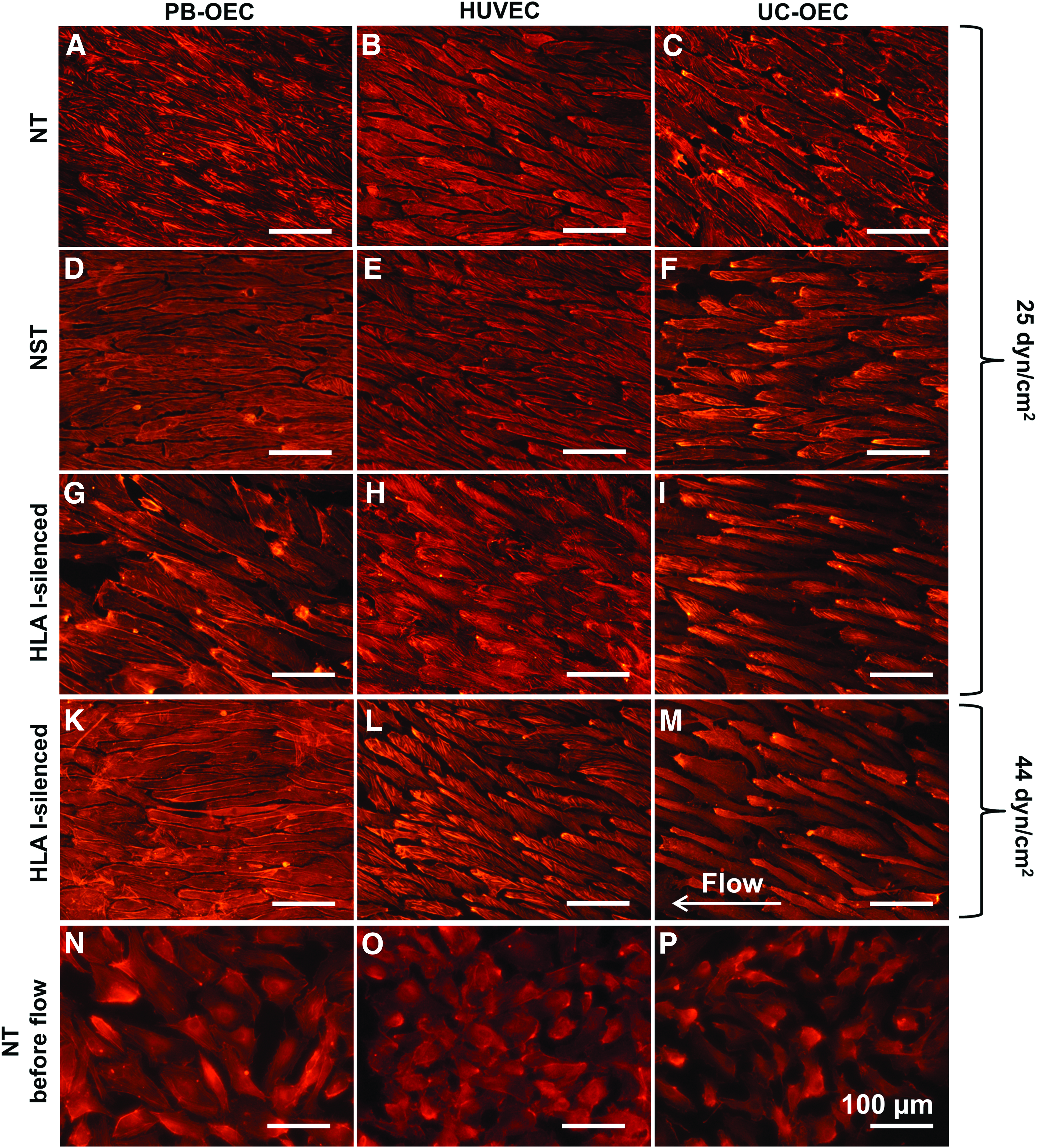
Alignment of NT, NST, and HLA I-silenced PB-OEC, HUVEC, and UC-OEC under laminar flow. EC from three different sources were transduced with lentiviral vectors encoding a β2-microglobulin-specific shRNA to downregulate HLA I expression. NT and NST cells transduced with a nonsense shRNA served as controls. Cells were either cultivated in flow chambers exerting low
Tube formation assay with HLA I-silenced EC and ASC in 3D fibrin gels
Nonsilenced EC are able to form capillary-like networks in fibrin gels. 29 In this study, the effect of HLA I silencing and/or lentiviral transduction on this hallmark of endothelial functionality was examined. In a first step, tubes of monocultures of NT, NST, and HLA I-silenced cells were assessed with respect to tube area, number of junctions, tube length and number of tubes (Fig. 9A–M). No differences between NT, NST, and HLA I-silenced cells were observed in PB-OEC monocultures, indicating that neither the transduction procedure itself nor the elimination of HLA I molecules interfered with tube formation (Fig. 9N).
