
Abstract
Histone methylation is regarded as an important epigenetic event during stem cell differentiation. Jumonji AT-rich interactive domain 1A (Jarid1a) is a histone demethylase that specifically catalyzes demethylation of dimethyl or trimethyl histone H3K4me3, which is normally associated with transcriptionally active genes. Runt-related transcription factor 2 (Runx2) has been identified as a key transcription factor in the early stage of osteogenesis. A better understanding of this epigenetic mechanism that governs osteogenic differentiation of bone mesenchymal stem cells (BMSCs) can provide new insights into BMSC-based bone tissue engineering. To define the function and regulatory mechanisms of Jarid1a in the osteogenic differentiation of BMSCs, we compared the expression of Jarid1a between undifferentiated and osteoinductive BMSCs. The expression of osteogenic transcriptional factors in BMSCs after Jarid1a knockdown was explored using western blot. To determine whether Jarid1a was associated with Runx2 during osteogenic differentiation, endogenous coimmunoprecipitation (co-IP) experiments were performed with osteoinductive BMSCs extracts. Then, we systematically evaluated the function of si-Jarid1a in enhancing BMSCs osteogenesis and the therapeutic potential of si-Jarid1a-modified BMSCs in a rat calvarial bone defect model with β-tricalcium phosphate scaffolds. Knockdown of Jarid1a by small interfering RNA enhanced osteogenic differentiation of BMSCs in vitro. Knockdown of Jarid1a significantly improved the mRNA and protein expression of bone-specific factors. Furthermore, co-IP in BMSCs lysate suggested that Jarid1a was physically and functionally associated with Runx2. The repair potential of bone defect was dramatically improved by Jarid1a-knockdown BMSCs, including increased bone volume, increased bone mineral density, and decreased scaffold residue in vivo. Altogether, this study explores the functional and regulatory role of Jarid1a in osteogenic differentiation and bone regeneration of BMSCs, and provides a new approach for bone defect repairing using epigenetic modification in vitro and in vivo.
Introduction
B
How to effectively promote BMSCs' osteogenic differentiation has become a core issue in the bone repair or regeneration. Previous studies have focused on modulating the inductive conditions or integrating osteo-induced genes into the original genome to facilitate osteogenic differentiation.5–7 Recent studies also suggest that epigenetic regulation of the genome irrelevant to the DNA sequence plays an important role in the maintenance and determination of stem cell fate. 8 It has been demonstrated that the histone methylation is regarded as a major mechanism of epigenetic modulation, especially in the lineage determination and differentiation of stem cells.9,10 However, there have been few reports on the epigenetic mechanism contributing to BMSCs' osteogenic differentiation and bone regeneration.
Jumonji AT-rich interactive domain 1A (Jarid1a), also known as lysine-specific demethylase 5A (KDM5A) or retinoblastoma binding protein 2 is one of these histone demethylases that catalyze the removal of dimethylation and trimethylation at histone H3 lysine 4 (H3K4) and regulate the methylation pattern of histones.11,12 Therefore, it would be of interest to clarify its functional roles in maintenance and determination of stem cell fate. Some studies have shown that inhibition of Jarid1a can improve stem cell differentiation with an increase of trimethylated H3K4 (H3K4me3) levels at the promoter regions and activation of related genes.12–14 However, the role of Jarid1a in osteogenic differentiation and bone repairing potential of BMSCs is not fully understood.
In this study, to define the function and regulatory mechanisms of Jarid1a in the osteogenic differentiation of BMSCs, the expression of osteogenic transcriptional factors in BMSCs after Jarid1a knockdown was explored using western blot. To determine whether Jarid1a was associated with Runx2 during osteogenic differentiation, endogenous coimmunoprecipitation (co-IP) experiments were performed with osteoinductive BMSCs extracts. Then we explored the osteogenesis of si-Jarid1a modified stem cells in vivo. The results would improve our knowledge about the epigenetic mechanisms governing the BMSCs' osteoblastic lineage differentiation, which would benefit the development of bone tissue engineering or cell therapy based on BMSCs.
Materials and Methods
Cell culture
Sprague Dawley rats were obtained from the Shanghai Animal Experimental Center, and all procedures were approved by the Animal Research Committee of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (No. 201449). Bone marrow was obtained from the femur and tibia of 4-week-old male Sprague Dawley rats according to the protocol.
15
Primary BMSCs were incubated in a humidified 5% CO2 atmosphere at 37°C. For proliferation assays, proliferation medium (PM) consisted of α-minimum essential medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Gibco). For osteoinductive culture, differentiation medium (DM) was comprised of α-minimum essential medium (MEM) supplemented with 10% FBS, 10−8 M dexamethasone, 10 mM β-glycerol phosphate, and 50 mg/mL
Transfection of small-interfering RNAs
The oligonucleotides, including small-interfering RNAs of Jarid1a (si-Jarid1a), Runx2 (siRunx2), and negative control (si-NC), were synthesized by GenePharma Co., Ltd. (Shanghai, China). For transient transfections, 40 nm siJarid1a, siRunx2, or si-NC was mixed with X-treme GENE HP DNA transfection reagent (Roche, Basel, Switzerland) in Opti-MEM medium, and the mixture was directly added to cells in six-well plates at a density of 3 × 105 cells/well. For long-term detection, siRNAs were repeatedly transfected every 3 days. The oligonucleotide sequences were as follows: si-Jarid1a 5′-GCTGTACGAGAGTATACAC-3′ and siRunx2 5′-UGCCUCUGCUGUUAUGAAA-3′.
