
Abstract
Induced pluripotent stem cells (iPSCs) have been shown to differentiate to functional cardiomyocytes (CM) with high efficiency through temporally controlled inhibition of the GSK3/Wnt signaling pathways. In this study, we investigated the ability of temporally controlled release of GSK3/Wnt small-molecule inhibitors to drive cardiac differentiation of iPSC without manual intervention. Porous silica particles were loaded with GSK3 inhibitor CHIR99021 or Wnt inhibitor IWP2, and the particles containing IWP2 were coated with 5 wt% poly(lactic-co-glycolic acid) 50:50 to delay release by ∼72 h. iPSCs reprogrammed through mRNA transfection were cultured with these particles up to 30 days. High-performance liquid chromatography suggests a burst release of CHIR99021 within the first 24 h and a delayed release of IWP2 after 72 h. Annexin V/propidium iodide staining did not show a significant effect on apoptosis or necrosis rates. Cultured cells upregulated both early (Nkx 2.5, Isl-1) and late (cTnT, MHC, Cx43) cardiac markers, assayed with a quantitative real-time polymerase chain reaction, and began spontaneous contraction at 3.0 ± 0.6 Hz at 15–21 days after the start of differentiation. CM had clear sarcomeric striations when stained for β-myosin heavy chain, and showed expression and punctate membrane localization of gap junction protein Connexin43. Calcium and voltage-sensitive imaging showed both action potential and calcium transients typical of immature CM. This study showed that the cardiac differentiation of pluripotent stem cells can be directed by porous silica vectors with temporally controlled release of small-molecule inhibitors. These results suggest methods for automating and eliminating variability in manual maintenance of inhibitor concentrations in the differentiation of pluripotent stem cells to CM.
Introduction
C
Induced pluripotent stem cells (iPSC) represent a renewable and scalable cell source for cardiac tissue engineering applications. 4 Small-molecule inhibition of the GSK3/Wnt inhibitory pathways in iPSC resulted in cardiac differentiation efficiencies of up to 98%. 5
Current procedures for differentiating iPSC into CM involve manual media changes and inhibitor addition. A method of limiting handling interactions would be ideal for clinical translation of iPSC to cardiac cells. Our ultimate goal is to create a self-differentiating material that requires minimal handling interactions. In this study, we combined iPSC with particles that regulated the release of inhibitors of the GSK3 and Wnt pathways.
Porous silica particles (pSi) were tailored to release small-molecule inhibitors at desired timepoints. Drugs loaded into pSi alone without any coatings are released in a burst release, with all the drugs dispersed within 24 h. 6 pSi have a high loading efficiency, retain drug bioactivity, and can be coated with biocompatible polymers. 7 Polymers such as poly(lactic-co-glycolic acid) (PLGA) have been used widely in drug delivery platforms because of their tunable release kinetics based on co-polymer ratios and exposure of the hydrolysable chemical backbones.8–10 pSi have been shown to load and deliver hydrophobic small molecules for therapeutic purposes.11,12
Amniotic fluid-derived stem cells (AFSC) have been widely researched for treatment of congenital heart diseases13–15 ; however, AFSC cannot directly differentiate into functional beating CM. 13 Our laboratory has previously reprogrammed AFSC by modified mRNA transfection and subsequently differentiated AFSC-iPSC into functional beating cardiac cells. 16 Using small-molecule-loaded pSi, AFSC-iPSC could effectively differentiate into cardiac cells. The temporal exposure to the GSK3/Wnt inhibitors was shown to be critical for efficient cardiac differentiation of iPSCs, 4 with GSK3 inhibition at day 0 and Wnt inhibition at day 3 of differentiation. Therefore, our pSi differentiation platform had two formulations of particles aiming at mimicking this inhibitory cascade, with the GSK3 inhibitor loaded within pSi particles alone and the Wnt inhibitor loaded into pSi and further coated with PLGA to delay release kinetics. Differentiation was evaluated in vitro for cardiac differentiation. Cells were cultured in transwells to maintain consistent inhibitor exposure released from particles as well as to maintain cellular waste/nutrient exchange. Differences between cardiac cells derived from direct exposure to GSK3/Wnt inhibitors and those exposed to particles containing the same inhibitors were evaluated through differentiation efficiency, phenotypic/genotypic analysis, and electrophysiology.