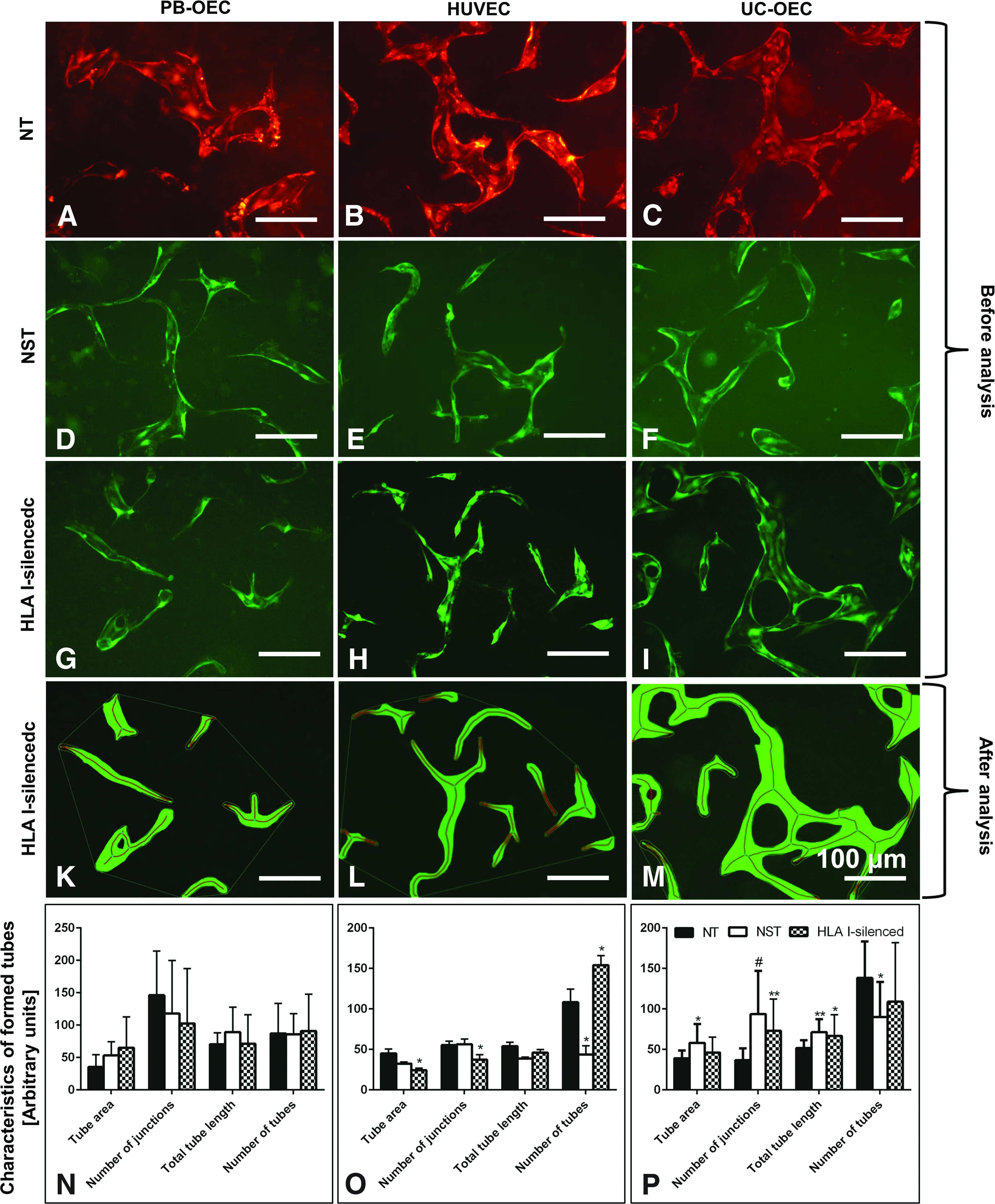
Tube formation of NT, NST, and HLA I-silenced GFP-expressing PB-OEC, HUVEC, and UC-OEC in 3D fibrin gels before
With HUVEC, only few significant differences were observed: HLA I-silencing resulted in a 1.85-fold decreased tube area (p = 0.031), a 1.49-fold decreased number of junctions (p = 0.032), and a 1.42-fold increased number of tubes (p = 0.031), indicating a slightly impaired tube formation capacity. The lentiviral transduction decreased the number of tubes 2.5-fold (p = 0.029; Fig. 9O). More changes were present with NST UC-OEC forming tubes with a 1.49-fold increased tube area (p = 0.024), 2.55-times more junctions (p ≤ 0.0001), and 1.38-fold longer tubes (p = 0.002), but 1.54-times less tubes (p = 0.018).
