
Abstract
Tissue engineering techniques provide a great potential to de novo construct a histological bladder. Smooth muscle regeneration is extremely important for the functional recovery of engineered neobladder. However, many challenges remain for the use of bladder smooth muscle cells (SMCs) as the cell sources. Recent evidences showed that smooth muscle progenitor cells (SPCs) in the peripheral blood have the capacity of differentiating into SMCs, while their use for bladder regeneration has not yet been reported. The aim of our study was to investigate the effect and mechanism of autologous SPCs on bladder regeneration in a rabbit model. In this study, autologous SPCs were isolated and cultured from the peripheral blood, labeled with CM-DiI, and then seeded into a porcine bladder acellular matrix (BAM) to construct a SPC-BAM complex, which was finally implanted to substitute the partial bladder with an equivalent size. In the results, SPCs demonstrated the phenotype of stem/progenitor cells, expressed SMs markers (alpha-smooth muscle actin [α-SMA], desmin, calponin, SM22α, and smooth muscle myosin heavy chain [SMMHC]), and displayed carbachol-induced contraction. Compared with cell-free BAM, the SPC-BAM was able to improve histological regeneration (smooth muscle regeneration, vascularization, and nerve formation) and functional recovery (urodynamic function and smooth muscle contraction) of the engineered neobladder. Cell tracing indicated that seeded SPCs could survive and directly integrated into the regenerated neobladder. In addition, SPCs could also promote proliferation and migration of rabbit bladder SMCs through the paracrine platelet-derived growth factor-BB (PDGF-BB). In conclusion, our study first demonstrated that SPCs from the peripheral blood could enhance histological regeneration and functional recovery of the tissue-engineered neobladder through both the direct integration and indirect paracrine effect, supporting the use of SPCs as the cell sources for tissue engineering of the bladder.
Introduction
T
Generally, there are two major approaches of bladder reconstruction by tissue engineering. The first approach involves a cell-free scaffold that serves as a support matrix to allow the natural process of regeneration to occur in vivo. When the scaffold, such as small intestinal submucosa or bladder acellular matrix (BAM), was used for bladder reconstruction in the cell-free approach, the urothelium could be well regenerated. However, only a low density of poorly organized smooth muscle tissue could be found in the central zone of the regenerated bladder. Bladder function was not well improved due to the graft shrinkage and scar formation in the regenerated bladder.6–8 Despite the improvement of the smooth muscle regeneration by the addition of bioactive factors into the scaffold, the bladder structure and function were still unsatisfied.9,10 It is of great importance to develop approaches to enhance smooth muscle regeneration so that a fully structural and functional bladder can be engineered.
The second approach utilizes a cell-based technique to in vitro construct a cell-seeded scaffold, which was then implanted for bladder reconstruction. Accumulated evidences indicated that cell-based tissue engineering is superior to the cell-free approach for bladder regeneration.11–13 Currently, autologous SMCs from bladder biopsy in patients were used to engineer a new bladder tissue in clinical applications.14,15 However, several limitations remain for the use of bladder SMCs as the cell sources.
First, isolation and culture of SMCs from the patients with muscle-invasive bladder cancer might have a high risk of contamination with tumor cells. Second, SMCs from the diseased bladder demonstrate abnormal phenotypic and functional characteristics, and might be inappropriate for being used as cell sources in tissue engineering.16,17 Third, the bladder SMCs might be dedifferentiated after a large expansion and lose their structure and function. 18 In addition, the harvest of bladder tissue is an invasive procedure that can cause infection, bleeding, and patient discomfort. At present, the need is real and urgent for development of new cell sources for bladder smooth muscle regeneration.
Recent evidences showed that smooth muscle progenitor cells (SPCs), which are considered the precursors of SMCs, can directly differentiate into functional SMCs.19–22 Transplantation of SPCs has been studied in tissue engineering and regenerative medicine applications. Liu et al. reported that SPCs were more efficient to enhance vascular regeneration in tissue-engineered blood vessels than mature SMCs. 23 Report also showed that the transplantation of SPCs, in a unilateral hindlimb ischemia model, could induce the production of angiotensin-1 and formation of a stable vascular network. 24 As SPCs can be easily harvested from the peripheral blood through a very minimally invasive procedure, circulating SPCs are regarded as the idea cell sources for engineering smooth muscle tissue. The current study, therefore, was performed to study the effect and mechanism of circulating SPCs on bladder regeneration. In this study, autologous SPCs were isolated and cultured from rabbit peripheral blood, and then seeded into the scaffold, porcine BAM, to construct a SPCs-BAM complex, which was finally implanted to substitute the partial bladder with an equivalent size. Histological and functional evaluations were performed to investigate the effect of SPCs on bladder regeneration. The mechanism of SPCs on smooth muscle regeneration was also studied. To the best of our knowledge, this was the first study investigating both the effect and mechanism of autologous SPCs on bladder regeneration.
Materials and Methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Affiliated Drum Tower Hospital, Nanjing University School of Medicine. The investigation was performed according to the institutional and national guidelines for laboratory animals. Totally, 25 male New Zealand white rabbits weighing 2.5–3.0 kg were used in this study. Five of them were used for SPC culture and characterization. The remaining 20 rabbits were used for bladder reconstruction with tissue engineering approaches. All the rabbits were housed individually in a temperature-controlled cage for a 12-h light–12-h dark cycle with well feeding.
Isolation and culture of SPCs and rabbit bladder smooth muscle cells
Rabbit peripheral blood (20 mL) was collected through cardiac puncture and mononuclear cells (MNCs) were separated from the buffy coat by density gradient centrifugation (Ficoll-Paque, density = 1.077 g/mL; GE Healthcare) as described previously. 25 MNCs were resuspended in the Endothelial Cell Growth Medium-2 (EGM-2; Lonza), which is the endothelial cell basal medium-2 (EBM-2) supplemented with the entire growth factor BulleKit and 5% fetal bovine serum (FBS), and then seeded on 60-mm diameter dishes precoated with 0.1% gelatin (Sigma-Aldrich). The medium was refreshed everyday for 3 days and was then changed with the SPC growth medium to facilitate cell differentiation. SPC growth medium was prepared by the addition of platelet-derived growth factor-BB (PDGF-BB, 40 ng/mL; PeroTech) and transforming growth factor-β1 (TGF-β1, 5 ng/mL; PeroTech) into EGM-2, with the absence of vascular endothelial growth factor (VEGF). The SPC growth medium was refreshed everyday until the emergence of SPCs and was then refreshed every 2 days.
Rabbit bladder smooth muscle cells (RBSMCs) were isolated and cultured according to a protocol modified from our previous studies.11,15,26,27 Briefly, the smooth muscle layer of the rabbit bladder was incubated in 0.25% Trypsin/0.038% EDTA solution at 37°C for 20 min, and then finely minced with scissors, followed by treating with 0.1% type I collagenase (Sigma-Aldrich) at 37°C for another 20 min. The cells were collected, washed, and cultured in Dulbecco's modified Eagle's medium/Ham's F12 (DMEM/F12; Gibco) medium supplemented with 10% FBS (Gibco). The medium was refreshed every other day.
