
Abstract
Extracellular vesicles (EVs) are nanometer-scale particles that are secreted by cells and mediate intercellular communication by transferring biomolecules between cells. Harnessing this mechanism for therapeutic biomolecule delivery represents a promising frontier for regenerative medicine and other clinical applications. One challenge to realizing this goal is that to date, our understanding of which factors affect EV uptake by recipient cells remains incomplete. In this study, we systematically investigated such delivery questions in the context of breast cancer cells, which are one of the most well-studied cell types with respect to EV delivery and therefore comprise a facile model system for this investigation. By displaying various targeting peptides on the EV surface, we observed that although displaying GE11 on EVs modestly increased uptake by MCF-7 cells, neuropeptide Y (NPY) display had no effect on uptake by the same cells. In contrast, neurotensin (NTS) and urokinase plasminogen activator (uPA) display reduced EV uptake by MDA-MB-231 cells. Interestingly, EV uptake rate did not depend on the source of the EVs; breast cancer cells demonstrated no increase in uptake on administration of breast cancer-derived EVs in comparison to HEK293FT-derived EVs. Moreover, EV uptake was greatly enhanced by delivery in the presence of polybrene and spinoculation, suggesting that maximal EV uptake rates are much greater than those observed under basal conditions in cell culture. By investigating how the cell's environment might provide cues that impact EV uptake, we also observed that culturing cells on soft matrices significantly enhanced EV uptake, compared to culturing on stiff tissue culture polystyrene. Each of these observations provides insights into the factors impacting EV uptake by breast cancer cells, while also providing a basis of comparison for systematically evaluating and perhaps enhancing EV uptake by various cell types.
Introduction
E
Materials and Methods
Plasmid construction
All plasmids were constructed using standard molecular biology methods. Full plasmid construction details are described in the Supplementary Methods section (Supplementary Data are available online at www.liebertpub.com/tea). Source materials include pDisplay-pHuji, obtained from Addgene (plasmid No. 61556), deposited by Robert Campbell, 16 and pCD-UPRT-EGFP gifted by Okay Saydam (Medical University of Vienna). Maps for plasmids generated in this study are available on request.
Cell culture
HEK293FT cells (Thermo Fisher Scientific), MCF-7 cells (gift from Sadie Wignall, Northwestern University), and MDA-MB-231 cells (gift from Tom O'Halloran, Northwestern University) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 4 mM
Cell line generation
To package lentiviral vectors, HEK293FT cells were plated at ∼8 × 105 cells/mL in 10-cm dishes (8 mL medium). Six to eight hours later, once cells were attached to the plate at ∼70% confluency, cells were transfected with 3 μg pMD2G, 8 μg psPAX, 10 μg of viral vector (pGIPZ backbones), and 1 μg DsRed-Express2 as a transfection control. Medium was changed ∼16 h later. Lentivirus was harvested from the conditioned medium 28 h later. Medium was cleared of cells by centrifugation at 500 g for 2 min at 4°C and filtered through a 0.45-μm-pore filter (VWR). Lentivirus was concentrated from filtered supernatant by ultracentrifugation at 100,420 g for 90 min at 4°C in a Beckman Coulter Optima L-80 XP ultracentrifuge with an SW 41 Ti rotor. Concentrated lentivirus was used to transduce ∼1.5 × 105 HEK293FT or MDA-MB-231 cells. Transduced cells were flow sorted on a BD FACSAria II flow cytometer to select for the top 50% of GFP-positive cells to create cell lines MH134 (HEK293FT CD63-CD-UPRT-EGFP) and MH135 (MDA-MB-231 CD63-CD-UPRT-EGFP).
EV production, isolation, and characterization
EV-depleted medium was made by supplementing DMEM with 10% exosome-depleted FBS (Gibco), 1% penicillin/streptomycin, and 4 mM
Immunoblotting
For western blot analysis, cells were lysed in RIPA buffer, and protein concentration was determined by BCA assay (Pierce). EVs were not lysed, and loads were normalized by vesicle count as determined by NanoSight. EVs and cell lysates were heated in Laemmli buffer at 70°C for 10 min. Samples were run on 4–15% gradient polyacrylamide gel (Bio-Rad). Proteins were transferred to a PVDF membrane (Bio-Rad) at 100 V for 45 min. Membranes were blocked in 5% milk for 1 h at room temperature, blotted with the rabbit anti-FLAG antibody (ab1162; Abcam) diluted 1:1000, and incubated overnight at 4°C. Primary antibodies were detected with the horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary antibody (Thermo Fisher Scientific).