Quantitative real-time polymerase chain reaction analysis
Total RNAs were isolated with TRIzol reagent (Invitrogen, Carlsbad, CA), and the first-strand complementary cDNA was synthesized using the PrimeScript™ RT Reagent Kit (TaKaRa). Quantifications of all gene transcripts were performed by real-time polymerase chain reaction (PCR) using a Power SYBR Green PCR Master Mix and a 7500 Real-Time PCR Detection System. The gene-specific primers osteopontin (OPN), bone sialoprotein (BSP), osterix (OSX), osteocalcin (OCN), and special AT-rich sequence-binding protein 2 (Satb2) were introduced as previously described. 16 The relative mRNA level was expressed as fold change relative to controls after normalization to the expression of GAPDH, respectively.
Western blot analysis
Protein lysates were generated using radioimmunoprecipitation assay lysis buffer (Thermo Fisher Scientific), and the concentration of proteins was determined using a BCA protein assay (Thermo Fisher Scientific). A total of 20 μg proteins were separated on a 7% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis gel and transferred to PVDF membranes (Millipore, Billerica, MA) as previously described. After blocked with 5% nonfat milk, the membranes were incubated with primary antibodies for Jarid1a, Runx2, OPN, OCN, BSP, and Satb2 (all from Abcam, Inc.) respectively. Secondary antibodies were labeled with infrared (IR)-dyes. Signals were visualized using an Odyssey Infra-red Imaging System (LI-COR Biosciences, Lincoln, NE). β-actin was used as the control to normalize the loading materials. The gray value of every band was read with Odyssey and the ratio of target protein to β-actin for each sample was calculated.
Immunocytochemistry
BMSCs were seeded onto 12-well plates, transfected with si-Jarid1a or si-NC, and cultured in PM and DM for 4 days. Then the cells were fixed in 4% paraformaldehyde and incubated in blocking buffer (phosphate-buffered saline containing 10% normal goat serum, 0.3% TritonX-100 and 0.1% NaN3) for 1 h, as reported. Following incubation with primary antibodies against OSX and OCN (all from Abcam) overnight at 4°C, the cells were incubated with secondary antibodies for 1 h at room temperature. The cell nuclei were counterstained with propidium iodide (PI) (BD Biosciences). The immunoreactive cells were visualized and imaged using a fluorescent microscope (Leica Microsystems, Germany). The percentages of positive cells were determined by the ratio of immunopositive BMSCs to PI stained nuclei. In total, 500–1000 cells were counted in random fields for each group.
Alkaline phosphatase activity and mineralization assays
BMSCs transfected with si-Jarid1a and si-NC were seeded onto 12-well plates at a density of 1.0 × 105 cells/well and cultured in PM and DM respectively. After the cells were fixed in 4% paraformaldehyde at day 7, alkaline phosphatase (ALP) staining was performed according to the manufacturer's instructions (Rainbow, Shanghai, China). After fixation in 95% ethanol for 20 min at day 14, the cells were incubated with 40 mM alizarin red staining (ARS) solution (Sigma) for 20 min at room temperature. A semiquantitative analysis of ALP activity was performed as previously described. 16 In brief, the protein concentration was determined using a BCA Protein Assay Kit. ALP activity was determined at 405 nm using p-nitrophenyl phosphate (Sigma) as the substrate, and ARS staining was detected at 590 nm using cetylpyridinium chloride (Sigma) by a microplate reader (Bio-Tek, Winooski, VT). Finally, the ALP and ARS levels were normalized to the total protein content.
Co-IP assay
After BMSCs were cultured in DM for 7 days, cells lysates were prepared by incubating the cells in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 10%glycerol, 0.5% Triton X-100, 2 mM EDTA, 25 mM glycerophosphate, 100 mM NaF, 200 mM Na3VO4, 1 mM PMSF, 1 μg/mL leupeptin, and aprotinin) as before. 17 The insoluble material was removed via centrifugation at 14,000 g for 15 min, total proteins were incubated with Runx2 or Jarid1a antibodies for 12 h with constant rotation, and the recovered supernatant was immunoprecipitated with antibody-coated Dynabeads (Invitrogen) according to the manufacturer's protocol. The immunoprecipitates were then washed four times with 1 mL lysis buffer and then resuspended in SDS sample buffer for western blotting analysis. Five percent of total cell lysates were used as input.
Lentiviral vector construction and transduction
GFP-labeled plasmid vectors containing si-NC and si-Jarid1a (5′-GCTGTACGAGAGTATACAC-3′) were obtained from Obio Technology (Shanghai), and 293T cells were maintained in DMEM supplemented with 10% FBS. Lentiviruses were produced by transfecting 293T cells with 10 μg of plasmid encoding si-NC or si-Jarid1a, 5 μg of Pax plasmid, and 5 μg of VSVG plasmid using Lipofectamine 2000. The culture medium was then changed the next day, and the supernatant was harvested after 48 h. The lentiviruses were filtered and concentrated by ultrafiltration, and aliquots were stored at −80°C. For transduction, cells were incubated with the virus and 8 mg/mL polybrene for 24 h. Lentiviruses (si-NC, si-Jarid1a) were added to reach an multiplicity of infection of 50 for 24 h.