The SPCs and RBSMCs were maintained at 37°C in a humidified atmosphere with 5% CO2 until subconfluence for subculture. Both the cells used in this study were at passage 2–5.
Real-time reverse transcription–polymerase chain reaction
For real-time reverse transcription–polymerase chain reaction (RT-PCR), the primer pairs were designed according to data from GenBank and determined by nucleotide blast standard search to avoid cross-reactivity with other known sequences (Table 1). The total RNA was extracted by using Trizol reagent (Invitrogen) and converted into complementary DNA (cDNA) with Prime Script® RT Master Mix (TaKaRa) according to the manufacturer's protocol. Real-time PCR of every gene was performed in triplicate in a 20 μL volume reaction system with the SYBR® Premix Ex Taq™ (Tli RnaseH Plus; TaKaRa). The thermal cycling conditions were 30 s at 95°C, followed by 40 cycles of 5 s at 95°C and 34 s at 60°C, using the Applied Biosystems 7500 real-time RT-PCR system. The real-time RT-PCR was repeated in both the SPCs and RBSMCs at passage 4 from five rabbits, and the relative amount of each gene normalized to the internal GAPDH was evaluated.
α-SMA, alpha-smooth muscle actin; SMMHC, smooth muscle myosin heavy chain.
Immunofluorescent staining
SPCs and RBSMCs at passage 4 were seeded onto glass slides precoated with 0.1% gelatin and cultured until 50–60% confluence. After fixation, permeability, and block, the cells were then stained with primary antibodies against α-SMA, desmin, calponin (Sigma-Aldrich), SM22α (Abcam), and SMMHC (Chemicon) at 4°C overnight, followed by staining with secondary antibody conjugated with Alexa Fluor 633 at room temperature for 30 min. Finally, the cell nuclei were counterstained by 4′-6-diamidino-2-phenylindole (DAPI; Molecular Probes). An IgG-matched isotype served as control for each antibody. The stained cells were examined using a fluorescent microscope (Zeiss).
Flow cytometric analysis
The SPCs at passage 4 were trypsinized, washed thrice, resuspended in phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA; 2 × 105 cells in 100 μL), and then incubated with primary antibodies at room temperature for 40 min in the dark. The conjugated and nonconjugated primary antibodies (Table 2) were used for direct and indirect flow cytometric (FCM) analyses, respectively. The conjugated primary antibodies were FITC-CD29, FITC-CD90, FITC-CD34, FITC-CD14, FITC-α-SMA, and PE-kinase insert domain receptor (PE-KDR). The nonconjugated primary antibodies were CD31, CD45, calponin, and PDGF receptor beta (PDGFR-β).
PE-KDR, PE-kinase insert domain receptor.
A fixation/permeabilization kit (BD Biosciences) was applied for intracellular staining (α-SMA and calponin). For indirect FCM analysis, cells incubated with nonconjugated antibodies were washed twice and then incubated with Alexa Fluor 488-conjugated secondary antibodies at room temperature for 30 min in the dark. All the labeled cells were washed twice, resuspended, and finally analyzed with FACSCaliber (BD Biosciences). IgG-matched isotypes were set for each procedure. FCM analysis was repeated in five rabbits and the average of positive cells was calculated.
Cell contractility assay
Carbachol-induced contractility of SPCs and RBSMCs was performed as a modified protocol previously described.21,28 Briefly, the SPCs and RBSMCs at passage 5 were cultured until 70–80% confluence, washed twice with PBS, and then stimulated 1 mmol/L carbachol for 1 min. Contractility of SPCs and RBSMCs was observed under an inverted microscope (Nikon). Photographs were taken before and after carbachol treatment in the same field.
Conditioned medium of SPCs and RBSMCs
SPCs and RBSMCs at passage 2–4 were cultured in 25 cm2 flasks until 90% confluence, washed twice with PBS, and refreshed with 2 mL of serum-free DMEM/F12. The cells were then cultured for 24 h, and the conditioned medium (CM) with secreted growth factors was centrifuged and collected. Serum-free DMEM/F12 was considered non-CM. Half of the CM or non-CM was treated with PDGF-BB antibody (0.1 μg/mL; R&D Systems) for 30 min to neutralize PDGF-BB. All the CM or non-CM with or without PDGF-BB antibody was used in the following cellular biological experiments. CM was prepared from SPCs and RBSMCs in five rabbits.
Cell proliferation assay
Cell proliferation assay was performed as our previous protocol.26,29 Briefly, RBSMCs at passage 2–4 (4 × 103/well) were seeded in quintuplicate onto 0.1% gelatin-coated 96-well plates. After attachment, the cells were serum starved for 48 h in basal medium (DMEM/F12 containing 0.5% FBS and 0.5% BSA) for cell cycle synchronization. Then, the cells were treated with the CM or non-CM with or without PDGF-BB antibody. The wells without cells served as blank controls. After culture for 48 h, 10 μL of Cell Counting Kit-8 (CCK-8; Dojindo) was added into each well, followed by incubation for another 3 h. Finally, the absorbance was measured at 450/620 nm on a microplate reader (Tecan). The cell proliferation assay was repeated five times.
Cell migration assay
Cell migration assay was performed using Hanging Cell Culture Inserts (8 μm pore size; Millipore) in 24-well plates as our previous protocol.26,29 Briefly, the undersurfaces of the insert filters were precoated with 0.1% gelatin for 30 min at 37°C. Subconfluent RBSMCs were serum starved overnight and released in the basal medium at the cell density of 5 × 105/mL. The bottom chambers were filled with the CM or non-CM with or without PDGF-BB antibody (1300 μL/well) and the upper chambers were filled with cell suspension (200 μL/well). The basal medium without cells was added to the upper chambers (200 μL/well) for using as blank controls (three wells for each condition). After incubation for 10 h, the filters were fixed and stained with 0.2% crystal violet (Sigma-Aldrich) for 20 min at room temperature. After several washes with PBS, nonmigrated cells on the upper surfaces were gently removed. Finally, the crystal violet in the migratory cells adherent on the under surface was eluted with 10% acetic acid. The absorbance of the blue dye mixture was measured at 570 nm (Tecan) and normalized to the non-CM group for quantitative analysis. This cell migration assay was repeated five times.
Enzyme-linked immunosorbent assay
Level of PDGF-BB in the CM was determined by enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) according to the manufacturer's instruction. The ELISA experiment was repeated five times.
Cell labeling
SPCs were labeled with the CellTracker™ CM-DiI (Molecular Probes) to track their fate after transplantation according to the manufacturer's instruction. Briefly, SPCs (cell density 1 × 106/mL) were incubated with 2 μg/mL CM-DiI for 5 min at 37°C and 15 min at 4°C, and finally washed twice for the subsequent experiments.
Cell-seeded BAM
A porcine BAM was prepared as our previous protocol described.9,27 After being labeled with CM-DiI, SPCs were seeded on a sterilized BAM at a concentration of 5 × 106 cells/cm2 and incubated for 30 min for cell attachment. The SPCs-BAM construct was then cultured in SPC growth medium for 1 week. The medium was changed every day. Cell growth in the BAM was evaluated before transplantation. A small part of the SPC-BAM construct was retrieved, embedded, and frozen at −20°C. Frozen tissue sections (6 μm) were stained with DAPI to confirm the cell distribution in the scaffold.
