
Abstract
Hypertrophic scar (HS) is a dermal fibroproliferative disease characterized by fibroblast over-proliferation, overproduction, and deposition of the extracellular matrix. Growing evidence demonstrated that adipose-derived stem cells (ASCs) secrete a plethora of trophic and antifibrotic factors, which suppress inflammation and ameliorate fibrosis of different tissues. However, few studies investigate their effect on repressing HS activity. This study evaluated the suppressing effect of ASCs on HS fibroblast bioactivity and the possible mechanism via a coculture model. HS-derived fibroblasts (HSFs) and ASCs were isolated from individual patients. HSFs or HSFs treated with transforming growth factor-β1 (TGF-β1) were cocultured with ASCs and the change of HSF cellular behaviors, such as cell proliferation, migration, contractility, and gene/protein expression of scar-related molecules, were evaluated by cell counting assay, cell cycle analysis, scratch wound assay, fibroblast-populated collagen lattice (FPCL) contractility assay, real-time quantitative polymerase chain reaction, ELISA, and western blotting assay. After 5 days of ASC coculture treatment, the expression levels of collagen I (Col 1), collagen III (Col 3), fibronectin (FN), TGF-β1, interleukin-6 (IL-6), interleukin-8 (IL-8), connective tissue growth factor (CTGF), and alpha-smooth muscle actin (α-SMA) in HSFs decreased significantly while the expression levels of decorin (DCN) and MMP-1/TIMP-1 (matrix metalloproteinase/tissue inhibitor of MMP) ratio increased significantly. Besides, after 5 days of exogenous TGF-β1 stimulation, the expression levels of Col 1, FN, TGF-β1, IL-6, CTGF, and α-SMA in HSFs increased significantly. Impressively, all these increased gene expression levels were reversed by 5 days of ASCs coculture treatment. Additionally, the proliferation, migration, and contractility of HSFs were all significantly reduced by ASC coculture treatment. Furthermore, the protein levels of TGF-β1 and intracellular signal pathway-related molecules, such as p-smad2, p-smad3, p-Stat3, and p-ERK, were downregulated significantly in HSFs after 5 days of ASCs coculture treatment. This study demonstrated that coculture of HSFs with ASCs not only inhibited proliferation, migration, and contractility of HSFs but also decreased the expression levels of HSF-related or TGF-β1-induced molecules. Additionally, the antifibrotic effect on HSFs was likely mediated by the inhibition of multiple intracellular signaling. The results of this study suggest the therapeutic potential of ASCs for HS treatment, which is worth of further investigation.
Introduction
H
It has been reported that mesenchymal stem cells (MSCs) could inhibit fibrotic tissue formation by secreting a number of cytokines and antifibrotic factors.10,11 Adipose-derived stem cells (ASCs) are one type of MSCs, which have the advantages of autologous, nonimmunogenic, self-renewal, multidifferentiation potential, easy access, available for large quantity, and less donor site morbidity and thus are an ideal therapeutic cell source.12,13 Recently, increasing evidence demonstrates that ASCs have potential of serving as an antiscarring or antifibrotic agent.
For example, implantation of autologous fat graft for the treatment of gluteal muscle contracture could achieve satisfactory aesthetic results. 14 It was also shown that intralesional injection of ASCs could suppress HS in a rabbit ear model. 15 Another study demonstrated that ASC transplantation acquired smaller infarct size and less scar formation in a mouse model of acute myocardial infarction. 16 ASCs could also attenuate pulmonary fibrosis induced by repetitive bleomycin administration. 17 Furthermore, in vitro studies confirmed that ASCs could attenuate collagen production and enhance dermal fibroblast functions.18,19
Transforming growth factor (TGF)-β1 is a well-known profibrotic cytokine widely involved in fibroblast activation, proliferation, and ECM production in HS formation. 20 HS and their derived fibroblasts express excessive TGF-β.21,22 and its receptors 23 when compared with normal skin and normal fibroblasts. Additionally, TGF-β1-related signaling pathway plays an important role in HS pathogenesis involving a multitude of cytokines, which regulate a broad array of intracellular responses in HS including ECM production and deposition, fibroblast proliferation, angiogenesis, and inflammatory cell infiltration.24,25 Because of this, manipulation of TGF-β function or blocking its associated signal transduction may hold putative therapeutic strategy for HS. 26
To date, the inhibitory effects of ASCs on HS-derived fibroblasts (HSF) activity and its potential molecular mechanisms are not well defined. Therefore, in this study, we investigated whether coculture of ASCs with HSFs could potentially inhibit HSF functions and the possible biochemical mechanism that may involve in the inhibitory effect of ASCs on various cellular behaviors such as cell proliferation, migration, contractility, and gene/protein expression of scar-related molecules.
Materials and Methods
Harvest and culture of human ASCs
Protocols for the handling of human tissue and cells were approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine. Human adipose tissues were donated by the patients for research purposes only with written informed consent. Human adipose tissue was harvested from abdomen or inner thigh portion of patients by standard liposuction procedures. The samples were washed extensively with chloramphenicol once and phosphate-buffered saline (PBS) twice and then digested by 0.075% collagenase type I (Sigma-Aldrich) dissolved in Dulbecco's modified Eagle's medium (DMEM; Hyclone) containing 10% fetal bovine serum (FBS; Biosum) at 37°C for 1 h with vigorous shaking. Equal volume of DMEM containing 10% FBS was added to neutralize the collagenase activity. The cell suspension was centrifuged at 1200 g for 10 min. The pellet was resuspended in DMEM containing 10% FBS, penicillin (100 U/mL), and streptomycin (0.1 mg/mL), and then incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2. These primary cells were defined as passage 0 and were passaged with 0.05% trypsin-EDTA (Gibco) once 80–90% of confluency reached. Cells were cultured to passage three for the use of all experiments. To reduce individual variation, ASCs were respectively extracted from each of nine patients' fat tissues, three cell samples were randomly selected and mixed as one pooled cell sample and total three pooled cell samples were generated for various experiments.