Animal experiments
All procedures were approved by the Animal Research Committee of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (No. 201449). The pie-shaped (Φ5 mm × 2 mm3) porous β-tricalcium phosphate (β-TCP) scaffolds with an average pore size of 500 μm and 75% porosity were provided by Shanghai Bio-lu Biomaterials Co., Ltd. All surgical procedures were performed on 12-week-old male Sprague Dawley rats as described previously. 18 A 1.0–1.5 cm sagittal incision was made on the scalp, and the calvarium was exposed by blunt dissection. Two critical size defects were created in each rat by a 5 mm-diameter electric trephine (Nouvag AG, Goldach, Switzerland). After the BMSCs transduced with si-NC or si-Jarid1a were seeded onto the scaffolds, the cell-seeded scaffolds were implanted into the defects after 24-h culture in vitro. Eighteen rats were bred and randomly allocated into the following graft study groups: (1) β-TCP (n = 6), (2) β-TCP with BMSCs/si-NC (n = 6), and (3) β-TCP with BMSCs/si-Jarid1a (n = 6). The rats were able to function normally after this procedure. Microcomputed tomography (micro-CT) analysis and histological observation at 8 weeks postoperatively were used as before. 18 The percentages of new bone volume relative to tissue volume (BV/TV) and the bone mineral density (BMD) were also determined using the analysis software. The sections were stained with van Gieson's picrofuchsin to identify new bone formation. Red indicated new bone formation, and TCP showed black. The surface areas of the newly formed bone and TCP residue were measured using Image Pro Plus™ (Media Cybernetics, Silver Springs, MD) based on the entire bone defect area.
Statistical analysis
The experimental statistics used in this study are expressed as the mean ± standard deviation. Data were analyzed using Student's two-tailed t-test to compare the means of two groups or one-way ANOVA for comparison of means of multiple groups, using SPSS 17.0 software. p < 0.05 was considered statistically significant.
Results
Inhibition of Jarid1a enhances osteogenic differentiation
The Jarid1a protein expression was not affected by culturing in differentiation medium (DM) for 2 days (Fig. 1A, B). The protein levels of extracellular structural proteins OPN and BSP were increased during osteogenic induction (Fig. 1A, C, D). Although the expression level of the Jarid1a protein was obviously decreased by si-Jarid1a (*p < 0.05), the protein levels of OPN and BSP exhibited an approximately twofold increase in siJarid1a transfected BMSCs compared with control cells in DM culture (p < 0.05), but in PM culture, the levels of these proteins showed no significant difference (Fig. 1). These results suggested that Jarid1a may act as an important regulator of BMSCs' osteogenesis. To investigate the downstream targets of Jarid1a during osteogenic differentiation, our results indicated that BMSCs transfected with si-Jarid1a oligonucleotides at day 4 were fixed and immunostained with primary antibodies against osteogenic markers OSX and OCN (Fig. 2A). At the baseline, 19.8% ± 3.23% of DM cultured BMSCs expressed OSX and 30.5% ± 