Partial bladder substitute
Twenty rabbits received partial bladder substitute as our previously described protocol. 9 Briefly, the rabbits were anesthetized with ketamine (50 mg/kg) and droperidol (2.5 mg/kg) intramuscularly before the surgery and the anesthesia was then maintained with ketamine and droperidol intravenously throughout the surgical procedure. After a midline lower abdominal incision, the bladder was exposed, dissected freely, and filled with 70–80 mL of sterile normal saline through urethral catheter. About 30–40% of bladder at size of 4 × 5 cm was removed, avoiding injury to the ureters and trigone, and replaced with an SPC-BAM construct with an equivalent size in the experimental group or with a cell-free porcine BAM patch in the control group. The nonabsorbable 4/0 silk sutures were used to identify the junction between the bladder native and the surrogate area. After making sure that no urine leakage was found, the abdominal wall and skin were then closed without using any drainage. Penicillin (40 MU, BID, intramuscularly) was administered postoperatively for 3 days. Appetite, mental status, and defecation of the rabbits were observed postoperatively. Animals of each group were scheduled to be sacrificed at 1, 3, and 6 months postoperatively.
In this study, the bladder substitute area was divided into marginal zone and central zone to analyze the contractive function of smooth muscle tissue, neovascularity, nerve formation, and smooth muscle regeneration in the neobladder tissues. This method could be more reasonable in evaluating the histological regeneration and functional recovery of tissue-engineered bladder and more conducive in analyzing the spatial distribution of bladder regeneration. 9
Urodynamic evaluation
Urodynamic evaluation was performed before and 1, 3, and 6 months after surgery, according to a protocol from our previous study. 11 Briefly, prewarmed sterile saline was infused into the bladder at a constant rate (10 mL/min) after the bladder was emptied. The volume of instilled saline and intravesical pressure were simultaneously monitored. When the first fluid leakage was observed around the catheter, the leak point pressure (LPP) was recorded. The total volume of instilled saline at this point was recorded as the bladder capacity. Bladder compliance was calculated by dividing the bladder capacity by LPP.
Muscle tissue contractility
Retrieved neobladder tissues from both the marginal and central zones at 6 months postoperatively were used for evaluation of muscle tissue contractility, according to our previously described protocol with a modification. 9 Native bladder tissues were used as normal controls. Briefly, the bladder tissue was immediately placed in ice-cold Krebs-Henseleit buffer (Sigma-Aldrich) after retrieval and opened along the lateral edges. The full-thickness strip (2 × 10 mm) of the bladder tissue was fixed with a microtissue holder in an organ bath (Chengdu Instrument, Chengdu, China), which contains Krebs-Henseleit buffer at 37°C, bubbled with 95% O2 plus 5% CO2, and connected to a force displacement transducer. Coupled with an isometric force transducer, the strips were allowed to equilibrate for 30 min. The strips were washed every 15 min, preadjusted to 0.6 g tension, and stimulated by electrical field or drug. Electrical field stimulation was applied with the parameters consisting of 15 V, 2 ms in duration, 90 Hz frequency, and 2-min interval between 15-s stimulation. The contraction was recorded for 1 min. After electrical field stimulation, the same strip was equilibrated in 10 mL fresh Krebs-Henseleit buffer for 5 min. Carbachol was then added by using 100 μL microsyringe (Eppendorf) near the oxygen bubbler at the final concentration of 100 μM. The evoked contraction was monitored for 2 min. In this study, both the electrical field at the frequency of 90 Hz and carbachol at the concentration of 100 μM are able to induce maximum contractile strength. 9
Histological evaluation
Bladder specimens were retrieved from both the marginal and central zones at 1, 3, and 6 months postoperatively. Specimens were fixed in 10% buffered formalin, dehydrated, and embedded in paraffin. Tissue sections (4 μm) were used for hematoxylin and eosin (HE) and Masson's trichrome (MT) stainings. In immunohistochemical staining, sections were deparaffinized, blocked, and incubated at 4°C overnight with α-SMA, desmin, and CD31 and S-100 antibodies, respectively. Subsequent reaction was done by using Dako REAL™ EnVision™ Detection System, Peroxidase/DAB, and the sections were counterstained with hematoxylin. For cell tracing, immunofluorescent staining was performed by using the central zone of the neobladder in the experimental group at 1 month after surgery. Frozen sections (6 μm) were blocked, incubated with α-SMA at 4°C overnight, and followed by being stained with IgG Alexa Fluor 488. Finally, DAPI staining was performed to detect the cellular nucleus.
Microvessel density (MVD) and microvessel area (MVA) were evaluated according to our previously described protocol. 9 The microvessel with a luminal structure was identified by staining with CD31 antibody.
Statistical analysis
All data were shown as mean ± standard deviation. The comparisons between control and experimental group were statistically analyzed by the independent Student's t test. The relationships between groups were evaluated by analysis of variance (ANOVA) tests. When ANOVA indicated a significant difference, groups were further compared by using the Least Significant Difference test. Differences were considered statistically significant at p < 0.05.
Results
Characterization of SPCs
Primary SPCs emerged after about 4 days of culture (Fig. 1A). At about day 7, SPC colonies were identified as elongated sprouting cells radiating from a central core of cobblestone-like cells (Fig. 1B). After subculture, the SPCs became predominantly spindle-shaped cells with a typical “hill and valley” morphology (Fig. 1C), which was similar to the primary RBSMCs (Fig. 1D).
Morphology of SPCs and RBSMCs:
Real-time quantitative RT-PCR assay, immunofluorescent staining, and FCM analysis were performed to determine the phenotype of SPCs. The results demonstrated that SPCs expressed the messenger RNA (mRNA) of smooth muscle-specific markers, α-SMA, desmin, calponin, SM22α, and SMMHC (Fig. 2A). Immunofluorescent staining further confirmed the positive expression of these above markers (Fig. 2B). In FCM analysis, SPCs showed positive for CD29 (92.52% ± 10.95%), CD90 (89.78% ± 8.84%), PDGFR-β (75.76% ± 12.98%), α-SMA (99.86% ± 0.17%), and calponin (98.68% ± 1.76%), but negative for CD14 (1.21% ± 0.48%), CD45 (0.99% ± 0.52%), CD31 (0.28% ± 0.12%), CD34 (0.04% ± 0.02%), and KDR (4.03% ± 2.31%), indicating that SPCs expressed the markers for both the progenitor cells and SMCs (Fig. 2C). In addition, the spindle-shaped SPCs displayed contraction in response to carbachol treatment, which was similar to RBSMCs (Fig. 3).