Methods
Cell source
Cells were isolated from second-trimester human amniotic fluid and reprogrammed to iPSC through mRNA transfection as previously reported. 14 Pluripotency was assessed through RNA expression of pluripotency markers OCT4, nanog, and Tra-1-81 with teratoma formation in a nude mouse as previously reported. 16 Reprogrammed AFSC were then passaged onto a mouse embryonic fibroblast (MTI-Globalstem, Gaithersburg, MD) feeder layer (1.85 × 10 4 cells/cm2) for further expansion and maintained in knockout serum replacement iPSC media (Dulbecco's Modified Eagle's media/F12, 20% knockout serum replacement, 1% non-essential amino acids, 1% Pen/Strep, 0.1% beta-mercaptoethanol, 4 ng/mL basic fibroblast growth factor; Thermo Fisher Scientific, Waltham, MA). At 60–70% confluency, colonies were passaged to new mouse embryonic fibroblast feeder layers, every 7–10 days. All studies of primary human cells were approved by the Institutional Review Boards of both Baylor College of Medicine and Rice University, and subjects gave informed consent.
pSi synthesis
A tannic acid template and the Stöber process were used to synthesis pSi as per previous studies. 17 Briefly, 272 mg tannic acid (Sigma Aldrich, St. Louis, MO) was dissolved in 50 mL ethanol while continuously stirring. Then, 25 mL ammonium hydroxide (Sigma Aldrich) was added and stirred for 1 min. Tetraethyl orthosilicate (300 μL; Sigma Aldrich) was added to the mixture dropwise and stirred continuously for 3 h. The resulting particles were centrifuged and washed in 1:1 ethanol:water for a total of six washings. The particles were then resuspended in 15 mL of deionized water and filtered twice through 40 μm centrifuge tube filters. Particles were centrifuged again; the water was removed and then resuspended in 1 mL isopropyl alcohol. The particles were then dried overnight in a vacuum oven at 60°C and −36 Pa until dry. Once dry, 4 mg of pSi was rehydrated in 0.5 mL inhibitor loading solutions, consisting of CHIR99021 (Tocris bioscience, Avonmouth, Bristol, United Kingdom) or IWP2 (Tocris bioscience) re-suspended in dimethyl sulfoxide at 600 μg/mL for 20 min at 37°C with constant mixing. The particles were then washed and lyophilized for further use.
PLGA encapsulation of pSi
Particles loaded with IWP2 were further encapsulated with a PLGA coating to delay the release kinetics by using a Solid-in-Oil-in-Water (S/O/W) method. An oil solution consisting of dissolved 5 wt% 50:50 PLGA (Lactel Absorbable Polymers, Pelham, AL) in dichloromethane was homogenized with the prepared loaded pSi. The solid in oil emulsion was then added dropwise to a water solution consisting of 2.5% polyvinyl alcohol. The solution was allowed to stir continuously for 6 h, washed, and lyophilized until ready for use.
Particle characterization
Dynamic light scattering (DLS) and Z-potential of pSi were performed by using a Zetasizer ZEN3600 (Malvern, Worcestershire, United Kingdom). Moreover, samples were prepared for a scanning electron microscope (SEM) by drying overnight on a stage and sputter coating with a 5 nm thick layer of Pt/Pl. SEM images were taken by using Nova NanoSEM 230. PLGA-pSi and pSi size distribution was also measured with the software ImageJ (NIH Image). Transmission electron microscope samples were prepared by drying nanoparticles onto 300 mesh carbon-coated copper grids and then analyzed. Fluorescein isothiocyanate (FITC)-labeled pSi has been used to verify the presence of the silica inside the PLGA shell by confocal microscopy.
Inhibitor loading efficiency and release studies
All supernatants after initial loading of inhibitors were stored at −20°C. Supernatants were analyzed by reverse-phase high-performance liquid chromatography (HPLC). Inhibitor release studies were done by sampling particle suspension inhibitor release up to 6 days. Briefly, drug release solution was prepared by using 1 mL of 1 × phosphate-buffered saline (PBS) and 1 mL of 1% bovine serum albumin. The lyophilized pSi were suspended at a concentration of 3 mg/mL of drug release solution and incubated at 37°C with constant stirring. At 1, 3, 5, 12, and every 24 h thereafter, suspensions were centrifuged and 1 mL drug release solution was taken and replaced with fresh drug release solution. Agilent Zorbax Eclipse Plus C18 (100 × 4.6 mm, 3.5 μm pore particle size; Agilent Technologies, Santa Clara, CA) column was used for analysis. Samples flowed through the column at 1.0 mL/min and with a mobile phase consisting of acetonitrile in 0.1% trifluoroacedic acid: H20 in 0.1% trifluoroacedic acid (V/V). Samples were analyzed at 281 nm.