3.42% expressed OCN. The proportions of immunoreactive OSX-and OCN-expressing cells were clearly lower (10.1% ± 3.03% and 8.2% ± 2.41%) in PM cultured cells (p < 0.05) irrelevant to knockdown of Jarid1a, but dramatically higher (61.2% ± 7.21% and 65.2% ± 6.62%) in osteoinductive Jarid1a-knockdown cells (p < 0.05) (Fig. 2B, C). These results indicated that important osteogenic factors OSX and OCN were significantly upregulated in osteogenic differentiated BMSCs. Also, the absence of Jarid1a elevated the expression of osteogenic factors in osteoinductive BMSCs. Thus, the promotion of osteogenic differentiation via Jarid1a inhibition may depend on osteogenic inducing condition.
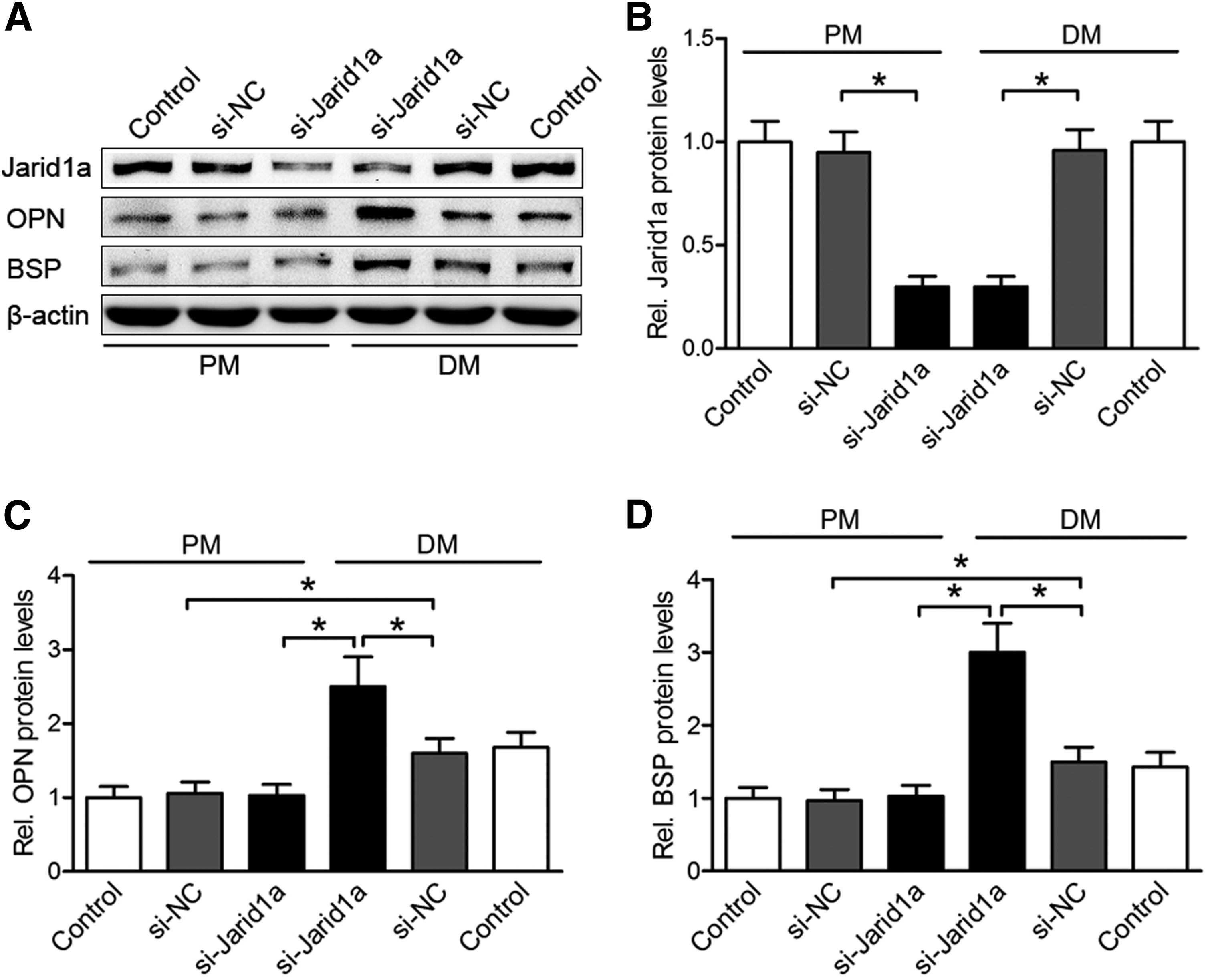
Knockdown of Jarid1a on osteogenic markers expression in the proliferation and differentiation of BMSCs.

The expression of bone-specific markers in Jarid1a-knockdown BMSCs, and the results of ALP and ARS staining.
The effect of Jarid1a-knockdown on ALP activity and mineralization
To characterize the function of Jarid1a in osteogenic differentiation of BMSCs, a combination of in vitro experiments were conducted. At day 7, osteogenic-induced BMSCs showed robust positive ALP staining relative to PM cultured cells, especially in Jarid1a-knockdown BMSCs (Fig. 2D). In addition, the mineralization in BMSCs culture at day 14, as exhibited by ARS, was compatible with the ALP staining results (Fig. 2D). The semiquantitative analysis of the ALP activity showed that the absorbance index values were not elevated in noninduced cells by Jarid1a-knockdown (p > 0.05), but significantly increased in osteoinductive cells at day 7 (p < 0.05) (Fig. 2E). The semiquantitative analysis of the ARS results showed that the absorbance index values from Jarid1a-knockdown cells were 1.9-fold higher than the control group at day 14 (Fig. 2F). These results suggested that Jarid1a acted as an important negative regulator of BMSCs' osteogenesis.
Jarid1a represses osteogenic differentiation without affecting Runx2 expression
The mRNA expression of bone-specific factors BSP, OPN, OSX, OCN, and Satb2 was increased in osteoinductive BMSCs compared with noninduced BMSCs at day 7 (p < 0.05), and these high levels of expression were dramatically enhanced by Jarid1a-knockdown in osteoinductive BMSCs, but showed no changes in noninduced cells at day 7 (Fig. 3A–E). Runx2 mRNA expression was increased in osteoinductive BMSCs compared with noninduced BMSCs (p < 0.05), but not improved by knockdown of Jarid1a (p > 0.05, Fig. 3F). These results were supported by Runx2 protein levels (p > 0.05, Fig. 4A, B). In addition, to determine whether Jarid1a inhibited osteogenic differentiation through Runx2 protein, DM-cultured BMSCs were cotransfected with si-Jarid1a and siRunx2 (small interfering RNA of Runx2). Runx2 plays an indispensable role in the expression of osteogenic markers OPN, BSP, and SATB2 by knockdown of Jarid1a (p < 0.05, Fig. 4C). Taken together, these results suggested that Jarid1a did not regulate Runx2 expression, but promoted osteogenic marker expression by knockdown of Jarid1a in a Runx2-dependent manner.