Similarly, HLA I-silenced UC-OEC formed tubes with 1.99-times more junctions (p = 0.003) and 1.29-fold longer tubes (p = 0.044), indicating more branched networks of NST and HLA I-silenced UC-OEC compared to nonsilenced cells (Fig. 9P).
It has been shown previously that the effect of co-culturing EC with a capillary-supporting cell type-like ASC improved tube formation significantly in terms of the number of junctions and tube length.11,30 In this study, we showed for the first time that these capillaries displayed not only cord-like structures but also contained definitely a hollow lumen (Fig. 10A panel d).

Tube formation of co-cultures with HLA I-silenced EC and ASC. 3D tube formation assays were performed using fibrin gels and HLA I-silenced PB-OEC, HUVEC, and UC-OEC in co-culture with ASC in a ratio of 1:0.5
To answer the question whether silencing of HLA I could interfere with these pivotal properties of EC, in the last step, we performed co-culture experiments with HLA I-silenced EC and ASC (Fig. 10A). Co-culture assays showed that the addition of ASC led to an improved tube formation of HLA I-silenced PB-OEC, HUVEC, and UC-OEC representing a highly branched and widely connected network. More precisely, HLA I-silenced EC from all three sources formed tubes with a remarkable increase in the number of junctions (4.14–16.38-fold; p ≤ 0.0001) and tube length (1.97–4.55-fold; p ≤ 0.0001), and a decreased number of tubes (1.18–4.11-fold; p ≤ 0.0001 for HUVEC, 10B). Thus, the addition of ASC considerably improved tube formation of HLA I-silenced cells as observed recently with nonsilenced HUVEC and PB-OEC. 11
This was confirmed by direct comparison of ASC co-cultures with HLA I-silenced EC and NT EC (Fig. 10C). Co-cultures of HLA I-silenced EC from all three sources showed essentially the same tube formation like NT EC in terms of all parameters. Only minor deviations were observed with PB-OEC, showing slightly increased tube length (1.28-fold; p ≤ 0.05, Fig. 10C panel d), and with HUVEC where the number of tubes was marginally lower (1.44-fold; p ≤ 0.05, Fig. 10C panel e). Thus, HLA I-silenced EC from all three sources showed a tube formation pattern similar to that of nonsilenced EC, which is a fundamental prerequisite for the application of HLA I-silenced EC for vascular tissue engineering.
Discussion
The ideal vascular graft is composed of a confluent EC monolayer on the luminal side and a network of capillaries on the adventitial side mimicking the vasa vasorum. HLA I silencing has evolved as a technique to reduce the immunogenic potential of various cell types, including EC, resulting in a decreased risk of rejection after transplantation in murine models.20,22,23 In addition, HLA I-silenced EC were shown to be protected against allogeneic cellular and humoral immune responses in vitro. 21 Thereby, the spectrum of available cells is potentially broadened from autologous to allogeneic EC, which could be used off the shelf for vascular tissue engineering applications—provided that the cells remain functional.
In this study, EC from three different sources were stably silenced for HLA I expression by lentiviral-mediated RNAi and characterized not only for the silencing efficacy but also for their functionality, including antithrombogenic, vessel tone regulating, and capillarizing properties. The major findings are as follows: (1) EC isolated from peripheral blood and umbilical cord blood and vein were effectively HLA I silenced to a suitable level that was previously shown to reduce their immunogenicity, (2) HLA I-silenced EC maintained expression of surface antigens like CD31 and VE-cadherin, as well as vWF and eNOS, and (3) HLA I-silenced EC were still capable to align under unidirectional flow and co-cultures of HLA I-silenced EC and ASC formed highly branched 3D capillary networks similar to nonsilenced EC.