Generation, characterization, and use of gelatin matrices
An array of gelatin stiffnesses was generated by adding 200 μL of gelatin dissolved in PBS to individual wells of a 48-well plate to produce surfaces coated with 1%, 2.5%, 5%, or 10% gelatin; here, % is defined as g gelatin per 100 mL solution. Plates were kept at 4°C overnight and then crosslinked for 1 h with 5 mM EDC (Sigma) and 6 mM NHS (Sigma) in deionized water. Rheology was performed using an Anton Paar MCR 302 rheometer with parallel-plate fixture. To determine the modulus of each hydrogel, gelatin formulations were cast into silicone wells measuring 15 mm in diameter, with hydrogel thicknesses of ∼2 mm, and crosslinked with EDC/NHS, as above. Hydrogels were biopsy punched to 8 mm outer diameter to fit the parallel-plate fixture. Normal force was set to 0.05 N, and the shear moduli were measured at 100 rad/s and 1% strain. Gelatin wells were washed three times with PBS before cell plating. Cells grown on gelatin were harvested with trypsin, filtered through an 80-μm nylon filter (EMD Millipore) to remove excess gelatin, and washed with a PBS/EDTA solution before flow cytometry.
EV delivery experiments
Recipient MCF-7, MDA-MB-231, or HEK293FT cells were plated 24–36 h before EV delivery at ∼2.5 × 105 cells/mL in 48-well plates (0.3 mL medium). Equal numbers of vesicles (as quantified by NanoSight) were added to each well within each experiment; in all experiments, between 5 × 109 and 2 × 1010 EVs were added per well. Where indicated, polybrene was added to cells at the time of EV delivery at a concentration of 6 μg/mL, a concentration that has been suggested for EV delivery by a commercial vendor, and cells were spinoculated at 300 g for 1 h at 24°C and then returned to the incubator. 18 Where indicated, equal numbers of concentrated EVs (∼100 μL) were combined with annexin V and incubated at 37°C for 15 min before delivery to cells at the final concentrations reported. Cells were harvested using a PBS/EDTA solution before flow cytometry. Fluorescence was quantified using a BD LSRII flow cytometer 2 h after EV delivery. FCS files were analyzed using FlowJo v10.1 (TreeStar). Mean fluorescence intensity (MFI) of cells receiving only a medium change (i.e., autofluorescence) was deemed background and was subtracted from the MFI of EV recipient cells. Statistically significant differences in MFI were evaluated using pairwise, two-tailed Student's t-tests with a significance threshold of p < 0.05. Independent repeats of main text figures are provided in the Supplementary Data to best capture observed sources of experimental variation.
Results
Targeting peptide display on EVs confers diverse effects on EV uptake
An important and open question in the field of EV-mediated delivery is how targeted EV uptake might be best achieved. One widely explored strategy is the engineered display of targeting peptides on the EV surface, which has been convincingly shown to enhance EV uptake by specific recipient cells, including using the rabies virus glycoprotein (RVG) peptide to target Neuro2A cells and the GE11 peptide to moderately increase EV uptake via the epidermal growth factor receptor (EGFR) on breast cancer cells.12,13 However, extending this approach to new targets has proven challenging, as has identifying the factors that currently limit the efficiency and specificity of EV-mediated delivery, and the reasons underlying these challenges are not yet clear. To address this gap in our understanding, in this study we systematically investigated the delivery of EVs to breast cancer cells, which are a well-studied and facile target cell type that comprises a model system enabling us to comparatively evaluate multiple strategies for achieving EV delivery to target cells.
We first evaluated the strategy of targeting via engineered display of peptides on EVs using four previously reported peptides that, in one way or another, achieved specific uptake via receptors expressed on breast cancer cells. We selected the GE11 peptide to target EGFR 13 and neuropeptide Y1 (NPY1) to target the neuropeptide Y1 receptor (NPY1R)19,20 on the estrogen receptor-positive breast cancer cell line MCF-7. We selected the neurotensin (NTS) peptide to target the neurotensin receptor (NTSR) 21 and a urokinase plasminogen activator (uPA) peptide to target the urokinase plasminogen activator receptor (uPAR)22, 23 on the triple negative breast cancer cell line MDA-MB-231. GE11 has been previously demonstrated to target EVs to MCF-7s and serves as a useful internal control, 13 while NPY and amino acids 1–135 of uPA have been shown to mediate targeting of nanoparticles to breast cancer cells but have not been investigated for EV targeting.19,20,22,23 NTS has not been specifically used to achieve targeting to breast cancer, although NTSR1 is upregulated in breast cancer cells,21,24 suggesting that targeting EV uptake via NTS/NTSR1 may be possible.