Cellular toxicity to particles
pSi without any inhibitors loaded were added to iPSC cultures at a density of 2.5 ng/cell to assess apoptosis and necrosis. Annexin V and propidium iodide staining were used to quantify apoptosis/necrosis. Cells were lifted at 0 and 72 h, washed, and double stained with Annexin V-FITC and propidium iodide (Affymetrix ebioscience, Santa Clara, CA). The cells were then analyzed through flow cytometry (BD Acurri C6 Plus; BD biosciences, San Jose, CA). To assess the effect of transwell culture on cell survival, live/dead staining was done according to the manufacturer's protocol (Thermo Fisher). Cells were imaged by using fluorescent microscopy.
Differentiation of iPSC using dissolved GSK3/Wnt inhibitors
By adapting previously published protocols, 5 AFSC-iPSC were differentiated into cardiac cells by temporally inhibiting the GSK3 and Wnt signaling pathways over a span of 5–7 days. On reaching ∼70% confluency, undifferentiated colonies were dissociated in collagenase type 2 (Worthington Biochemical Corp., Lakewood, NJ) for 5 min, then manually dislocated from the feeder layer, dispersed into single-cell suspension, and finally plated as a monolayer of cells onto Matrigel (BD Biosciences) at 260,000 cells/cm2. Cells were expanded for 3 days in mTeSR1 media (STEMCELL Technologies, Cambridge, MA). Medium was then changed to RPMI 1640 (Thermo Fisher) and the GSK3 inhibitor, CHIR99021, was added at a concentration of 6 μM, representing differentiation day 0. After 24 h, medium was replaced with fresh RPMI 1640. At day 3, the Wnt inhibitor, IWP2, was added to RPMI/B27 without insulin at a concentration of 2.5 μM. At day 7, insulin and ascorbic acid were added to the RPMI 1640 media. The occurrence of beating colonies was monitored through phase-contrast microscopy after day 7.
Differentiation of iPSC using pSi-released inhibitors
To test the effectiveness of inhibitor release from loaded pSi, dissociated iPSC were plated at 260,000 cells/cm2 onto Matrigel-coated 12-well polyethylene terephthalate ThinCert cell culture inserts (Greiner Bio-One, Monroe, NC) with 0.4 μm pore size (Fig. 3A). This dosing for pSi differentiation was calculated based on the two-dimensional (2D) differentiation with dissolved inhibitors. The calculated dosages for the 2D differentiation with dissolved inhibitors are ∼67.5 μg CHIR99021 and 25 μg IWP2 per million cells, and, therefore, representative amounts of both particle formulations were used based on calculated loading efficiencies: ∼1.5 mg CHIR99021-loaded pSi and 4.0 mg IWP2-loaded PLGA-pSi were suspended in 24 mL of RPMI 1640. At day 0 of differentiation, 1 mL of particle suspension was added to each seeded transwell and an additional 1 mL RPMI 1640 surrounded the insert. Every 48 h, 1 mL of fresh RPMI1640 replaced the media surrounding the insert. After day 8, insulin and ascorbic acid were added to the RPMI 1640 and 1 mL of media was added to each of the inserts and surroundings.
Flow cytometry
Cells were detached at day 12 into suspension with Accutase (ThermoFisher) and stained with a fluorescently conjugated antibody for cardiac troponin T (BD Biosciences) at a dilution of 1:100. BD Acurri C6 Plus software (BD Biosciences) was used for all flow cytometry data collection. FlowJo software (Tree Star, Inc., Ashland, OR) was used for data analysis.
Gene expression analysis
Gene expression was quantified by using a quantitative real-time polymerase chain reaction (qRT-PCR). To assess the upregulation of early- and late-stage cardiac markers, cell samples were collected at 0, 1, 5, 10, and 20 days after the start of differentiation. At each timepoint, RNA samples were collected by using an RNA collection kit following the manufacturer's protocol (Affymetrix, Santa Clara, CA). mRNA samples were then reverse transcribed to DNA by using a cDNA kit following the manufacturer's protocol (Applied Biosystems, Foster City, CA). The resulting DNA samples were analyzed with qRT-PCR by using a proprietary assay system following the manufacturer's protocol (Affymetrix). All samples were assayed for expression of Oct4, Isl1, Nkx2.5, cTnT, connexin 43, myosin heavy chain, and GAPDH as a house keeping gene by using DNA primers (Thermo Fisher). The expression of each gene was first normalized to the level of GAPDH. Relative fold changes were determined by calculating ΔΔCt compared with day (−3) or day 0. Biological triplicates for each group were assessed, and results were reported as mean ± standard deviation.
Immunofluorescence
Cell cultures were fixed in 4% paraformaldehyde (Alfa Aesar, Ward Hill, MA) at 4°C for 20 min. Fixed cells were permeated with Triton X100 (Sigma-Aldrich) for 5 min at room temperature. Next, cells were incubated with specific antibodies (Abcam, Cambridge, United Kingdom) for cardiac markers myosin heavy chain and connexin 43, then in DyLight-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Cells were imaged by using an epifluorescence microscope (DMI 6000B; Lieca Microsystems, Wetzlar, DE).