Mechanisms of graft rejection
HLA I is expressed on most nucleated cells and platelets in a highly polymorphic manner. 31 In consequence, HLA histoincompatibility is the major cause for graft rejection and thus the most relevant hurdle in transplantation.32–34 Basically, there are three HLA I-associated mechanisms responsible for graft rejection35,36: (1) CD8+ cytotoxic T cell-mediated HLA I allorecognition and apoptosis, (2) HLA I-associated antibody-mediated complement-dependent and cellular cytotoxicity resulting in cell lysis, and (3) NK cell-mediated cell lysis in the absence of HLA I molecules. Since both the presence and the complete absence of HLA I are detrimental regarding alloimmune responses, previously we have demonstrated that a residual HLA I expression of at least 10% is required to prevent NK cell cytotoxicity. In addition, downregulation of HLA I expression above 60% showed to efficiently protect the target cells against alloimmune responses.20,21,23 Lentiviral vector-mediated RNAi was previously shown to be an effective method to downregulate, but not to completely knock out the expression of HLA I in several cell types, including UC-OEC, in vitro and in vivo.18–23 In this study, this technology was extended to EC from two other sources where HLA I expression was downregulated between 47% and 67% (Fig. 1B). Although this displays a lower efficacy than observed previously with UC-OEC, 21 based on our aforementioned studies, it is still expected that these two EC types remain protected from cellular and humoral immune responses.
Risks associated with lentiviral vectors
Despite its high effectiveness, the lentiviral vector-mediated transduction is associated with a number of safety concerns such as pseudorandom insertional mutagenesis37,38 leading to unpredictable gene disruptions. This potentially leads to a loss of function of a respective gene or to disturbed regulation such as tumorigenesis as shown here. 39 In this study, treatment of two patients suffering from X-linked severe combined immunodeficiency with retroviral vector-mediated gene-corrected CD34+ cells developed uncontrolled T cell proliferation with symptoms of leukemia. Gene analysis revealed the insertion of the vector in proximity to the LMO2 proto-oncogene promotor leading to aberrant transcription of LMO2. 39 Thus, every viral vector-mediated gene transfer requires a thorough characterization of genetically modified cells regarding their morphological and functional properties. This study investigated, to our knowledge, for the first time, the functionality of lentiviral vector-mediated HLA I-silenced EC from three sources regarding specific surface markers, response to flow dynamics, and capability to form capillaries.
Characterization of lentiviral vector-mediated transduced EC: surface markers
A hallmark for a fully functional endothelium is the expression of the glycoproteins CD31 and VE-cadherin regulating, among others, its barrier function that allows a selective exchange of molecules and leukocytes between the blood stream and the underlying tissue. 40 In particular, CD31 was identified to be responsible for cell elongation, migration, and gel invasion in the tube formation process, whereas VE-cadherin was found to stabilize tubes by particularly strong cell–cell adherence junctions and support intercellular lumen formation. 41 The expression of vWF and eNOS by EC is necessary for an undisturbed blood flow due to their homeostatic and vasoprotective functions. The absence of vWF promotes the proteolytic degradation of factor VIII leading to the development of a blood clotting disorder. 42 eNOS synthetizes nitric oxide (NO) regulating vessel dilation, whereby reduced NO activity increases the risk for developing thrombosis and atherosclerosis. 43 As shown by immunohistochemistry (Figs. 2–5) and quantitative polymerase chain reaction (Supplementary Fig. S1), all these molecules were fully expressed before and after lentiviral transduction, indicating that not only NT EC but also NST and HLA I-silenced EC were capable to fulfill the required barrier function and promote antithrombogenicity and vessel tone regulation.
Flow cytometry confirmed in a quantitative way that, lentiviral transduction and/or HLA I silencing do not affect the expression of EC-specific molecules like CD31, CD144/VE-cadherin, and CD309/VEGF-R2 (Fig. 6A), the expression of a subset of markers identifying specific EC subpopulations (Fig. 6B, C) and the uptake of Dil-ac-LDL (Fig. 7). Thus, NST and HLA I-silenced cells maintained their morphological and functional properties.
Response to flow dynamic
The adherence of EC under blood flow-mediated shear stress is a characteristic property of functional EC. Sensing of shear stress is conferred by a mechanotransductory complex at sites of cell–cell junctions consisting of CD31, CD144/VE-cadherin, and VEGF-R2 (CD309/VEGF-R2), triggering various intracellular signaling cascades leading to horizontal EC alignment. 44 This elongated phenotype along with pronounced actin bundles in the cytoplasm is a prerequisite for a number of functions like the stimulation of eNOS expression, antithrombotic activity, and prevention of leukocyte adhesion. 45
Conversely, impairment of the horizontal organization, as present in diabetic patients, alters eNOS activation and thus promotes atherosclerosis and peripheral vascular disease. 46 In this study, the ability of HLA I-silenced EC to align under unidirectional flow was unaffected compared to NT and NST EC as shown by the presence of long, outstretched EC with distinct actin bundles (Fig. 8). This is likely to be connected to the full expression of CD31 and VE-cadherin as shown before, which are important parts of the shear stress sensing apparatus.