We first developed and validated an approach for comparing various EV targeting strategies within a single experimental framework. Proteins for displaying targeting peptides on the EV surface were engineered by genetically linking the peptides of interest to the portion of the PDGFR transmembrane domain that abuts the extracellular leaflet of the plasma membrane, which is a strategy that has previously been shown to orient peptides on the exterior EV surface. 13 EVs were then harvested from HEK293FT cells overexpressing the targeting constructs. Expression of such engineered display proteins in EVs was verified via western blot, using FLAG as a model targeting peptide (Supplementary Fig. S1). In contrast to our previous investigation of targeting peptides fused to the endosomal Lamp2b protein, 25 in which displayed peptides were degraded during EV biogenesis unless the peptide was protected with engineered glycosylation sites, unglycosylated constructs achieved better expression in EVs in the PDGFR system. Since direct evaluation of peptide display is not feasible for our candidate targeting peptides, we hypothesized that stability trends would follow those exhibited by FLAG (as previously observed for Lamp2b fusions 25 ), and thus, unglycosylated PDGFR display was used for all subsequent EV display experiments. To enable evaluation of EV uptake via flow cytometry, EV-producing HEK293FT cells were engineered to stably express a fusion protein that targets EGFP to the luminal face of exosomes and other EVs 11 (CD63-CD-UPRT-EGFP; for simplicity, hereafter, we refer to this construct as CD63-GFP). We confirmed that following incubation of nontargeted GFP-labeled EVs with MCF-7 and MDA-MB-231 cells for 2 h, cell-associated GFP was quantifiable by flow cytometry (Supplementary Fig. S2).
Having validated our platform, we next investigated whether our library of targeting peptides conferred enhanced EV uptake. Consistent with prior studies, GE11-displaying EVs conferred a modest increase in uptake by MCF-7 cells (Fig. 1), although this increase was near enough to the signal-to-noise threshold that the effect did not achieve statistical significance in all experiments (Supplementary Fig. S3). NPY display did not confer any effect on uptake of EVs by MCF-7 cells (Fig. 1B and Supplementary Fig. S3). In contrast, display of either NTS or uPA on EVs conferred a modest but consistent and significant decrease in EV uptake by MDA-MB-231 cells (Fig. 1B and Supplementary Fig. S3). To investigate whether these may be explained by variations in the avidities with which various targeted EVs bind target cells, cells were incubated with EVs on ice for 15 min to allow binding to occur in the absence of EV uptake. No meaningful difference in binding was observed for any of the four targeting peptide-displaying EVs compared to control EVs (Supplementary Fig. S4). This pattern was unchanged when incubation time was extended to 1 h and temperature was increased to 4°C, which are conditions used in prior work to quantify EV binding to cells via GPI-anchored nanobodies. 26 Altogether, these data suggest that targeting peptide display on EVs may well impact EV uptake, although the effects are generally modest and sometimes more complicated than simply target binding-enhanced uptake.