Harvest and culture of HSFs
Protocols for the handling of human tissue and cells were approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine. Excised human HS samples were donated by patients who received scar resection with informed consent. For HSF isolation, the tissue samples were washed extensively with chloramphenicol once and PBS twice, cut into 2 × 2 × 2 mm3 fragments followed by enzyme digestion with 0.2% collagenase type II (SERVA, GER) dissolved in DMEM containing 10% FBS at 37°C for 4 h with vigorous shaking. After centrifugation, the pellet was resuspended in DMEM containing 10% FBS, penicillin (100 U/mL), and streptomycin (0.1 mg/mL). Then, cells were seeded onto 10 cm culture dishes at a density of 1 × 104 cells/cm2 and incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2. The cells were passaged once reached 80–90% of confluency and then subcultured at the same density for two more passages. Passage three cells were harvested for the use of all experiments. To reduce individual variation, human HSFs were respectively extracted from each of nine patients' fat tissues, three cell samples were randomly selected and mixed as one pooled cell samples and total three pooled cell samples were generated for various experiments.
Coculture of human ASCs and HSFs
A six-well transwell culture dish (0.4 μm inserts; Corning) was used to develop the coculture system. Passage three ASCs were harvested with 0.25% trypsin-EDTA treatment, resuspended in DMEM containing 10% FBS and antibiotics (penicillin/100 U/mL, streptomycin/0.1 mg/mL), and then inoculated on the upper chamber of the transwell culture dishes at a density of 1 × 104 cells/cm2. Passage three HSFs were similarly harvested and inoculated on the lower chamber with the same density and culture medium as those of ASCs. Culture media of both chambers were changed every 2 days. The cocultured cells were set as an experimental group. As a control group, passage three HSFs were inoculated on the lower chamber of six-well culture dishes at a density of 1 × 104 cells/cm2 in DMEM plus 10% FBS and antibiotics.
To further explore the effect of ASCs on attenuating TGF-β, at day 1 of the experiment, both experimental and control cells were cultured without or with TGF-β1 at a concentration of 2 ng/mL of culture medium.
Cell proliferation assay
During the culture process as above described, HSFs were harvested with 0.25% trypsin-EDTA treatment at days 1, 3, 5, and 7, centrifuged at 1000 g for 5 min, and then resuspended in DMEM. Ten microliters of cell suspension solution were added to a hematocytometer for cell counting under an inverted-phase contrast microscope (Olympus). The assay was performed in triplicate and repeated in four pooled cell samples at each desired time point.
Cell cycle analysis
At the 5th day of cell culture, HSFs of both coculture group and control group were respectively harvested with the treatment of 0.25% trypsin-EDTA. The cells were then centrifuged at 1200 g for 5 min, washed with PBS twice, and centrifuged again. The pellet was resuspended in a mixed solution containing 0.3 mL of PBS and 0.7 mL of ice-cold absolute ethyl alcohol with gentle agitation for fixation and then the samples were incubated at 4°C overnight. Thereafter, the cells were centrifuged and stained in 1 mL ice-cold PBS solution containing 20 μL of propidium iodide (1 μg/mL; Sigma), 1 μL of Triton X-100 (0.1%; Sigma), and 0.2 mg of RNase (1 mg/mL; Sigma) for 30 min. The cells were then analyzed using a flow cytometer (Beckman Coulter) equipped with ModiFit LT v2.0 software. The analysis was repeated in three pooled cell samples.
RNA isolation and real-time quantitative polymerase chain reaction
Total RNA extraction and reverse transcription were performed as previously described. 27 Briefly, total RNA was extracted from the cells of coculture group and control group at day 5 and 10 or from the cells of both groups without or with TGF-β1 treatment at day 2 and 5 using a TRIzol Reagent (Invitrogen). cDNA was reversely transcribed from 2 μg of total RNA per sample with the use of AMV reverse transcriptase (Promega). A 20 μL reaction solution composed of 2 μg total RNA, 4 μL 5 × buffer, 2 μL dNTP, 1 μL oligo-(dT), 0.5 μL AMV reverse transcriptase, 0.5 μL RNase inhibitor, and ddH2O was filled up to total volume of 20 μL for the reaction. The mixture was incubated at 30°C for 10 min, 45°C for 60 min, 98°C for 5 min, and 5°C for 5 min.
The designed primers for quantitative polymerase chain reaction (qPCR) analysis were listed in Table 1. cDNA was amplified using a Power SYBR Green PCR master mix (Applied Biosystems) in a real-time thermal cycler (Stratagene) and measurement was conducted in triplicates for each sample. qPCR conditions were set as follows: denaturation at 95°C for 30 s, primer annealing at temperatures listed in Table 1 for 30 s, and elongation at 72°C for 45 s with total 40 cycles. GAPDH gene was used as an internal control. qPCR assay was performed in triplicate and repeated in three pooled cell samples.
PCR, polymerase chain reaction.
In vitro scratch wound assay
As previously described, 28 an in vitro scratch wound assay was used to evaluate cell migration. Briefly, HSFs derived from coculture group and control group were cultured in six-well culture plate along with DMEM containing 10% FBS until 100% confluency, then a scratch was created on each well using a 200 μL pipette tip (PipetTipFinder, LLC) to make a scratch in the middle of the culture dish. The scratched wound was about 0.45–0.50 mm in width per well. Afterward, the cultures were switched to serum-free medium for 24 and 48 h. Digital photograph of each wound was acquired under an inverted-phase contrast microscope (Olympus) immediately, 24 and 48 h after the scraping. Wound closure (cell migration) was investigated by measuring the wound area using the commercial software Image pro-plus version 6.0 (Media Cybernetics) and public domain image processing program. Results were presented as the percentage of the initial wound area using the following formula: Cell migration rate (%) = [Gap24 h (or Gap48 h) − Gap0 h]/Gap0 h × 100%. Photographs of each wound were acquired in three random views and the mean cell migration rate of each sample plus standard deviation was presented as the final result. The assay was repeated in three pooled cell samples.