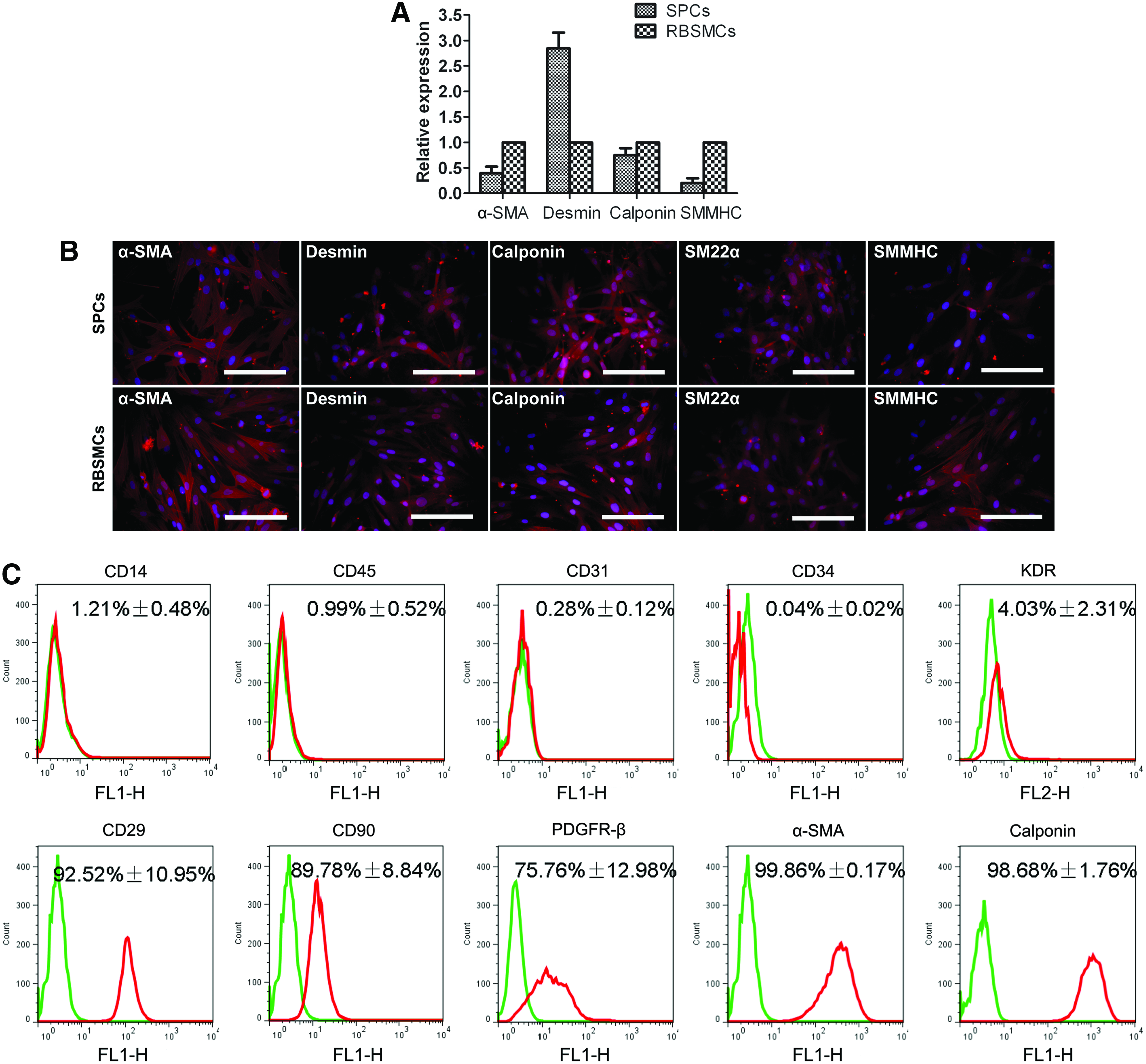
Characterization of SPCs:
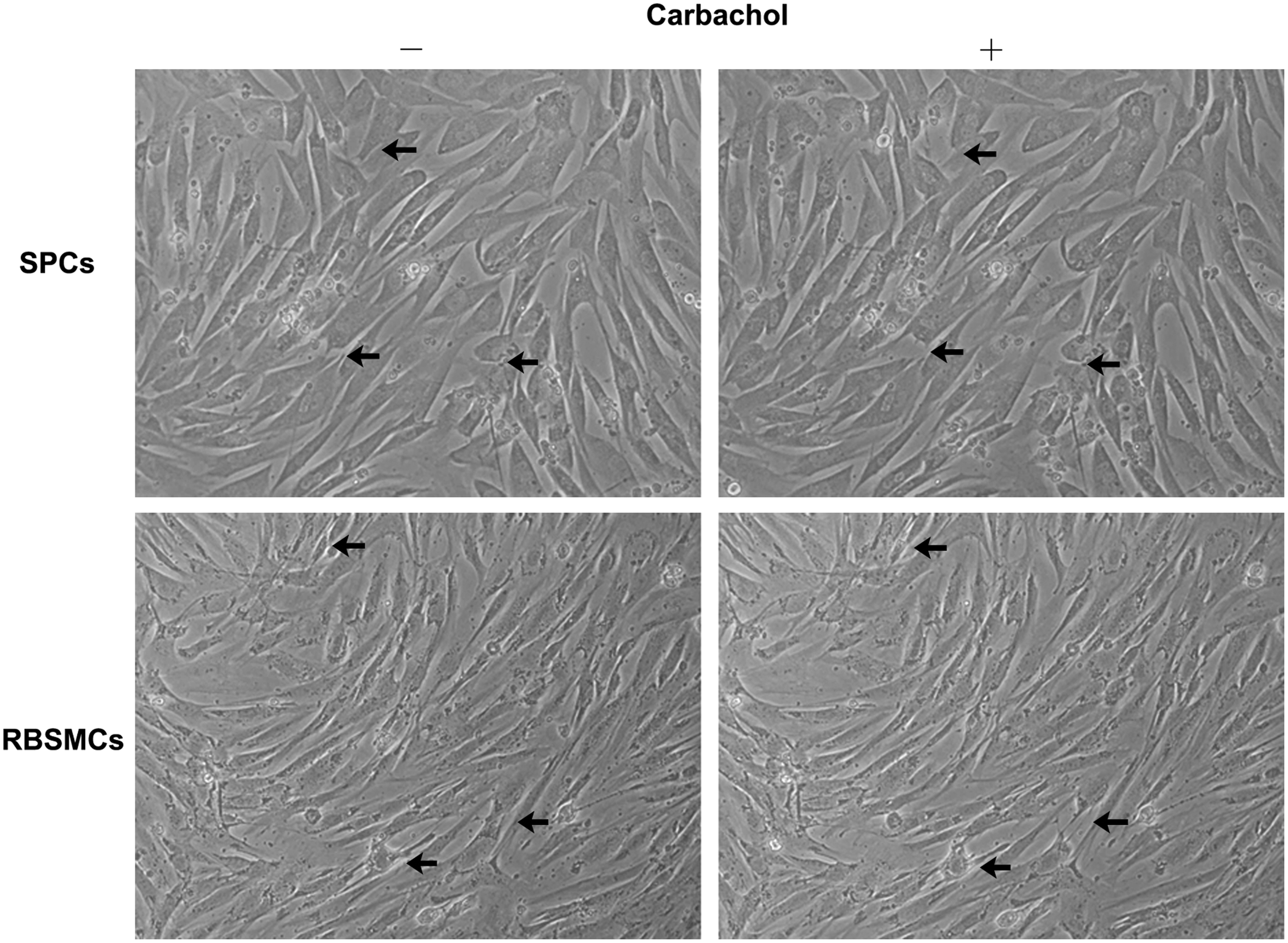
Carbachol induced contraction of SPCs and RBSMCs at passage 5.