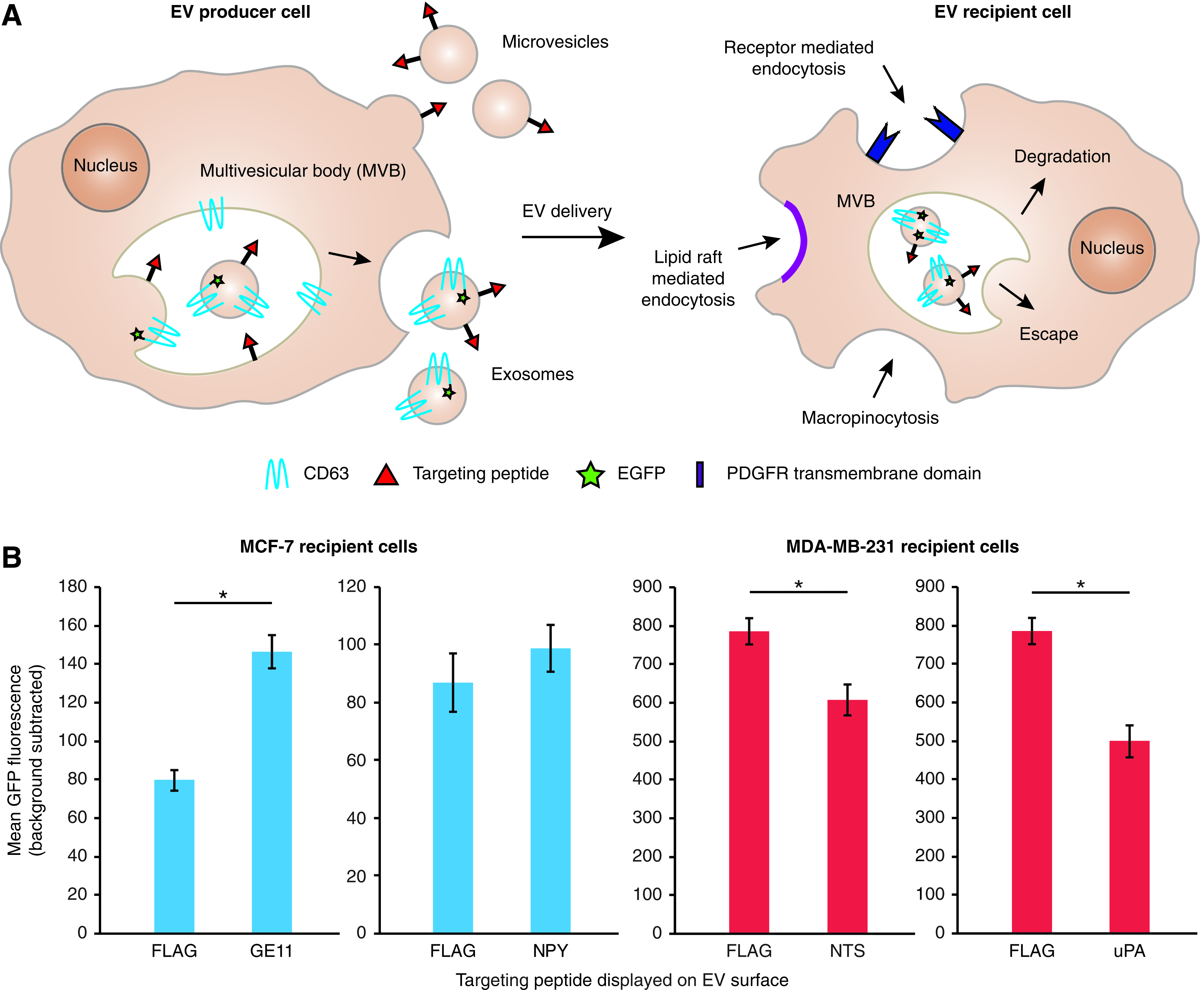
Evaluation of targeting peptide display on EVs on recipient cell uptake.
Upregulation of the targeted receptor does not increase specific uptake of targeting peptide-displaying EVs
To investigate whether low receptor expression level on target cells may limit peptide-mediated specific EV uptake, we leveraged the fact that MCF-7 cells may be treated with estradiol to increase expression of NPY1R.19,27 Cells treated with 1 nM estradiol for 48 h indeed modestly upregulated NPY1R expression, as expected (Supplementary Fig. S5A). Delivery of EVs to estradiol-treated MCF-7 cells resulted in an overall increase in EV uptake, but not specific increase in EV uptake of NPY EVs relative to the control EVs (Supplementary Fig. S5B). To further address the possibility that receptor sites were saturated, cells were treated with a low dose of EVs, just below the threshold where nonspecific EV uptake is detectable in this system (Supplementary Fig. S5C). As before, this approach did not yield any increase in targeted EV uptake relative to the control EVs. We next hypothesized that high levels of nonspecific uptake of EVs by scavenger receptors may mask the effect of targeted EV uptake in this system. To investigate this possibility, EVs were pretreated with annexin V before delivering EVs to cells, since annexin V-mediated blockade of phosphatidylserine on EVs has been shown to reduce their uptake.28,29 However, no blockade of EV uptake was observed, even at high doses of annexin V (Supplementary Fig. S5D), suggesting that in this system, phosphatidylserine-binding receptors are not the primary source of nonspecific EV uptake.
EV source does not impact EV uptake
Some preliminary evidence has suggested that cells may exhibit higher levels of EV uptake if the EVs are produced by the same or a similar cell line. 30 However, this premise has not been systematically investigated in a manner that separates differences in basal levels of EV uptake among different cell lines from the effect of the EV origin on uptake. To address this possibility in our system, we evaluated the delivery of EVs from various cell sources to MCF-7 and MDA-MB-231 cells, including various pairs of source and recipient cells (Fig. 2A). Independent of EV source, MDA-MB-231 cells consistently exhibited more EV uptake than did MCF-7 cells, and HEK293FT cells exhibited no observable EV uptake, suggesting that recipient cell type is the major factor in determining EV uptake among these cases (Fig. 2B and Supplementary Fig. S6A). Although uptake of HEK293FT-derived EVs appeared higher across recipient cell lines, one explanation could be that the EVs from this cell line contain more GFP than do EVs derived from MDA-MB-231 cells. To investigate this hypothesis, GFP-labeled EVs from each source were adsorbed to latex beads and analyzed by flow cytometry. MDA-MB-231-derived EVs contained less GFP than did their HEK293FT-derived counterparts (Supplementary Fig. S6B), which appears to account for the apparent difference in EV uptake by recipient cells. Altogether, these data suggest that while the choice of EV recipient cell strongly influences the level of EV uptake, we did not find evidence that EV source affects EV uptake in this system.