Fibroblast-populated collagen lattice contractility assay
Fibroblast-populated collagen lattice (FPCL) contractility assay was performed as previously described. 26 Briefly, according to the manufacturer's protocol, collagen lattices were polymerized in a 24-well transwell culture dish. To form the collagen solution, 200 μL of rat tail tendon collagen solution (5 mg/mL; Shenyou Biotechnology Co., Ltd.) were added to 12 μL of NaOH (0.1 M) and mixed immediately by pipetting. Then, 23 μL 10× PBS was added into the mixture solution. Thereafter, 760 μL of cell suspension (containing 3 × 104 cells) were added to the mixture solution, gently mixed, and added into the lower chamber of the 24-well transwell culture dish with 500 μL volume per well. Collagen lattices were placed at room temperature for 30 min to form the gel, and then 1 mL of DMEM containing 10% FBS and antibiotics was added to each well. Afterward, the cell-contained gels were detached from the culture dishes as the floating state for self-contraction in each well. At the same time, for coculture group, ASCs of passage 3 were inoculated on the upper chamber of the 24-well transwell culture dishes at a density of 1 × 104 cells/cm2 with 1.5 mL of DMEM containing 10% FBS and antibiotics. For the blank control group, 1.5 mL of DMEM containing 10% FBS and antibiotics (without cell) was added to the upper chamber of the 24-well transwell culture dishes. Digital images of the floating lattices were acquired immediately, 24, 48, and 72 h postdetaching. To quantify the contracture ability, the surface areas of the gels were measured using image software (Macropath 5; PRO) at each time point. Contraction rate of FPCL was normalized to the bottom area of the well. The assay was repeated in three pooled cell samples.
ELISA analysis for TGF-β1 protein expression
HSFs from coculture group and control group were cultured in DMEM containing 10% FBS until the 5th day. Cells were then serum starved for 12 h, and then cultured in serum-free medium for 24 h. Thereafter, the medium was collected for ELISA analysis of TGF-β1 (Excell Bio) according to manufacturer's protocol. Absorbance was measured at 450 nm. ELISA analysis was performed in triplicate and repeated in three pooled cell samples.
Western blotting analysis
After 5 days of coculture with ASCs as indicated, total protein was extracted from HSFs of both groups with RIPA lysis buffer as described previously. 29 Protein extracts were denatured by heat at 100°C for 5 min and electrophoretically separated on a 12% SDS-PAGE (Bio-Rad). Proteins were transferred to PVDF membrane, blocked with 5% milk/Tris-buffered saline, incubated with primary antibodies as below described, followed by incubation with appropriate HRP-conjugated secondary antibodies (Jackson ImmunoResearch). The protein bands were eventually visualized using an enhanced chemiluminescence (ECL) detection kit (Amersham). The primary antibodies of rabbit against human p-Erk1/2 (Thr202/Tyr204), rabbit against human Erk1/2, mouse against human Phospho-Stat3-Y705, rabbit against human Stat, rabbit against human Phospho-Smad2 (Ser465/467), rabbit against human Phospho-Smad3 (Ser423/425), rabbit against human Smad2/3 (BD Biosciences), and rabbit against human β-actin (Sigma-Aldrich) were purchased from Cell Signaling Technology. The experiment was repeated in three pooled cell samples.
Statistical analysis
All data are presented as mean ± standard derivation. The differences between coculture and control groups were analyzed with Student's t-test. p-Value less than 0.05 was considered statistically significant. SPSS software, version 19.0 (SPSS, Inc.) was applied in this statistical analysis.
Results
ASCs inhibited proliferation and blocked cell cycle of cocultured HSFs
With coculture of ASCs, the cell proliferation rate of HSFs became apparently slower when compare with that of blank control HSFs. As shown in Figure 1A, the cell density was relatively lower in experimental group than in control group at days 3, 5 and 7.

Coculture of HSFs with ASCs inhibited HSF proliferation.
To further quantitatively analyze, cells were planted into 24-well plates at a density of 5000 cells/well. As shown in Figure 1B, a logarithmic phase appeared from day 1 to 7. At day 3, the mount of HSFs in the control group (3.5 ± 0.21 × 104) was significantly more than that in the coculture group (2.7 ± 0.12 × 104) (p < 0.05). At day 5, the mount of HSFs in the blank control and coculture groups was 6.5 ± 0.24 × 104 and 4.9 ± 0.19 × 104 respectively with significant difference between two groups (p < 0.01). At day 7, total cell number reached 8.2 ± 0.22 × 104 and 5.9 ± 0.21 × 104 respectively in control and coculture groups with significant difference between them (p < 0.01).
Cell cycle analysis demonstrated that coculture group presented less percentage of HSFs in G2/M phases at day 2 (3.16% ± 0.56%) and day 5 (4.13% ± 0.49%) compared to day 2 (5.93% ± 0.69%) and day 5 (7.38% ± 0.54%) of control group with significant difference (p < 0.05). In addition, coculture group also presented less percentage of HSFs in S phase at day 5 (2.91% ± 0.21%) than that of control group (5.29% ± 0.68%) with significant difference (p < 0.05). Additionally, cell percentage (92.8% ± 0.69%) of coculture group in the G0/G1 phases was also significantly higher than that of control group (87.5% ± 1.2%) at day 5 with significant difference (p < 0.05).