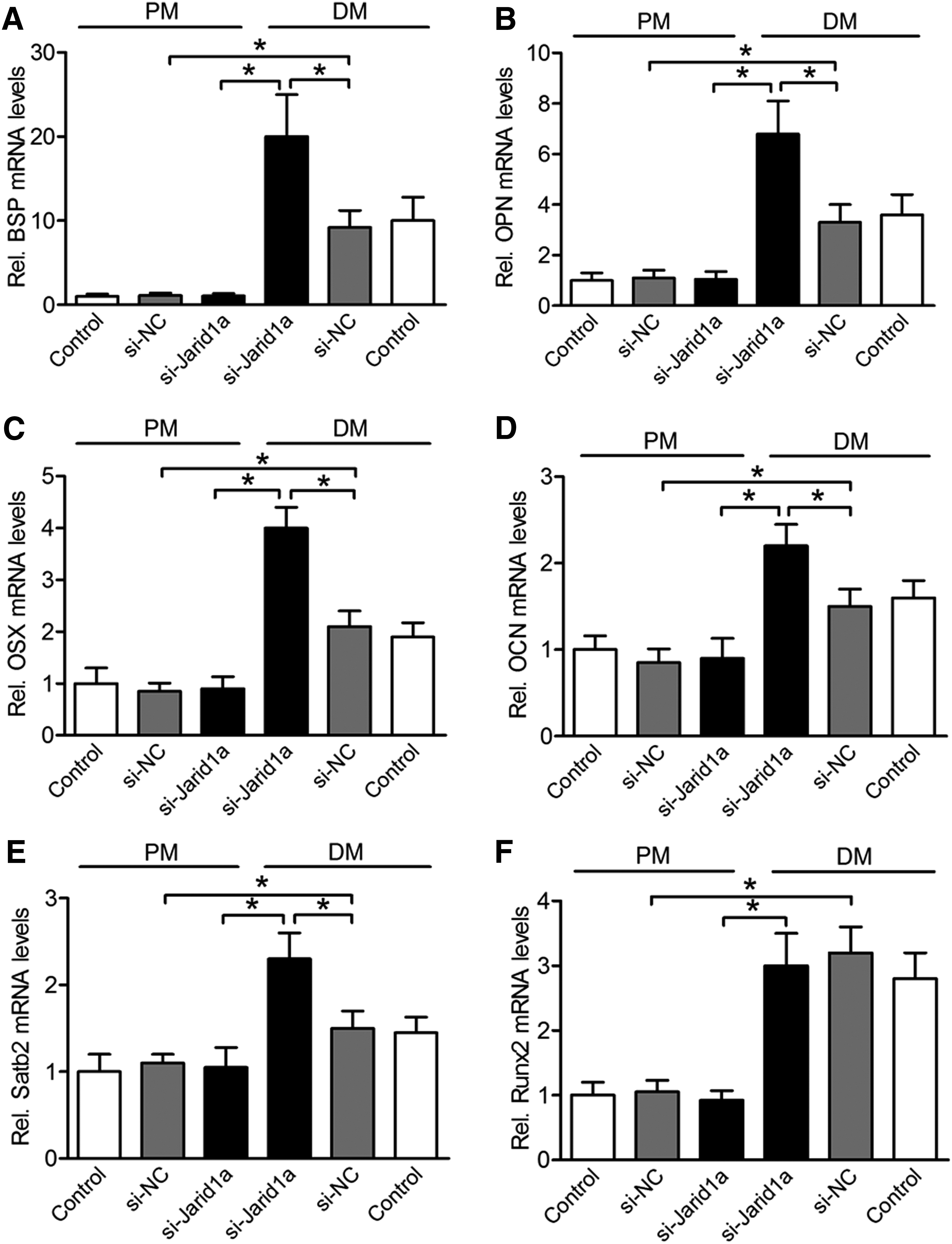
The mRNA expression of bone-specific factors by Jarid1a knockdown in PM- or DM-cultured BMSCs at day 7.
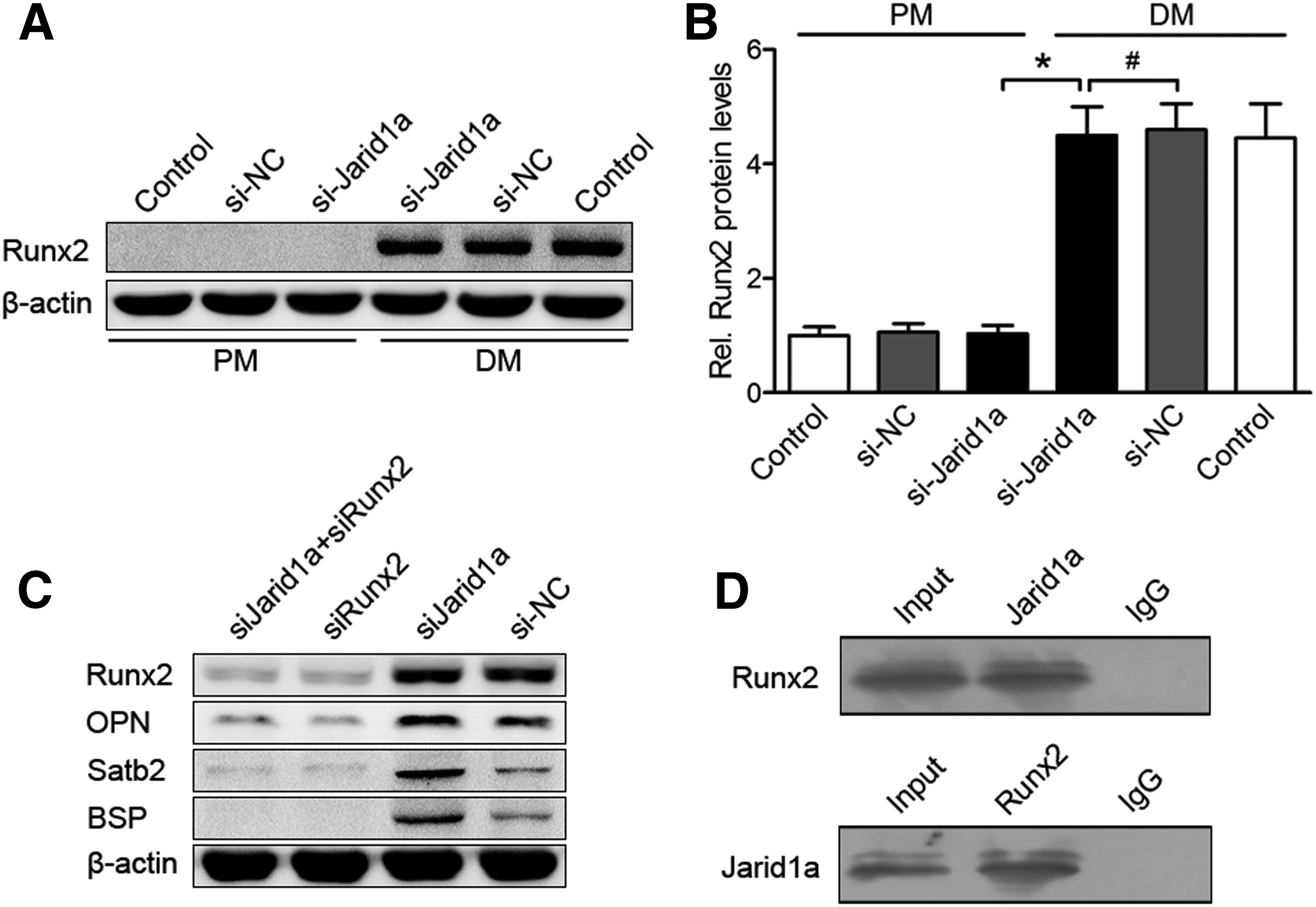
The relationship between Jarid1a and Runx2.