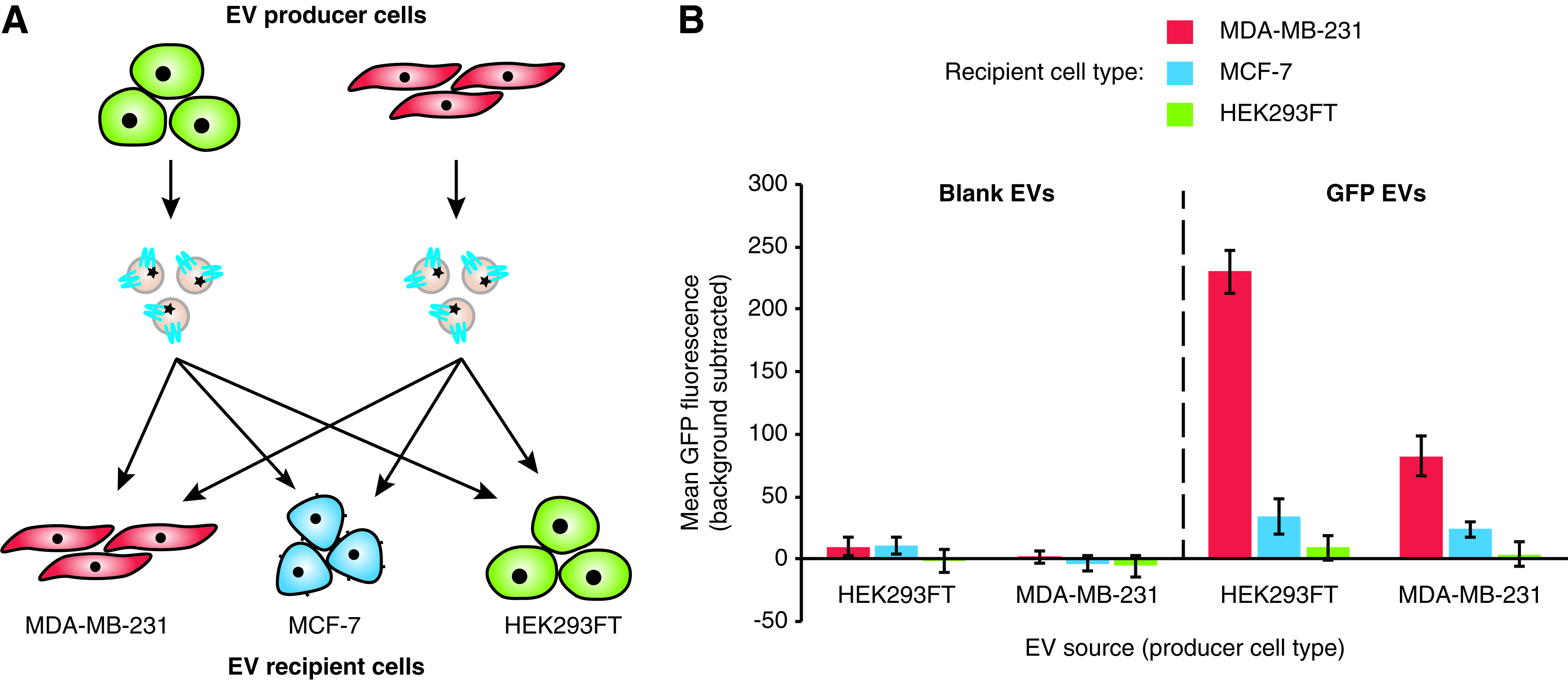
Effect of producer–recipient cell type pairing on EV uptake.
Polybrene and spinoculation synergistically increase EV uptake
As a point of comparison, we next sought to estimate the maximum possible rate of EV uptake in these cells using several methods developed for enhancing gene delivery. In particular, we investigated EV delivery under conditions of spinoculation and/or incubation with polybrene (Fig. 3A). Such techniques are commonly used to enhance lentiviral/retroviral delivery by mitigating charge repulsion between the vector and the cell surface, in the case of polybrene, 31 and by presumably inducing cytoskeletal rearrangements via low-level centrifugal forces, in the case of spinoculation. 18 To evaluate these factors in our system, cells were treated with EVs under conditions of polybrene addition and/or spinoculation (Fig. 3B and Supplementary Fig. S7). While spinoculation alone resulted in only a modest increase in EV uptake, polybrene treatment alone conferred a much more pronounced increase in EV uptake. Interestingly, the combination of polybrene and spinoculation had a synergistic effect on EV uptake, achieving an increase in uptake of ∼30-fold in MCF-7 cells and 5- to 10-fold in MDA-MB-231 cells. These results demonstrate that the maximal possible rate of EV uptake exhibited by these cells far exceeds that which is achieved with the peptide targeting strategies evaluated to date.