Human ASCs inhibited cell migration of cocultured HSFs
As shown in Figure 2, at 24 h, 61.7% ± 1.7% of the scratched area was filled by migrated HSFs of control group. By contrast, 36.9% ± 3.1% of the scratched area was filled by the migrated HSFs of coculture group, which was significantly different from that of control group (p < 0.01, Fig. 2B). After 48 h of culture, 84.5% ± 1.1% of the area was filled by the migrated HSFs of control group, whereas 43.2% ± 3.2% of the area was filled by the migrated HSFs of coculture group with significant difference between two groups (p < 0.01, Fig. 2B), suggesting that cocultured with ASCs could significantly inhibit the migration of HSFs.
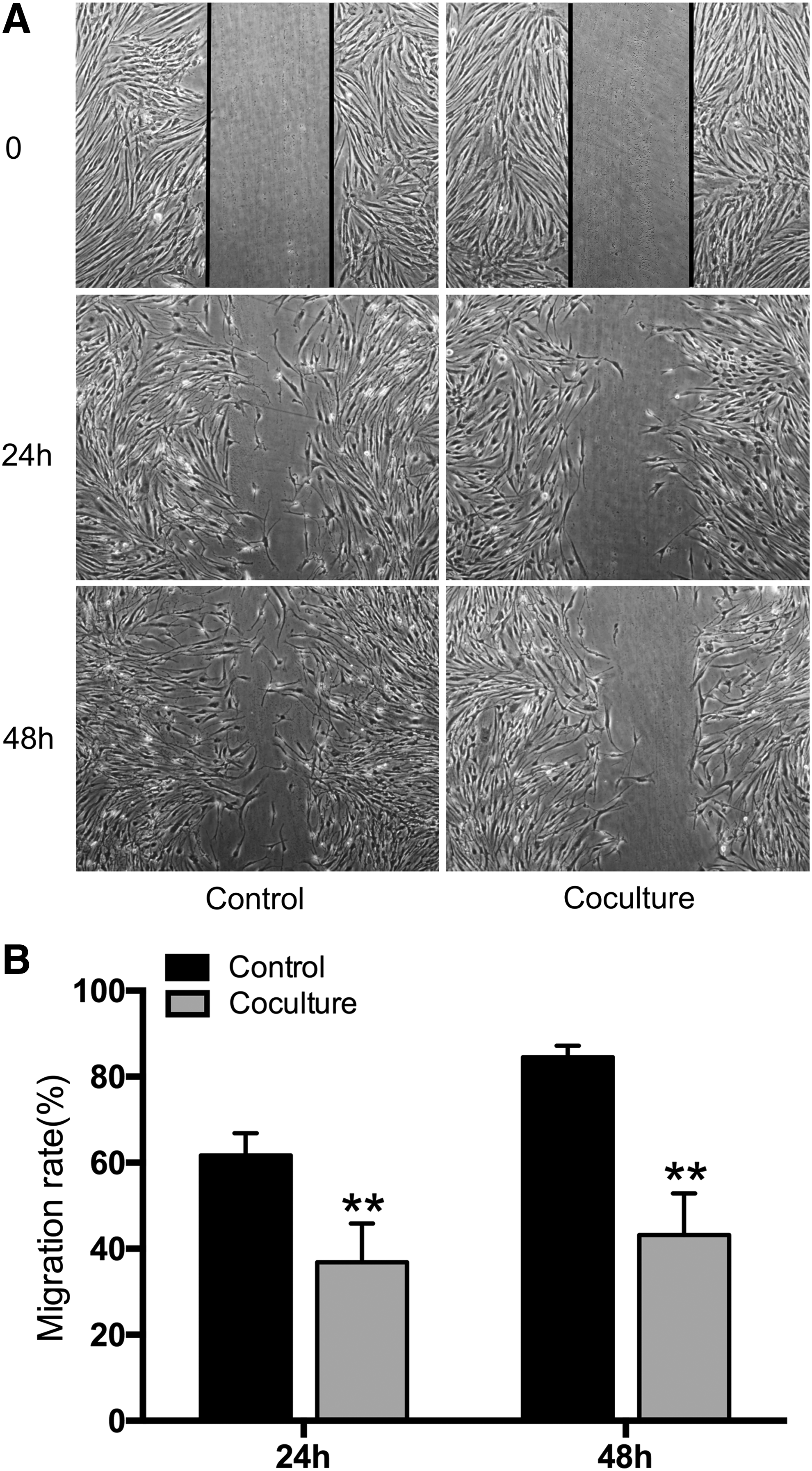
Coculture of HSFs with ASCs inhibited HSF migration.
ASCs attenuated the contraction of FPCL
A FPCL model was used to evaluate the inhibitory effect of ASCs on HSF contractility. As shown in Figure 3, at 24 h postrelease of the gel, cell containing floating collagen lattices self-contracted, which led to area reduction to 72.3% ± 2.9% of original area in control group. By contrast, coculture with ASCs led to less contraction of floating lattice with 95.7% ± 0.9% of original area, which is significantly different from that of control group (p < 0.01, Fig. 3B). At 48 h, the collagen gels contracted more and the average areas were 61.4% ± 3.5% and 93.2% ± 0.7% of original area respectively for control and coculture groups with significant difference between them (p < 0.01, Fig. 3B). At 72 h, the average areas of control and coculture groups respectively reached 45.6% ± 2.5% and 79.3% ± 1.3% of original area with significant difference (p < 0.01, Fig. 3B).