Paracrine effect of SPCs
The CM was prepared and the paracrine effect of the growth factors secreted from SPCs and RBSMCs was evaluated through in vitro cellular biological approaches as our previously published protocol. 29 The results indicated that SPCs were able to promote the proliferative viability and migratory activity of RBSMCs through paracrine effect.
In cell proliferation assay, the proliferation viability of RBSMCs in the SPC CM group (109.8% ± 6.5%) was significantly enhanced compare with the RBSMC CM group (97.3% ± 5.5%) and non-CM group, while the values in non-CM group served as 100% (p < 0.05). The proliferation viability of RBSMCs was significantly inhibited by the addition of PDGF-BB antibody to SPC CM to neutralize the activity of PDGF-BB (99.0% ± 3.8%, p < 0.05) (Fig. 4).
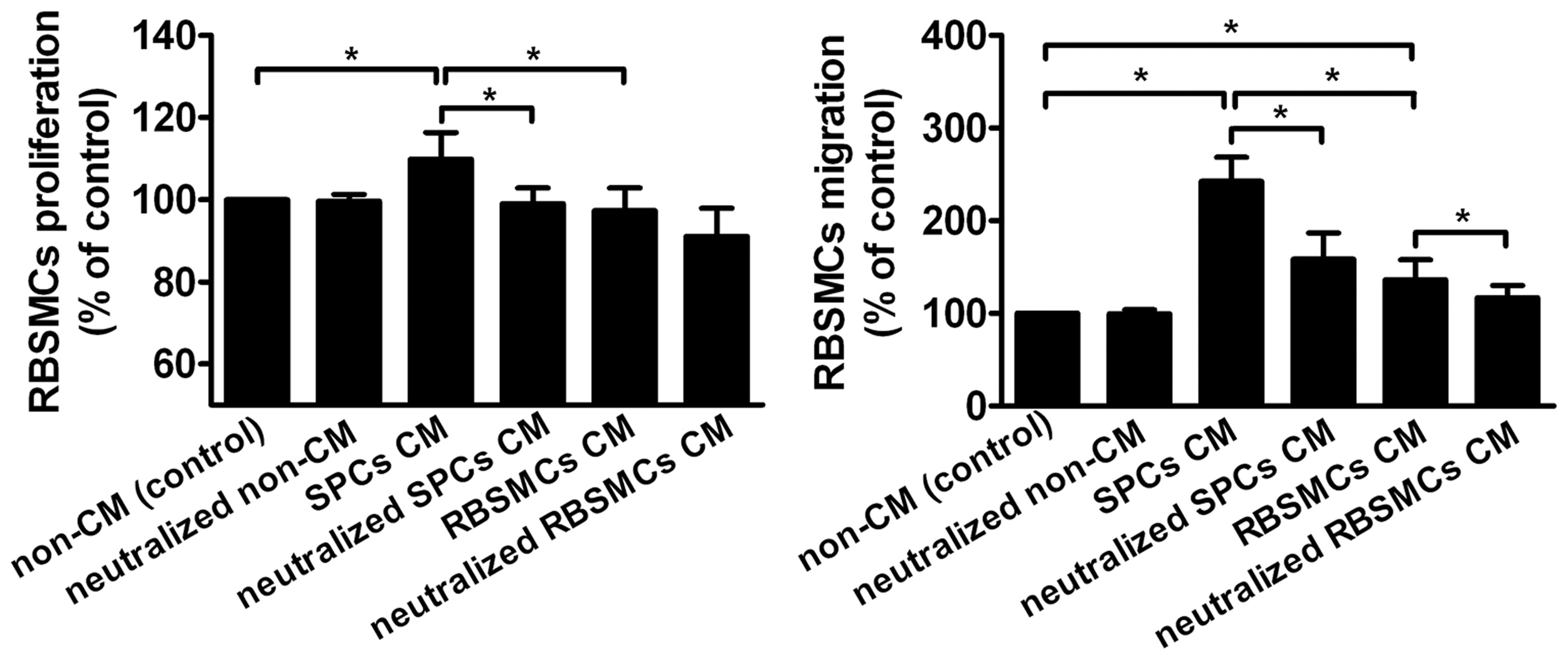
SPCs promoted proliferation and migration of RBSMCs (*p < 0.05).
In cell migration assay, the migratory activity of RBSMCs was significantly enhanced in both SPC CM and RBSMC CM groups compared with non-CM group, while the values in non-CM group served as 100% (p < 0.05). However, the migratory activity of RBSMCs in SPC CM group was significantly higher than RBSMC CM group (242.5% ± 26.1% vs. 136.2% ± 21.6%, p < 0.05). After the CM was added with PDGF-BB antibody to SPC or RBSMC CM to neutralize the activity of PDGF-BB, the migratory activity of RBSMCs was significantly reduced in both groups (158.6% ± 28.1% in neutralized SPC CM group and 116.8% ± 13.4% in neutralized RBSMC CM group, respectively) (Fig. 4).
ELISA was performed to investigate the secreted growth factors by SPCs. This study mainly investigated the secretion of PDGF-BB, as our previous study demonstrated it was the most potent factor for stimulating the activities of bladder SMCs. 26 Results showed that SPCs secreted significantly higher PDGF-BB than RBSMCs (178.2 ± 26.7 pg/mL vs. 35.7 ± 12.6 pg/mL, p < 0.01).
Macroscopic findings of regenerated neobladder
Of the 20 rabbits, 1 rabbit in the experimental group died after cardiac puncture for peripheral blood collecting and 2 rabbits in the control group died at 1 week and 3 months after surgery because of urinary leakage and stone formation, respectively. The other 17 rabbits survived their scheduled sampling time. After urodynamic studies were performed, the survived animals were anesthetized for the acquirement of bladder tissue and sacrificed at 1, 3, and 6 months postoperatively. Mild adhesions of the bladder to adjacent fat could be found in the two groups through gross examination. Graft shrinkage in the control group was more serious than those in the experimental group at 1, 3, and 6 months (16.3% ± 2.5%, 29.5% ± 3.5%, and 44.0% ± 4.4% vs. 10.3% ± 1.5%, 19.7% ± 3.1%, and 24.7% ± 2.5%, p < 0.05). Bladder stone formation was found obviously in five animals of control group, accompanied with local inflammatory hyperplasia of epithelium, while none presented in the experimental group. However, central graft calcifications were found in three of control group and four of experimental group (Fig. 5).