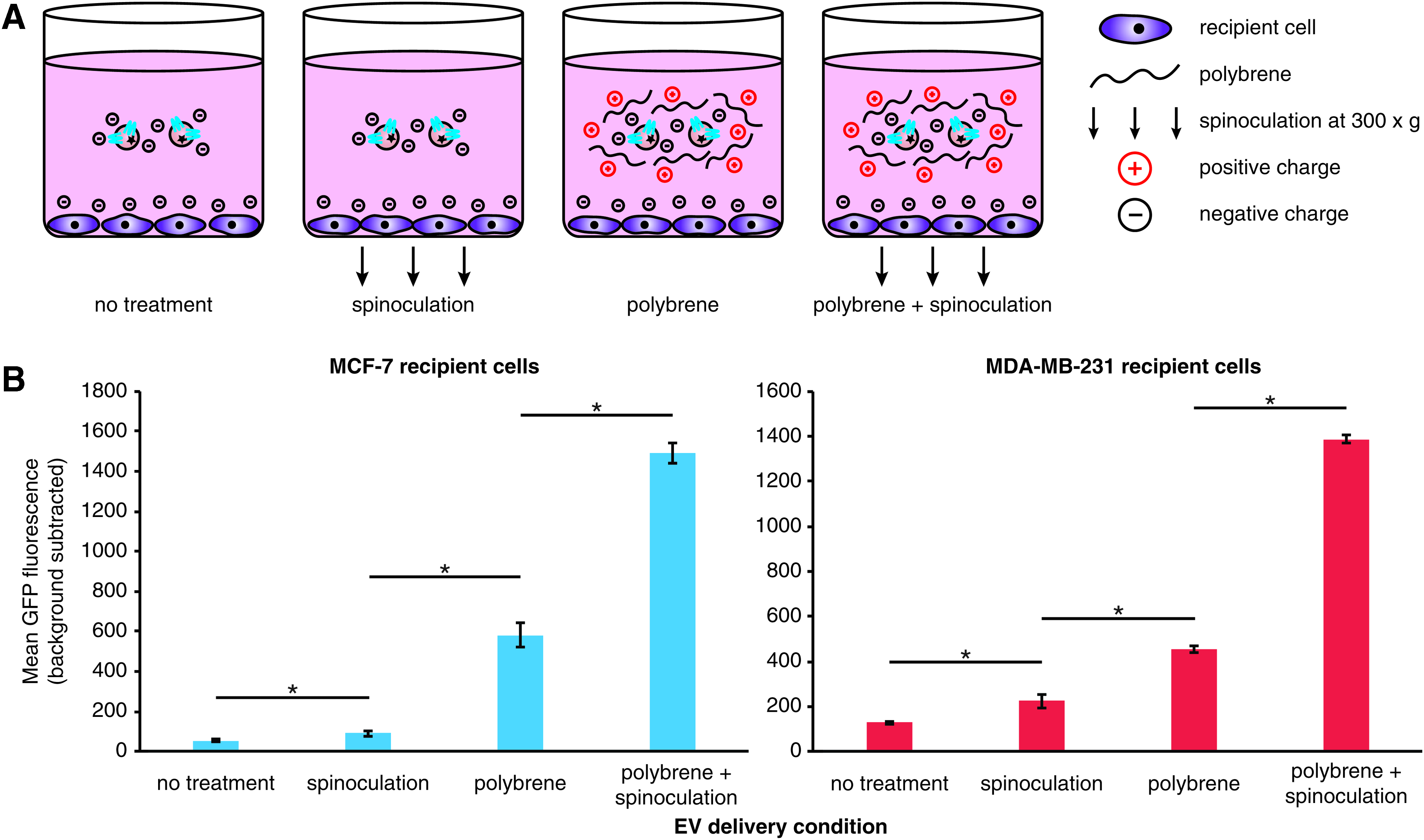
Enhancement of EV uptake by treatment with polybrene and spinoculation.
EV uptake is enhanced by growing cells on softer matrices
Given the pronounced influence of external factors such as polybrene and spinoculation on EV uptake, we next investigated whether environmental cues conveyed via the culture substrate may also modulate EV uptake. In particular, we investigated the influence of substrate mechanical stiffness. Stiffness of the cell culture matrix influences the growth and cell fate of breast cancer cells, with a stiffness of ∼5 kPa conferring maximal growth and maintenance of stem-like surface marker expression for MCF-7 and MDA-MB-231 cells.32,33 To investigate whether these factors impact EV uptake, MCF-7 and MDA-MB-231 cells were grown on hydrogels including various concentrations of gelatin to vary stiffness (Fig. 4A). We selected concentrations of 1%, 2.5%, 5%, and 10% gelatin to produce storage moduli ranging from 2 to 24 kPa (Fig. 4B). Notably, when compared to cells grown on tissue culture polystyrene (TCPS, which has a storage modulus of ∼3 GPa), 34 cells grown on gelatin demonstrated significantly higher levels of EV uptake for both cell lines, although there was little observable difference in uptake rates across gelatin concentrations (Fig. 4C and Supplementary Fig. S8). This exciting and novel observation of substrate-induced enhancement of EV uptake was most pronounced in MCF-7 cells, resulting in a sixfold increase when cells were cultured on gelatin, compared to a twofold increase in uptake for MDA-MB-231 cells. This trend is consistent with the polybrene+spinoculation study (Fig. 3), in that the cell line with a lower basal EV uptake rate exhibited a greater potential for uptake enhancement.