Jarid1a directly interacts with Runx2
To further explore the underlying mechanism of Jarid1a-mediated inhibition of BMSCs' osteogenic differentiation, we investigated the physical association between Jarid1a and Runx2, the key regulator of osteogenesis, by co-IP experiments. IP with antibodies against Jarid1a followed by immunoblotting with antibodies against Runx2 demonstrated that Jarid1a was coimmunoprecipitated with Runx2 (Fig. 4D up). Reciprocally, IP with antibodies against Runx2 and immunoblotting with antibodies against Jarid1a also revealed that Runx2 was coimmunoprecipitated with Jarid1a (Fig. 4D down). Collectively, these results suggested that Jarid1a was physically associated with Runx2 in vivo and played a role in Runx2-regulated osteogenic gene expression.
In vivo regulation of bone regeneration by si-Jarid1a
To observe new bone formation within the defects, the morphology of the newly formed bone was reconstructed using micro-CT at 8 weeks after explantation of the skull. In the si-Jarid1a group, new bone formation was observed, and the appearance of the implant became smoother. However, different sizes of gaps were observed in the peripheral area of the implant in the si-NC and β-TCP groups (Fig. 5A). From the sagittal view, the new bone formation in the si-Jarid1a group was greater than that in the si-NC and β-TCP groups (Fig. 5A). A quantitative