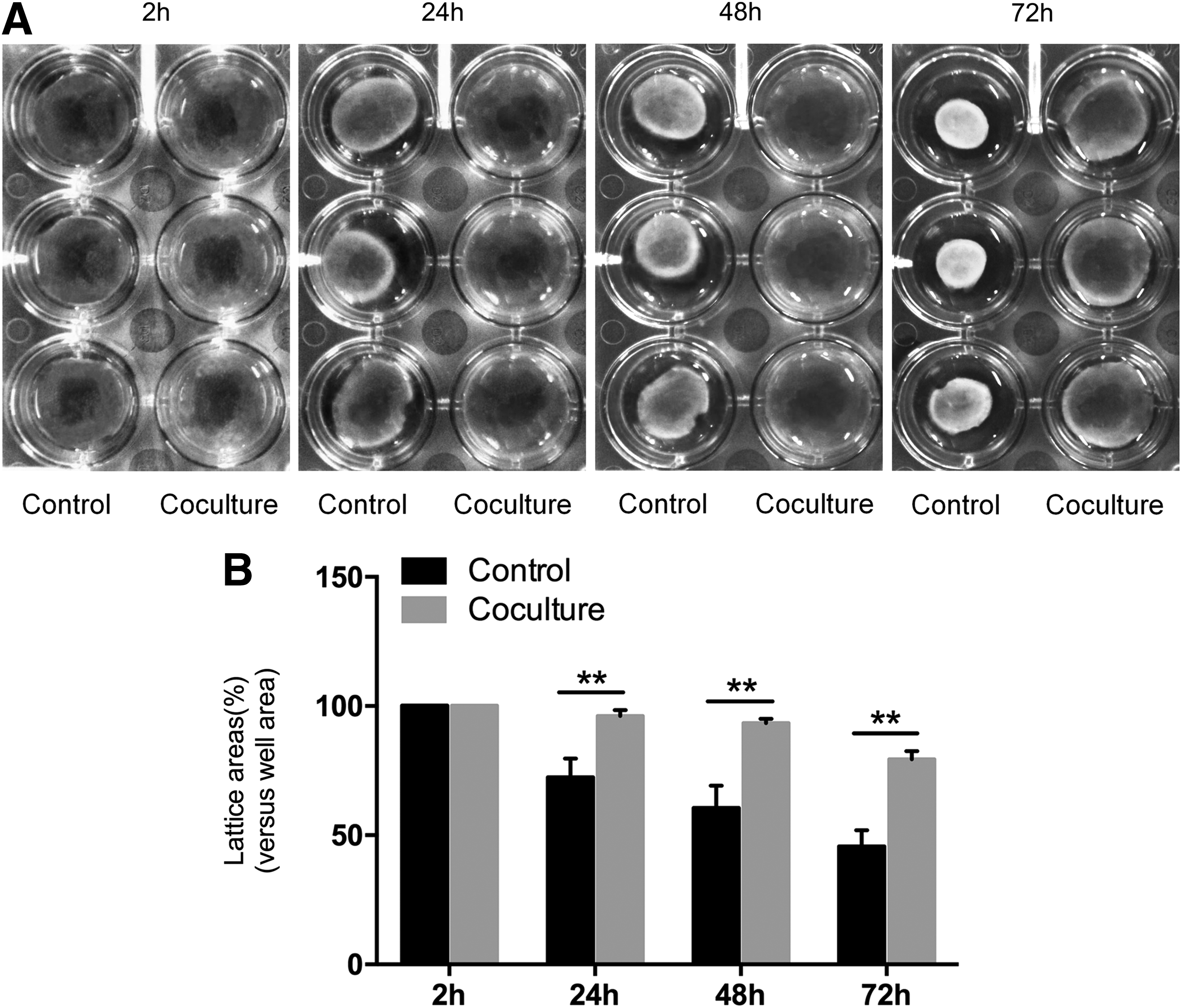
Coculture of HSFs with ASCs attenuated the contraction of HSF-populated collagen lattice (FPCL).
ASCs inhibited ECM gene expression in cocultured HSFs
After 5 days of coculture with ASCs, the gene expression levels of ECM molecules were significantly inhibited for collagen I (Col 1) (0.8 ± 0.07-fold of control, p < 0.05, Fig. 4A), Col 3 (0.62 ± 0.13-fold of control, p < 0.05, Fig. 4B), and fibronectin (FN) (0.45 ± 0.09-fold of control, p < 0.01, Fig. 4C). No significant difference in DCN gene expression was found (p > 0.05, Fig. 4F). Besides, the significant inhibitory effect was also found in FN at day 10 (0.73 ± 0.09-fold of control, p < 0.05, Fig. 4C). Additionally, increased MMP-1/TIMP-1 (matrix metalloproteinase/tissue inhibitor of MMP) ratio of their gene expression levels (1.18 ± 0.06-folds of control, p < 0.05, Fig. 4D) was also observed at day 5. Alpha-smooth muscle actin (α-SMA), a marker for myofibroblasts, was also significantly inhibited for its gene expression in the coculture group (0.49 ± 0.13-fold of control, p < 0.05, Fig. 4E) at day 5.