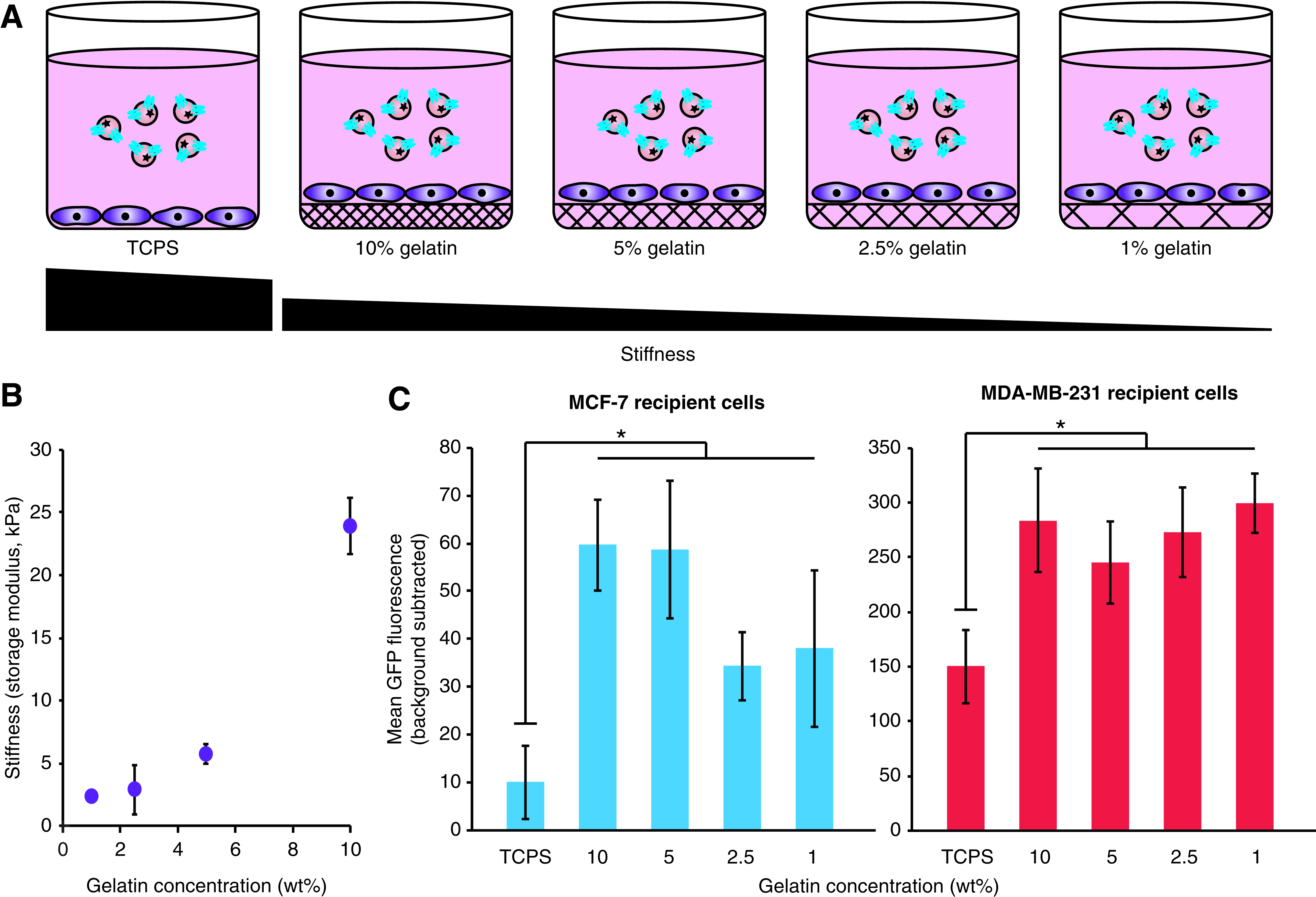
Dependence of EV uptake on cell growth matrix stiffness.
Discussion
As EV-based therapies advance in clinical development, there is an increasing need to understand the factors affecting their uptake by various recipient cells. This study elucidates several key factors that modulate EV uptake efficiency, at least within the context of breast cancer, and which may ultimately guide the design of effective EV therapeutics.