analysis using the micro-CT analysis system showed that the BMD of the si-Jarid1a group (0.57 ± 0.073 g/cm3) was markedly higher than that of the si-NC group (0.44 ± 0.061 g/cm3) (p < 0.05) or β-TCP group (0.35 ± 0.039 g/cm3) (p < 0.05) (Fig. 5B). In addition, the percentages of BV/TV in the three groups (11.25% ± 3.72% in the β-TCP group, 20.12% ± 4.71% in the si-NC group, and 32.42% ± 6.91% in the si-Jarid1a group) showed the same pattern as for the BMD levels (Fig. 5C). These results suggested that Jarid1a negatively regulated ossification and the knockdown of Jarid1a enhanced bone regeneration.
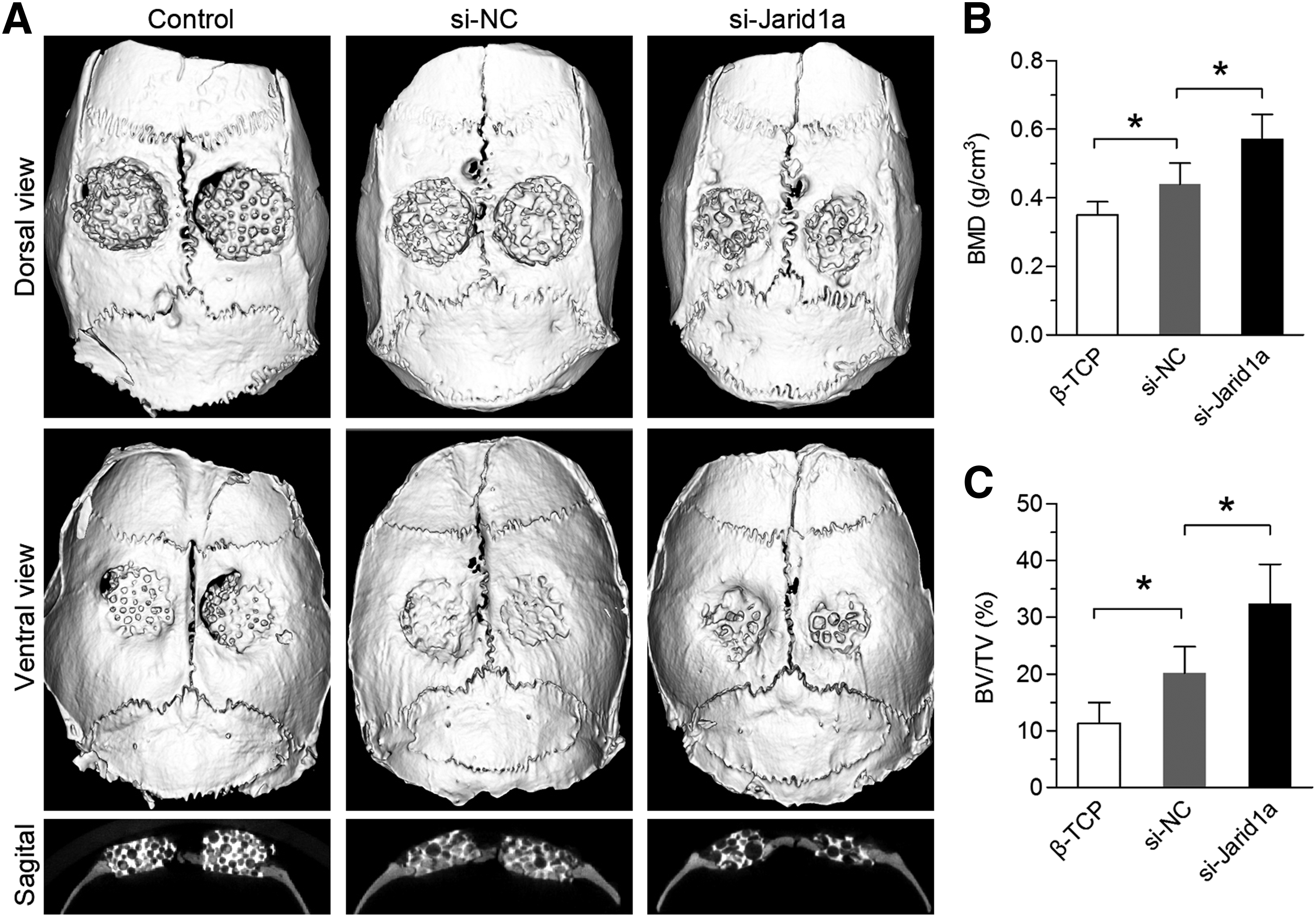
micro-CT evaluation of the repaired skull at 8 weeks postimplantation.
Histological analysis of bone regeneration
Histological evidence based on van Gieson staining of nondecalcified sections further supported the micro-CT findings (Fig. 6A). Based on the light microscopy, the percentages of new bone area at week 8 were 11.03% ± 1.81% in the β-TCP group, 24.05% ± 4.12% in the si-NC group, and 35.12% ± 4.82% in the si-Jarid1a group (Fig. 6B). The percentages of β-TCP residual area were 41.04% ± 6.2% in the β-TCP group, 32.1% ± 4.41% in the si-NC group, and 18.04% ± 2.42% in the si-Jarid1a group (Fig. 6C).
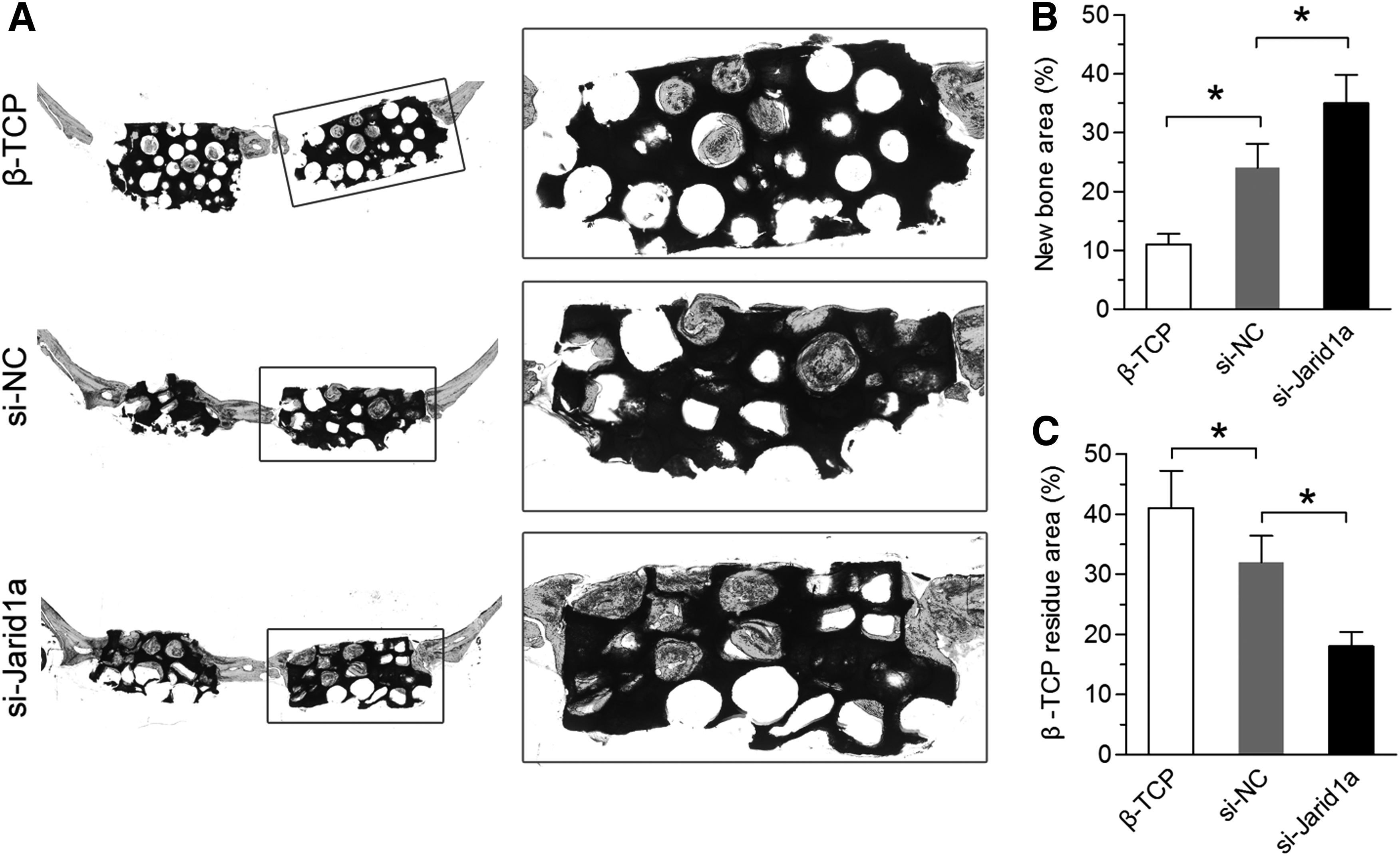
Histological analysis of newly formed bones and β-TCP residue in implants.