Rabbit bladder was injected with maximum normal saline preoperation until micturition
Functional recovery of regenerated neobladder
Urodynamic studies
Urodynamic studies were performed to evaluate the time course of functional status in the two groups at each time point. Preoperative bladder capacity and compliance demonstrated no significant difference between the experimental and control groups, and were considered 100% values. After surgery, bladder capacity and compliance in each group increased gradually as times passed. At 6 months after surgery, bladder volume and intravesical pressure curve showed the regenerated neobladder in experimental group executed a safe urine storage reservoir with larger capacity and lower pressure than in control group (Fig. 6A). Bladder capacity in the experimental group was higher than that of the control at 1, 3, and 6 months (of preoperative; 69.7% ± 4.2%, 86.5% ± 2.4%, and 98.1% ± 2.1% vs. 58.9% ± 3.5%, 68.1% ± 2.9%, and 71.1% ± 2.7%, p < 0.05). Bladder compliance of the experimental group was also superior to that in the control group at 1, 3, and 6 months (of preoperative; 60.1% ± 2.9%, 80.4% ± 3.7% and 95.7% ± 2.5% vs. 48.2% ± 3.2%, 63.2% ± 2.3% and 64.1% ± 2.8%, p < 0.05) (Fig. 6B).

Functional recovery of regenerated neobladder evaluated by urodynamic and organ bath studies.
Organ bath studies
The contractile ability of normal bladder tissue and the regenerated neobladder tissue retrieved at 6 months postoperation in both the experimental and control groups was evaluated with electrical field stimulation and carbachol interference (Fig. 6C). The tension of muscle strip from normal bladder tissue was considered 100%. Under carbachol interference, the muscle strip in both the marginal and central zone of the neobladder in the experimental rabbits exhibited stronger contraction than that in the control rabbits (of normal, 7.11% ± 4.41% vs. 25.94% ± 7.67% in central zone, p < 0.05; 21.76% ± 7.57% vs. 67.36% ± 8.82%, p < 0.05). Similarly, muscle strip in both the marginal and central zone of the neobladder in the experimental rabbits exhibited stronger contraction than that in the control rabbits under electrical field stimulation (of normal, 12.37% ± 9.28% vs. 42.27% ± 15.57% in central zone, p < 0.05; 34.02% ± 9.28% vs. 71.13% ± 18.56%, p < 0.05). However, the tension of muscle strip in the experimental rabbits was still weaker than that in the normal bladder (Fig. 6D).
Histological staining of regenerated neobladder
Urothelium formation
Multilayered urothelium was found in the marginal and central zones of the regenerated neobladder in the experimental and control groups at 1 month postoperation by HE and MT stainings (Fig. 7). At 6 months after operation, the lumen of the neobladder was still covered with multilayered urothelium.
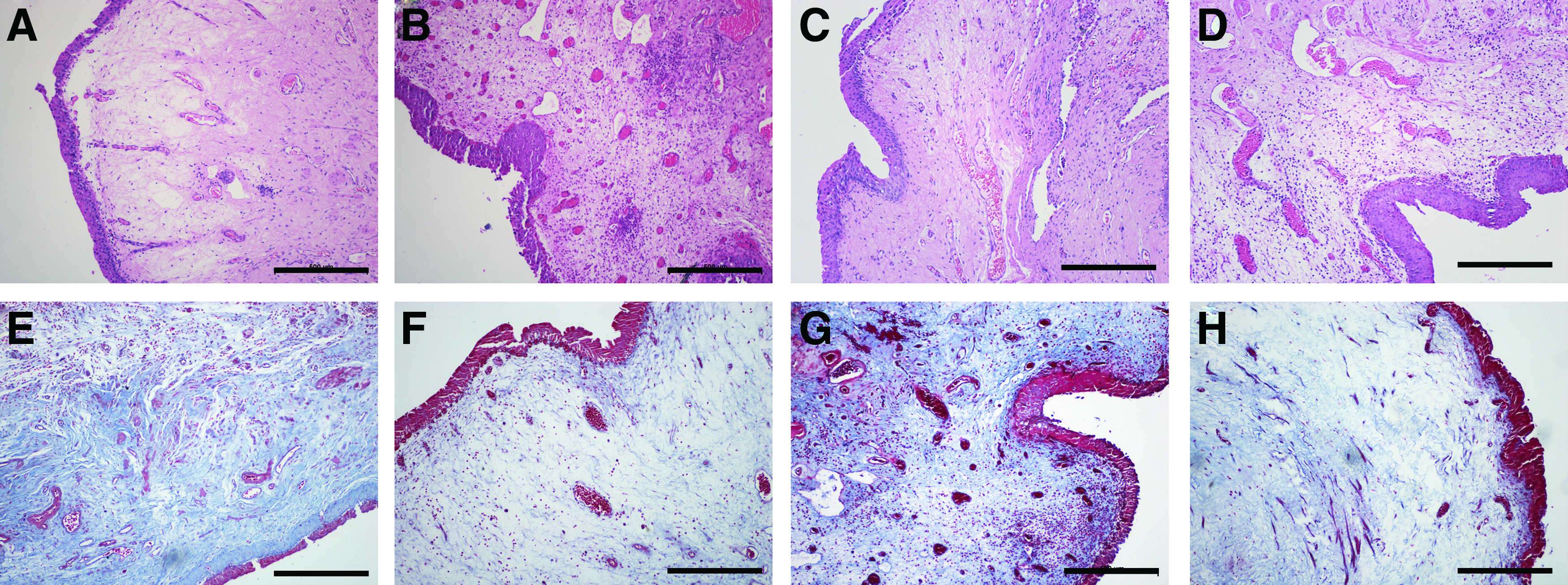
HE
Neovascularity
Vascular structures stained positive for CD31 were found in the two groups and gradually increased over time. MVD and MVA in both the central (Fig. 8) and marginal (Fig. 9) zone of the neobladder tissues in the experimental group were significantly higher than those in the control group at each time point (MVD: 15.0 ± 1.8, 23.0 ± 3.6, and 31.0 ± 3.1 vs. 8.0 ± 1.5, 11.0 ± 2.1, and 18.0 ± 2.5 in central zone, p < 0.05; 24.0 ± 2.6, 53.0 ± 4.6, and 62.0 ± 3.9 vs. 14.0 ± 2.1, 22.0 ± 2.3, and 29.0 ± 3.5 in marginal zone, p < 0.05; MVA: % of total area: 1.22% ± 0.27%, 2.51% ± 0.43%, and 3.97% ± 0.62% vs. 0.71% ± 0.13%, 1.05% ± 0.39%, and 2.08% ± 0.54% in central zone, p < 0.05; 3.31% ± 0.65%, 4.91% ± 0.51%, and 6.75% ± 0.87% vs. 1.08% ± 0.21%, 2.21% ± 0.49%, and 4.21% ± 0.61% in marginal zone, p < 0.05).