An exciting aspect of EV-mediated cargo delivery is the potential to engineer EVs to be specifically taken up by certain cell types by expressing targeting peptides on the EV surface. In this study, we investigated four targeting peptides displaying different effects on EV uptake, ranging from a modest increase to a significant decrease. Notably, none of the peptides selected based on previous success in nanoparticle targeting demonstrated increased uptake when displayed on EVs. One potential reason for this is that nanoparticle labeling with these peptides may achieve a much higher density of peptides on the particle surface than does harvesting EVs from cells transfected with the peptide constructs, such that binding avidity may be low in the context of EV targeting. However, the repeatable decrease in uptake observed when EVs were labeled with NTS and uPA raises some interesting questions. This apparent “antitargeting” phenomenon is unlikely to be due to either charge repulsion or entropic binding penalties, since the NTS and uPA peptides are expected to be slightly positively charged at physiological pH and are reasonably short in length. One potential explanation is that targeting EVs to receptors that may not be internalized as rapidly as scavenger receptors could result in a decrease in overall EV uptake. It is also possible that targeting some receptors (such as NTSR and uPAR, in this case) shuttles EVs into intracellular pathways that lead to rapid degradation, resulting in a decrease in cargo accumulation regardless of uptake rate, relative to nontargeted EVs. Further investigation is needed to fully determine the effect of EV labeling on uptake pathway and eventual fate of cargo within the recipient cell.
Contrary to some conventional wisdom, we found that EV source had a minimal effect on EV uptake, especially compared to the intrinsic uptake rate exhibited by the recipient cell type. From a practical standpoint, this finding may be promising by suggesting that a single, well-characterized, safe, and effective EV source may be used to generate EVs for a variety of applications.
We demonstrated that EV uptake may be significantly enhanced by chemical and physical manipulations, which may set an upper bound for delivery by other means. While the use of spinoculation and polybrene is not a translatable strategy, these experiments identify the degree to which key biophysical parameters impact EV uptake, which comprise insights that could be translationally relevant. For example, like nanoparticles, EVs may be formulated to facilitate delivery in vivo, 35 and our data suggest that formulations that mitigate electrostatic repulsion may substantially increase uptake in breast cancer cells (and probably other cell types). While it is not yet clear how spinoculation impacts cellular uptake of EVs, we hypothesize that cells undergoing cytoskeletal rearrangement (which may be stimulated by spinoculation 18 ) may increase EV uptake. This hypothesis is consistent with a recent report identifying filopodia as sites of active EV uptake. 36 Together, our data suggest that these effects may interact to modulate EV uptake, which could substantially impact EV-mediated delivery in vivo.
Finally, a particularly interesting observation is that growth substrate can substantially enhance EV uptake. One possible explanation is that growth of these breast cancer lines on soft gelatin leads to cytoskeletal rearrangements or more rapid membrane recycling. Alternatively, matrix-induced signaling may drive alterations in trafficking via specific intracellular compartments and pathways. A final intriguing possibility is that EV uptake may be modulated within the body by variations in extracellular matrix (ECM) stiffness, including both healthy and diseased-associated variations in ECM stiffness. Whether such an effect may ultimately exist and help to relate in vivo and in vitro observations remains to be seen. Should such a connection exist, it may guide the design of in vitro experiments to better mimic salient delivery phenomena in vivo, facilitating the development of EV-based therapies. In future work, it will be interesting to investigate how and whether the phenomena reported here extend to various types of EVs, including choices of EV producer cell type and of purification method and protocol, of which there is now substantial variety. Ultimately, better understanding the rules that govern EV uptake is critical to developing efficient EV therapeutics for applications in regenerative medicine and beyond.
Footnotes
Acknowledgments
The authors acknowledge support from the Northwestern Memorial Foundation and the Lynn Sage Cancer Research Foundation (to J.N.L.). This work was supported by the National Science Foundation's Graduate Research Fellowship Program (NSF GRFP) award DGE-1324585 (to D.M.S.) and award DGE-0824162 (to M.E.H.). This work was also supported by the Northwestern Institute for Cellular Engineering Technologies (iCET) seed grant, and NIH/NIEHS/NCATS UH3TR001207 (to E.S.G.). NanoSight analysis was performed at the Northwestern University Keck Biophysics Facility. Flow cytometry was performed at the Northwestern University Flow Cytometry Facility, which is supported by a Cancer Center Support Grant (NCI CA060553). Traditional sequencing services were performed at the Northwestern University Genomics Core Facility. Rheology was performed in the Analytical BioNanotechnology Equipment Core of the Simpson Querrey Institute at Northwestern University. The U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University provided funding to develop this facility, and ongoing support is being received from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.