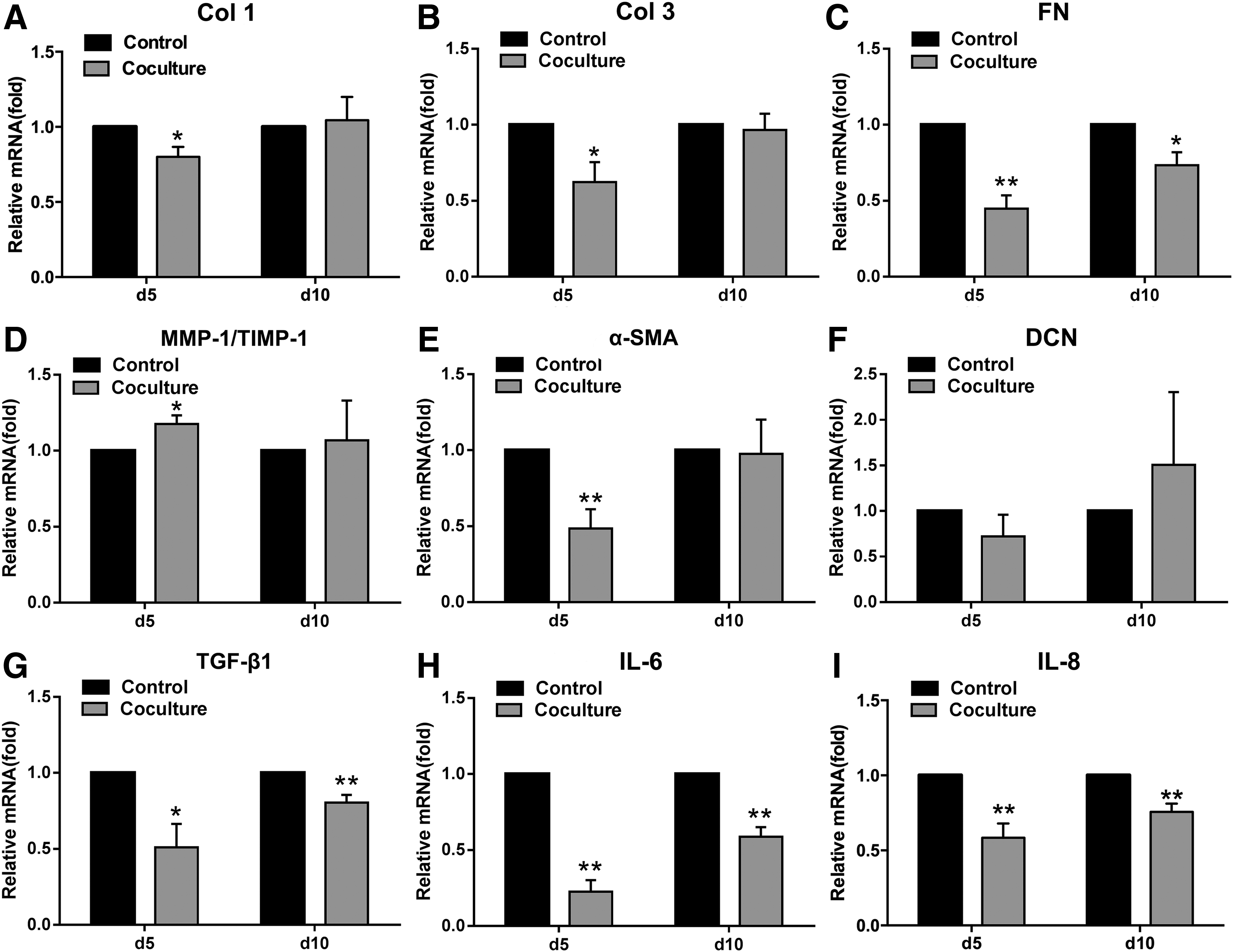
Coculture of HSFs with ASCs inhibited the gene expression of extracellular matrix and profibrotic factors. qPCR analysis of various gene expressions in HSFs without or with coculture of ASCs at day 5 and 10 for Col 1
ASCs inhibited gene expression of cytokines/growth factors in cocultured HSFs
After 5 days of coculture with ASCs, the expression levels of profibrotic genes of HSFs were significantly reduced compared to those of control HSFs, including TGF-β1 (0.51 ± 0.15-fold of control, p < 0.05, Fig. 4G), IL-6 (0.23 ± 0.08-fold of control, p < 0.01, Fig. 4H), IL-8 (0.58 ± 0.1-fold of control, p < 0.01, Fig. 4I), and connective tissue growth factor (CTGF) (0.87 ± 0.04-fold of control, p < 0.05, Fig. 5D). Similar inhibitory effect was also observed after 10 days of coculture with ASCs, including TGF-β1 (0.81 ± 0.05-fold of control, p < 0.01, Fig. 4G), IL-6 (0.59 ± 0.06-fold of control, p < 0.01, Fig. 4H), and IL-8 (0.76 ± 0.05-fold of control, p < 0.01, Fig. 4I).
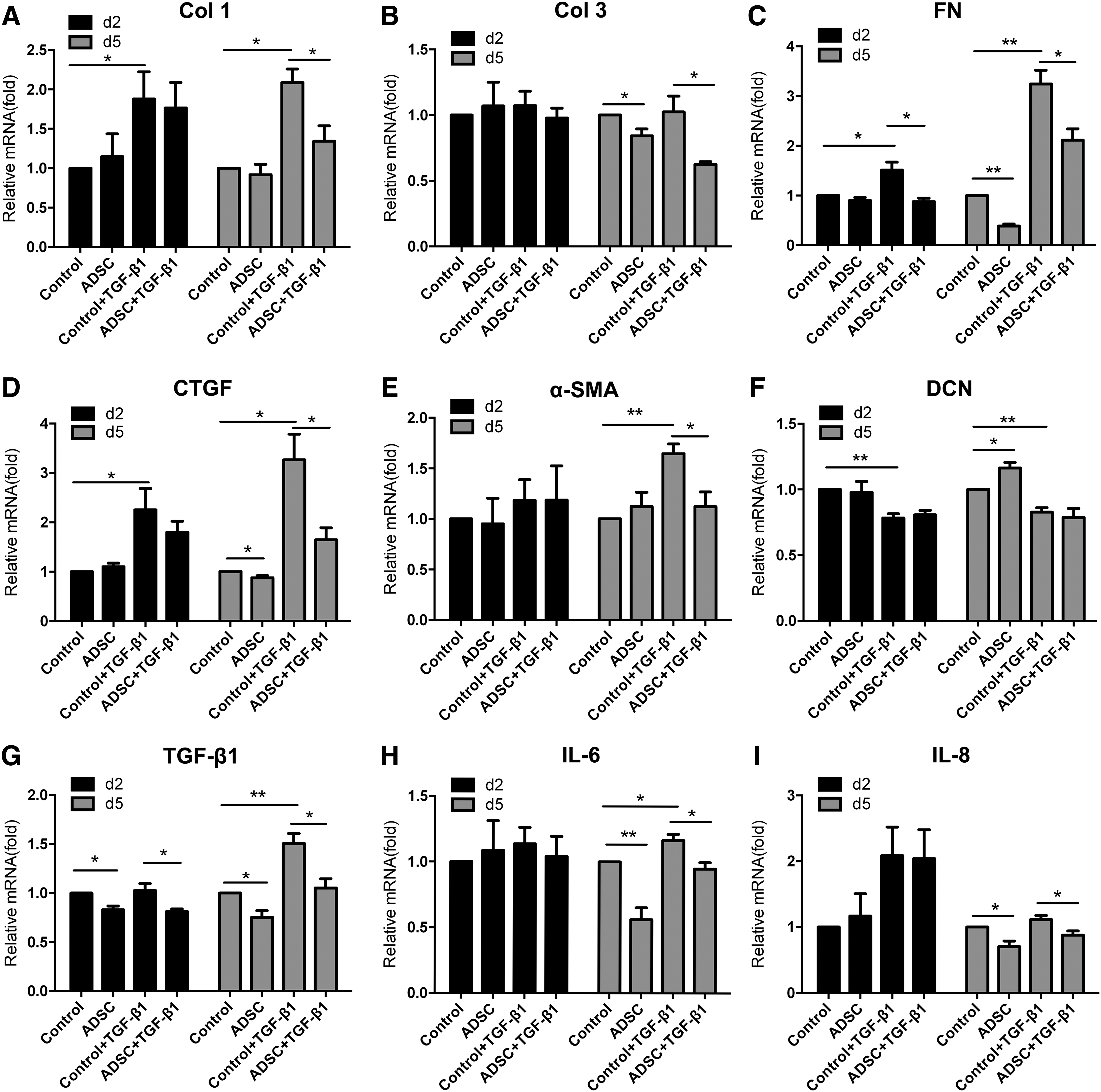
Coculture of HSFs with ASCs counteracted TGF-β1-mediated profibrotic effect. qPCR analysis of various gene expressions in HSFs without or with TGF-β stimulation of both control and coculture groups at day 2 and 5 for Col 1
ASCs attenuated TGF-β induced fibrotic response of HSFs
To further evaluate the possible anti-TGF-β effect mediated by cocultured ASCs, HSFs were cultured with or without exogenous TGF-β1. As shown in Figure 5, after 5 days of culture, exogenous TGF-β1 (2 ng/mL) could significantly induce the gene expression levels of Col 1 (Fig. 5A, p < 0.05), FN (Fig. 5C, p < 0.05), CTGF (Fig. 5D, p < 0.05), α-SMA (Fig. 5E, p < 0.05), TGF-β1 (Fig. 5G, p < 0.05), and IL-6 (Fig. 5H, p < 0.05). Importantly, cocultured ASCs could inhibit the expression of all these TGF-β1-induced genes with significant difference (p < 0.05). In addition, cocultured ASCs also inhibited the expression of other TGF-β1-induced genes including Col 3 (Fig. 5B, p < 0.05) and IL-8 with significance (Fig. 5I, p < 0.05). However, this inhibitory effect was not significant for most of the examined genes (p > 0.05) at time point day 2, except for TGF-β1 (Fig. 5G, p < 0.05) and FN (Fig. 5C, p < 0.05).
ASCs blocked intracellular signaling cascades in HSFs
As shown in Figure 6A, coculture with ASCs could significantly decrease TGF-β1 protein release from the HSFs (2.49 ± 0.43 pg/mL) when compared to that of control HSFs (6.02 ± 1.46 pg/mL, p < 0.05).

Coculture of HSFs with ASCs antagonized intracellular signaling in vitro. ELISA analysis shows that coculture of ASCs reduces TGF-β1 protein release