Discussion
Our data provided the evidence that histone demethylase Jarid1a physically and functionally interacted with Runx2 to repress BMSCs' osteogenic differentiation. Knockdown of Jarid1a enhanced osteogenic differentiation of BMSCs in vitro. ALP activity, matrix mineralization capacity, and bone-specific gene expression profiles revealed that inhibition of Jarid1a could promote osteogenic differentiation of BMSCs via regulation of osteogenic associated genes. Jarid1a-knockout BMSCs dramatically improved the repair of bone defect in vivo using tissue engineering technology. These preliminary observations are consistent with the current understanding of the repression effect of Jarid1a in adipose-derived stem cell differentiation. 14
Runx2 is an important osteoblast lineage-determining transcription factor involved in directing osteoblastic differentiation, and Runx2 appears to be the master gene in osteogenesis as it induces the expression of OCN, OSX, BSP, SATB2, and OPN, which are required in terminal osteoblast differentiation.2,19 Our data strongly suggested that Jarid1a played a major role in osteogenic differentiation and functioned in a Runx2-dependent manner, adding a new layer of control in the regulation of the epigenetic program of osteogenesis. Previous report showed Jarid1a as a potent transcriptional coactivator of Runx2 transcriptional activity suppressed bone marker gene expression in mouse calvarial osteoblasts. 20 But our results showed that the mRNA and protein levels of Runx2 were not affected by knockdown of Jarid1a in bone marrow stem cells of rats, and this was supported by a previous study. 14 These various results may be due to the species difference.
The role of Jarid1a in bone regeneration was evaluated by repairing a critical size bone defect model. Micro-CT showed that si-Jarid1a treatment could significantly enhance ossification in a calvarial model through the application of BMSCs. In contrast, only little new bone formation was observed in the si-NC and β-TCP groups. A quantitative analysis revealed that the new bone formation in the si-Jarid1a group was much higher than in the si-NC and β-TCP group, based on both BMD and BV/TV. In consistent with the above findings, a histological examination demonstrated that newly formed bone completely enriched the defect areas following the implantation of si-Jarid1a modified BMSCs, whereas there was only little new bone formation in the β-TCP group. This may be due to knockdown of Jarid1a promoting osteogenic differentiation of BMSCs, increasing the mineralization ability of osteoblasts, and enhancing the ability of BMSCs to produce new bone in vivo. Overall, our data demonstrated that Jarid1a plays an important role in bone regeneration by osteoinductive BMSCs.
In summary, our results demonstrate that Jarid1a inhibits osteogenic differentiation in a Runx2-dependent manner in vitro and knockdown of Jarid1a improves the potential of repairing bone defect by BMSCs in vivo. Our findings not only support the functional role of Jarid1a in osteogenic differentiation but also contribute to further understanding of the epigenetic regulation of Runx2-activated gene transcription. It may be possible to develop tissue selective Jarid1a inhibitors that can change epigenetic characteristics and promote osteogenic differentiation of BMSCs for the bone tissue engineering field. The limitation of the study is the lack of in vivo traces of transplanted BMSCs, which we will follow in subsequent studies.
Conclusion
In osteogenic differentiation, the demethylase Jarid1a negatively regulates BMSCs' differentiation by repressing the expression of osteogenic transcriptional factors. Jarid1a could directly interact with Runx2 and repress the expression of osteogenic transcriptional factors in a Runx2-dependent manner. Knockdown of Jarid1a enhances the potential of BMSCs in repairing bone defects in vivo. This study contributes to a better understanding of the molecular mechanisms of epigenetic regulation that govern the osteogenic differentiation of BMSCs, and also provides preclinical data which support the potential application of Jarid1a-knockdown BMSCs in promoting bone regeneration.
Footnotes
Acknowledgments
This work was supported by the National High Technology Research and Development Program (863 Program) (2015AA020311), the National Natural Science Foundation of China (81501605, 81570883, 81320108010), the Science and Technology Commission of Shanghai Municipality the Sailing Program (15YF1406800), and Chenguang Plan of Shanghai Education Development Foundation (16CG16).
Disclosure Statement
No competing financial interests exist.