Immunohistochemical staining of central zone of the neobladder with antibody against CD31 in control and experimental groups at 1, 3, and 6 months after surgery
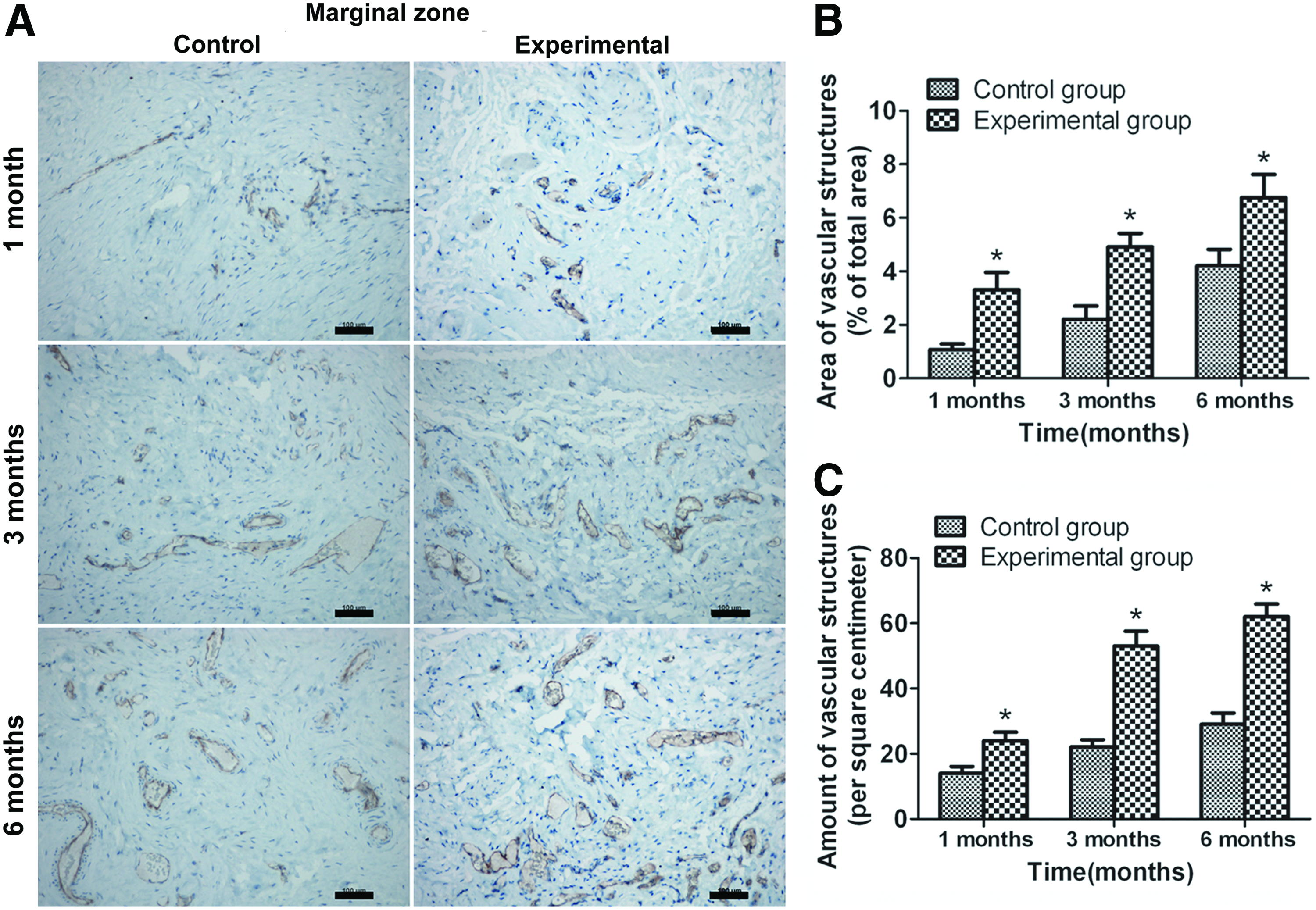
Immunohistochemical staining of marginal zone of the neobladder with antibody against CD31 in control and experimental groups at 1, 3, and 6 months after surgery
Nerve formation
To observe the nerve formation, retrieved bladder tissue was stained with S-100 antibody. In the marginal zone of neobladder, nerve fibers were found in both groups at 1 month after surgery, and increased at 3 and 6 months. S-100-positive nerve fiber in the experimental group was observed more abundantly than that in the control group (9.0 ± 1.8, 14.0 ± 2.1, and 22.0 ± 3.7 vs. 17.0 ± 2.9, 23.0 ± 3.6, and 35.0 ± 3.9, p < 0.05). However, in the central zone of neobladder, no nerve fiber was found at 1 month. In addition, although nerve fibers increased at 3 and 6 months, no significant difference was found between the two groups (p > 0.05) (Fig. 10).

Immunohistochemical staining of central zone
Smooth muscle formation
Regenerated bladder tissue was retrieved and stained with α-SMA and desmin antibodies to detect the formation of smooth muscle bundle. It was found that regenerated smooth muscle of the marginal and central zone gradually increased with time in the two groups. At 6 months after surgery, the formation of smooth muscle buddle of the neobladder in the experimental group showed more regular than that in the control group. Area of organized smooth muscle positive for α-SMA of the marginal and central zone in the experiment group was significantly higher than that in the control group at each time point (% of total area: 10.31% ± 1.31%, 16.19% ± 3.01%, and 26.48% ± 3.81% vs. 4.7% ± 0.61%, 8.18% ± 1.23%, and 12.86% ± 1.41% in marginal zone, p < 0.05; 5.98% ± 0.64%, 12.09% ± 1.75% and 18.03% ± 1.92% vs. 1.16% ± 0.41%, 7.13% ± 1.51%, and 11.56% ± 1.21% in central zone, p < 0.05) (Fig. 11). Meanwhile, area of organized smooth muscle positive for desmin of the marginal and central zone in the experiment group was also significantly higher than that in the control group at each time point (% of total area: 9.23% ± 1.19%, 15.09% ± 3.21%, and 24.38% ± 3.13% vs. 4.3% ± 0.57%, 7.28% ± 1.23%, and 11.96% ± 1.2% in marginal zone, p < 0.05; 5.12% ± 0.49%, 10.31% ± 1.41%, and 16.89% ± 1.68% vs. 1.09% ± 0.38%, 6.52% ± 1.29%, and 10.17% ± 1.08% in central zone, p < 0.05) (Fig. 12).

Immunohistochemical staining of central zone

Immunohistochemical staining of central zone
In addition, the central zone of the neobladder in the experimental group at 1 month was evaluated with immunofluorescent staining against α-SMA. The fluorescence of CM-DiI could be detected in the regenerated bladder tissue. Dual-positive fluorescence for CM-DiI and α-SMA indicated that SPCs may directly form smooth muscle in the neobladder after surgery (Fig. 13).

Fluorescence of CM-DiI-labeled SPCs (red)
Discussion
The main purpose of bladder tissue engineering is to construct a fully structural and functional neobladder. As is known, the urinary bladder is a muscular organ with the function of storing and excreting urine, which relies on the relaxation and contraction of bladder smooth muscle. Therefore, the full regeneration of smooth muscle layer is in urgent need of bladder reconstruction. However, despite a variety of methods being attempted by numerous investigators, the density and arrangement of regenerated smooth muscle were still unsatisfied.6,9,30,31 It is highly desirable to develop a useful method to accomplish the successful smooth muscle regeneration for tissue engineering bladder. Actually, one of the key problems for bladder regeneration is to seek an ideal SMC source that can promote the smooth muscle regeneration. SPCs are considered the precursors of SMCs and reported to have the capacity of enhancing vascular smooth muscle regeneration, 23 which indicates their potential applications for tissue engineering and regenerative medicine. However, the use of SPCs for bladder regeneration has not yet been reported. In this study, SPCs were isolated, characterized, and seeded onto the BAM to construct a SPC-BAM complex, which was then used to replace partial bladder. The tissue-engineered neobladder was evaluated by histological and functional studies. Both the role and mechanism of SPCs on smooth muscle regeneration were investigated.