To further investigate the potential mechanism, western blot was employed to examine the protein levels of related signaling molecules. As shown in Figure 6B, coculture with ASCs could significantly reduce the levels of phosphorylated Smad2 and Smad3, whereas the level of total Smad2/3 protein was not significantly reduced (Fig. 6B). In addition, reduced the levels of phosphorylated Stat3 (Fig. 6C) and p-Erk1/2 (Fig. 6D) were also observed without significant change of total protein levels of Stat3 and Erk1/2. These data indicate that ASCs may exert its antifibrotic effect via blocking related signaling process and reducing TGF-β autocrine production.
Discussion
HS is the result of abnormal growth of fibrous tissue characterized by over proliferation of fibroblasts, excessive ECM deposition, and severe contracture.1–3 Over deposition of ECMs in HS not only results from the overproduction, but also from reduced matrix degradation via aberrant expression of MMPs and their inhibitors (TIMPs). 30
Although not fully defined, the mechanism of HS may involve overproduction of profibrotic factors, which leads to fibroblast proliferation and ECM production. These potential factors include TGF-β1 and its downstream molecule like CTGF, IL-6, and IL-8. 28
The clinical treatment of HS remains a challenge due to the conventional therapies such as pressure, 8 silicone, 9 or steroid injection 5 are still relatively less effective for control scar prevention and contracture. Emerging therapy such as stem cell-based treatment may provide helpful solution in addition to current therapies.
MSC-based therapy is an emerging strategy for antiscarring and antifibrosis treatment via secreting a number of trophic functions.10,31 In this area, ASCs are a particularly preferable MSC source because they are more widely available and more easily obtained by less traumatic methods such as liposuction, and meanwhile they have fewer ethical issues and lower immunogenicity compared with other stem cell types.12,13 Recent studies have demonstrated that ASCs were potent in promoting wound healing and limiting scar formation,17,18,32 which make them an attractive therapeutic cell source for treating HS.
Despite increasing emerging evidence of ASC-mediated repressing effect on HS or tissue fibrosis in both animal study and clinical therapy,15–17,33,34 little effort has been made to understand the mechanism, particularly in molecule aspects of its efficacy on HSFs. Therefore, we performed this experiment by coculturing ASCs with HSFs to observe the effect of ASC on the proliferative and profibrotic phenotypes associated with HSFs.