Capillary formation in endothelial monocultures
Tube formation is a typical endothelial property which has been shown previously also for EC from umbilical cord veins- and blood or peripheral blood and different hydrogels.24,47,48 Here, we showed that tube formation by PB-OEC was not compromised by both transduction and the HLA I silencing procedure. Only HLA I-silenced HUVEC showed a marginally impaired tube formation capacity compared to NT cells whereas NST and HLA I-silenced UC-OEC showed improved tube formation with longer tubes and more junctions compared to controls (Fig. 9). Tube length is regulated by CD31 expression which induces EC elongation, migration and gel invasion 41 whereas branching is naturally conferred by specialized endothelial tip cells expressing increased amounts of VEGFR-2, VEGFR-3, platelet-derived growth factor-BB and others. 49
Thus, the observed changes could be related to the expression levels of these molecules which may be influenced by the strong expression of GFP used as marker for transduction efficiency rather than by the HLA I silencing. Since NST and HLA I-silenced cells had lower proliferation rates (Table 1), the influence of GFP expression on cellular metabolism including expression of certain molecules should not be underestimated. However, the effects of transduction and silencing on monoculture tube formation are of no consequence due to their relatively low biomechanical stability and high turnover rate 50 which prevents their use for vascular tissue engineering applications. Instead, co-cultures of EC and mural cells such as pericytes or vascular smooth muscle cells as present in natural vessels are of interest for the generation of stable vascular networks. 51
Capillary formation in co-cultures of EC and mural cells
It has been shown previously that ASC are also able to improve capillary networks built by HUVEC and PB-OEC with respect to branching points and tube length. 11 This is consistent with the findings of this study where tube formation of co-cultures with HLA I-silenced EC and ASC was not impaired regarding tube length or branching points compared to NT cells (Fig. 10). Thus, HLA I-silenced EC from each source can be used in combination with ASC for the generation of low immunogenic vascular networks.
ASC are naturally immune privileged due to their extremely low expression of HLA I and the complete absence of HLA II, 52 which makes them promising for allogeneic transplantation. The stabilizing effect of ASC was traced back to the increased expression level of VEGF-A in co-culture supernatants compared to EC-monoculture supernatants 30 and to the proangiogenic factors FGF and VEGF secreted by ASC. Moreover, direct cell–cell contacts between EC and ASC rather than proangiogenic factors were shown to be essential for the generation of widely branched and stable vascular networks. 47 Thus, this study allows the conclusion that HLA I silencing of EC did not interfere with expression of receptors for the aforementioned growth factors and adhesion molecules essential for tube formation.
In conclusion, HLA I silencing of EC and the genetic modification per se did not alter any of the tested morphological or functional properties, which are necessary for fully functional endothelialization and capillarization of vascular grafts. This suggests that, for vascular tissue engineering purposes, the sources for appropriate cells can be extended from autologous to allogeneic sources, which accelerate the generation of vascular grafts in acute cases. In particular, the use of UC-OEC would be favored over HUVEC and PB-OEC due to the relatively simple and quick isolation process yielding huge cell numbers.
With regard to an appropriate scaffold material for vascular grafts, a rather new approach using blood-derived fibrin is promising as it can be isolated from the patient's blood and thus used in an autologous manner. In the past, great success has been achieved by creating a fibrin-based vascular graft with the help of a specific molding technique, thereby overcoming fibrins limitation of poor stability.53–55
Moreover, xenogeneic vascular scaffolds generated by an intensified decellularization procedure have been proven to be less immunogenic than conventionally manufactured ones. 56 These vascular scaffolds all require full endothelialization to prevent thrombi formation and ideally also a prevascularization to be connected immediately to the external blood supply. The promising results of this study, however, have to be confirmed in animal studies to investigate the effect of vascular grafts seeded with HLA I-silenced EC on the host's immune system and their contribution to neovascularization. This study represents the prerequisite for these ambitious goals by paving the way for the usage of EC from easily accessible tissue sources with a low immunogenic potential.
Footnotes
Acknowledgments
This work has been carried out as an integral part of the BIOFABRICATION FOR NIFE Initiative, which is financially supported by the ministry of Lower Saxony and the Volkswagen Stiftung. (NIFE is the Lower Saxony Center for Biomedical Engineering, Implant Research and Development, a joint translational research center of the Hannover Medical School, the Leibniz University Hannover, the University of Veterinary Medicine Hannover, and the Laser Center Hannover.) Furthermore, part of the study was funded by the excellence cluster REBIRTH (from regenerative biology to reconstructive therapy, Unit 6.3).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.