Study reported that SPCs expressed PDGF-β, while the PDGF-β signaling plays pivotal roles in the differentiation of SPCs.
32
Simper et al. first demonstrated the culture of SPCs in human peripheral blood with the induction of PDGF-BB.
20
Another research reported that TGF-β1 combined with PDGF-BB might accelerate the establishment of functional SMCs.
22
Therefore, in this study, both PDGF-BB and TGF-β1 were used to isolate and culture SPCs from rabbit peripheral blood. Cultured SPCs expressed mesenchymal and SMC markers, and were capable to contract under carbachol treatment. Cell contraction is the vital physiological function of SMCs, which was important to maintain the function of muscular organ. The capability of SPs contraction may be due to the functional
According to our previous study, the regenerated bladder tissue was divided into marginal zone and central zone to, respectively, observe the histological and functional recovery of tissue-engineered neobladder, which could be more preferable to analyze the spatial distribution of bladder regeneration. 9 Results showed that multilayered urothelium could be well accomplished in both the central and marginal zone of the two groups at 1 month after surgery, which was in accordance with previous studies. 9 Meanwhile, the regenerated smooth muscle bundle and blood vessel in both the central and marginal zone of the experimental rabbits were significantly higher than that in the control ones. Our previous study indicated that delivery of exogenous growth factor might be limited to establish adequate blood supply for bladder regeneration, since the formation of vascular network only depended on the ingrowth of vessels from adjacent native tissue. 9 In this study, seeded SPCs significantly improved the revascularization rate of the regenerated bladder tissue. One of the reasons is that SPCs might participate in the vascular smooth muscle regeneration due to the important contribution of SPCs on vascular repair and remodeling. 33 The other might be that SPCs could produce the angiogenic bioactive factors and then promote the formation of vascular network.24,34
As is known, neuronal network is crucial to effectively regulate the function of urinary bladder. In this study, we also evaluated the regeneration of nerves fibers by staining with S-100, which is a neural marker expressed in peripheral nerve cells and could be observed in tissue-engineered bladder to detect neural cells. 35 In this study, S-100-positive nerves fibers significantly increased in the marginal zone of the experimental group, while the increase was not found in the central zone. Study showed that regenerated nerves fibers most likely originated from the host peripheral nerve after in vivo implantation with urine-derived stem cells expressing VEGF-loaded collagen hydrogels. 36 Due to the limitation of cell proliferation, satisfied nerve formation in the central zone of regenerated bladder is difficult when a large bladder area is reconstructed. Future studies should be committed to improve the neurogenesis of tissue-engineered neobladder. The application of neurotrophic factor or stem cells that could differentiate into neural cells may be a good method.37,38
It is reported that SMCs from native bladder biopsies could facilitate reconstitution of functional urinary bladder by tissue engineering.11,15 Study also showed that SMCs from peripheral blood had equivalent capability with SMCs from native bladder to establish a histological native-like urinary tissue. 39 In this study, urodynamic studies and radiographic cystograms were performed to observe the functional recovery of tissue-engineered neobladder.
In addition, the contractility of neobladder muscle tissue was evaluated by organ bath studies. Results showed that SPCs from peripheral blood demonstrated promotion effect on functional recovery of tissue-engineered neobladder and muscle contractility of neobladder tissue. Therefore, circulating SPCs can be used as the potential cell source for the construction of functional urinary bladder by tissue engineering.
Up to now, a variety of cell sources, including bladder SMCs,11,15 embryoid body-derived stem cells, 40 bone marrow-derived stem cells,41,42 adipose tissue-derived stem cells,35,43 human umbilical mesenchymal stem cells, 44 and so on, have been found to have the capability of facilitating smooth muscle regeneration in tissue-engineered bladder. However, the exact mechanism of this process was not well documented. Theoretically, stem cells seeded onto the scaffold could enhance tissue regeneration through direct and indirect approaches. Study indicated that, after a cell-based scaffold was implanted, seeded stem cells may differentiate into mature SMCs and then take part in the regeneration of smooth muscle layer in tissue-engineered bladder. 42 To distinguish the origination of the regenerated smooth muscle from the seeded stem cells or native bladder SMCs, cell tracing technique might be beneficial for this issue. 45 In this study, we labeled SPCs with CM-DiI, a widely used cell labeling fluorescent dye that could be detected up to 4 weeks after labeled cells being transplanted in vivo. 46 Results showed that CM-DiI-labeled cells could be detected in the regenerated bladder tissue at 1 month after surgery and expressed α-SMA, which suggested that the regenerated SMCs might partially derive from the implanted SPCs. However, the quantity of CM-DiI-labeled SMCs is still limited in the regenerated tissue. The main reason may be the static culture condition of SPCs in BAM that limit the ingrowth of SPCs into BAM. Since the dynamic culture condition is superior to the static condition in nutrition supply and cell metabolism, it is needed to study the effect of dynamic culture on cell growth in BAM in the future. 47
Studies showed that stem/progenitor cells could secret a variety of cytokines and growth factors that had trophic effect in tissue repair.48,49 Among these growth factors, PDGF-BB is reported to be the most potent factors for stimulating the activities of bladder SMCs. 26 Binding of PDGF-BB into the scaffolds could significantly promote the proliferation of SMCs. 9 In this study, SPC-seeded acellular scaffolds could significantly enhance the smooth muscle density in both marginal and central zone of regenerated neobladder. SPCs also significantly promoted the proliferation and migration of RBSMCs in vitro, which could be significantly inhibited by the addition of PDGF-BB antibody to SPC CM. In addition, SPCs were demonstrated to secrete significantly higher PDGF-BB than RBSMCs. Therefore, the main reason of SPC-based bladder regeneration may be partly due to the secretion of PDGF-BB. However, since many kinds of growth factors can regulate the activities of bladder SMCs, 26 future study is needed to evaluate whether SPCs could secrete other kinds of growth factors that may be involved in SPC-based bladder regeneration.
Conclusions
In this study, we successfully isolated and cultured SPCs from rabbit peripheral blood. SPCs expressed both mesenchymal and SMC markers, and possessed the capability of contraction. SPCs were seeded onto BAM to construct an SPC-BAM complex, which was then implanted to replace partial rabbit bladder. It is found that implanted SPC-BAM promoted smooth muscle regeneration, vascularization, and nerve regeneration, as well as functional recovery of tissue-engineered bladder. The implanted SPCs can be directly involved in the regenerated bladder smooth muscle tissues. In addition, SPCs can secret PDGF-BB, which promote proliferation and migration of RBSMCs, suggesting that SPCs may promote bladder regeneration through paracrine method (Fig. 14).

The role and mechanism of SPCs in the process of promoting smooth muscle regeneration of tissue-engineered bladder. Color images available online at www.liebertpub.com/tea
Footnotes
Acknowledgments
This study was supported by the National Natural Science Foundation of China (31500785, 31570993, and 31100702) and the Jiangsu Provincial Medical Youth Talent of the Project of Invigorating Health Care through Science, Technology and Education (QNRC2016071).
Disclosure Statement
No competing financial interests exist.