The results of this study show ASCs possessed specific anti-HS therapeutic effect by inhibiting HSF proliferation and partially blocking cell cycle (Fig. 1), inhibiting matrix gene expression (Figs. 4 and 5) and substantially inhibiting collagen lattice contraction (Fig. 3) in an in vitro experimental setting. It is also likely to enhance ECM degradation as they could modulate the MMP-1/TIMP-1 ratio (Fig. 4D). This ASC-mediated specific inhibition on HSF pathological phenotype will be crucial for the treatment of HS.9,24,35
In addition to inhibiting cell proliferation and matrix production, ASCs also significantly inhibited the expression of profibrotic factors and inflammatory cytokines such as TGF-β1, IL-6, and CTGF as shown in Figures 4 and 5. Additionally, α-SMA, a biomarker of HS was also significantly downregulated by cocultured ASCs (Fig. 4E), suggesting that ASCs may inhibit myofibroblast transformation mediated by TGF-β1 as indicated by previous studies.20,36 One of the HS clinical characters is tissue contracture majorly mediated by TGF-β-induced myofibroblast transformation. As shown in Figure 3, using a FPCL contraction assay, 37 coculturing of HSFs with ASCs could significantly inhibit the contractility of HSFs and the effect became more obvious with prolonged culture time period. Previous study showed that myofibroblast transformation was the major cause of HS tissue contraction, 38 which was enhanced by TGF-β1 with characterized enhanced expression of α-SMA. 39 Truly, implantation of ASCs or fat tissue was shown to decrease scar contracture, 14 our cellular experimental results (Figs. 4 and 5) further support this reported clinical phenomenon.
As multiple intracellular signaling are involved in HS development including Smad, Stat3, extracellular signal-regulated kinase (ERK)3,21,40–43 and TGF-β/Smad is the most important one for HS.40,41 The reduced phosphorylation of Smad2 and Smad3 (Fig. 6) may partially explain ASC-mediated reduction of TGF-β autocrine production, which in return reduce matrix expression, cell proliferation, and collagen contraction. Similar effect was also observed in ASC-mediated TGF-β/Smad signaling in vocal fold fibroblasts. 44
In addition to Smad signaling, the mitogen-activated protein kinase (MAPK) pathway was also found to be widely involved in HS pathogenesis.3,45 ERK was one of the MAPK members, which can be activated by TGF-β1 and participates in cell proliferation, differentiation, and apoptosis.43,46 This study showed that coculture with ASCs could also significantly reduce the phosphorylation of ERK in cultured HSFs (Fig. 6).
Stat3 is a transcription factor activated by tyrosine phosphorylation in human HS tissue sections 3 and Stat3 phosphorylation led to the activation of downstream targeting genes that control ECM production and cellular proliferation in HS lesions.3,47 Evidenced in Figure 6, reduced phosphorylation of Stat3 was also observed in HSFs cocultured with ASCs. Additionally, recent studies also showed that Stat3 could be activated by IL-6 trans-signaling pathways in HS, which mediated ECM production and dermal fibroblast proliferation. 42 Therefore, reduced expression of IL-6 by cocultured ASCs may also contribute to the reduced Stat3 signaling in HSFs.
In this study, a transwell coculture system is used, indicating that paracrine factors play an essential role. Whether cell–cell contact may have more important effect remains to be investigated. Although not tested, the possible candidate factors for anti-inflammatory and antifibrotic effect observed in this study may include hepatocyte growth factor (HGF), IL-10, and prostaglandin E2 as reported in the literature.30,48–53
HGF is regarded as an antifibrotic facilitator contributing to ASCs' antifibrosis function because it was found to be able to upregulate the expressions of MMP-1, −3, and −9 in injured sites in a PI3K/Akt/p70-dependent manner to promote apoptosis of myofibroblasts.54,55 Besides, the presence of HGF antibody could block the inhibitory effect of ASCs on myofibroblasts. 48
IL-10 is also likely the candidate as it was found to predominantly inhibit the synthesis of proinflammatory cytokines, such as IL-6 and IL-8.56,57 It was also suggested that expressions of pro- and anti-inflammatory cytokines are closely related to the development of HS. Therefore, ASC-mediated reduction of pro-/anti-inflammatory cytokine ratio may also contribute its anti-inflammatory and antifibrotic effects. ASCs may also exert anti-inflammatory effect via secreting Prostaglandin E2,30,53,58 which plays a key role in the immunosuppressive properties of ASCs. Accumulating evidence has suggested that PGE2 inhibits TGF-β1-induced activation and fibroblast proliferation, thereby reducing the production of α-SMA and collagens by elevating intracellular cAMP levels and promoting apoptosis in myofibroblasts by increasing the activity of the PTEN protein, which blocks the PI3K/Akt signaling pathway. 59 In addition to these potential mechanisms, a recent study also demonstrated that exosomes derived from human ASCs could accelerate cutaneous wound healing via optimizing the characteristics of fibroblasts, 60 suggesting that micro RNA, a common part of exosome contained molecules, may also involve in this particular regulation.
Conclusion
This study demonstrated that paracrine factors released from ASCs could significantly inhibit pathogenic phenotype of HSFs, including the reduction of cell proliferation and cell migration; downregulated gene expression of matrix molecules, growth factors, and inflammatory cytokines; and reduced cell contractility. These observed phenomena are likely mediated by attenuated multiple intracellular signaling. Further definition of the key released paracrine factors via proteomic or HPLC analysis and other techniques may hold potential therapeutic hope of ASCs in HS treatment.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation (31470943) and Innovation Program of Shanghai Municipal Education Commission (13ZZ090).
Disclosure Statement
No competing financial interests exist.