Calcium handling and membrane voltage potential
The calcium handling of spontaneously contracting cells was measured by imaging calcium-sensitive dye Indo-1 AM (Thermo Fisher) with an epifluorescence microscope (Olympus, Center Valley, PA) and a photomultiplier tube detection system and software (Ion Optix, Westwood, MA). Cells were washed with PBS at 37°C, replaced with Tyrode's buffer containing 2 μL of 2 mM Indo-1 AM, and allowed to incubate for 30 min at room temperature. The cells were then washed three times with fresh Tyrode's buffer and analyzed by using the Ion Optix system. The measured field of view was ∼50 μm × 50 μm in size. Detection wavelengths were recorded at 405 and 485 nm. Membrane voltage potential utilized the same procedure as calcium measurements but with voltage-sensitive dye Di-8-ANEPPS (Thermo). Detection wavelengths were recorded at 560 and 620 nm.
Peak shortening percentage analysis
Cells were recorded on spontaneous contraction from day 20 to 30 by using phase-contrast time-lapse video microscopy (Nikon Eclipse TE300, Melville, NY). Images were taken every 100 ms with a CoolSNAP HQ2 CCD camera (Photometrics, Tuscon, AZ) and analyzed with ImageJ. Minimum representative rectangles were used to outline individual contracting cells, and the lengths of the smallest and largest rectangles were used to calculate the peak shortening percentage.
Western blot
Western blot antibodies were purchased from Abcam, Inc.; electrophoresis, transfer blots, and enhanced chemo-luminescence (ECL) developing materials were purchased from Bio-Rad (Hercules, CA). Total protein lysates were collected from cells at 30 days of differentiation. Total protein concentration was quantified by a bicinchoninic acid kit (Thermo Scientific, Rockford, IL). Samples were normalized based on total protein concentration and run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels according to the manufacturer's protocols. Proteins were transferred to blotting membranes and then incubated overnight at 4°C with mouse monoclonal antibodies against MHC and cTnT (1:100 dilution in 10% milk in Tris Buffered Saline with 0.5% Tween 20 [TBST]) and mouse monoclonal antibodies against GAPDH (1:1000 dilution in 10% milk in TBST). The membranes were washed and incubated with secondary horseradish peroxidase antibodies (1:1000 in 10% milk in TBST) for 1 h at room temperature. The membranes were washed, then immersed in an ECL developing solution, and finally read by using Bio-rad ChemiDoc XRS+Statistics.
Statistical analyses were done in SigmaPlot (Systat Software, Inc., San Jose CA). Data were compared by using one-way analysis of variance followed by Tukey's test; p < 0.05 was considered significant. Results were presented as mean ± standard deviation, with number of samples/trials indicated in captions.
Results
pSi characterization and GSK3 and Wnt inhibitor release from pSi
The PLGA-pSi particles measured 8.24 ± 3.25 μm in diameter, and uncoated pSi particles measured 265 nm per DLS. Zeta potentials for pSi and PLGA-pSi were −13.1 ± 0.42 and −39.6 ± 1.42, respectively (Fig. 1F). CHIR99021 and IWP2 were loaded into silica particles by direct absorption from a 600 μg/mL solution. HPLC data showed a burst release profile with 81.1 ± 5.3% of CHIR99021 released within the first 24 h (Fig. 1G). After the initial 24 h, CHIR99021 continued to be released through day 7 with nearly 100% cumulative release at 144 h.

pSi characterization.
When IWP2 was encapsulated within the pSi and coated with 5 wt% PLGA 50:50, the results showed a cumulative release of 27.8 ± 7.2% within the first 48 h (Fig. 1). The particles were shown to have 40.2 ± 9.1% cumulative release at the end of 72 h. After the 144 h, IWP2 was shown to have a cumulative release of 56.4 ± 7.2%.
Cellular toxicity with pSi exposure
Forward scattering of annexin V-labeled cells measured by flow cytometry showed that 27.3 ± 2.1% of cells internalized uncoated pSi and 3.61 ± 1.4% of cells internalized PLGA-pSi after 3 days in culture (Fig. 2A). Uncoated pSi and PLGA-pSi were tested for cellular toxicity through an annexin V and propidium iodide assay (Fig. 2B). After 3 days, annexin V staining of iPSC exposed to pSi loaded with both GSK3 and Wnt inhibitors (12.5 ± 1.9%) showed no significant difference compared with the cells alone (10.1 ± 3.3%), suggesting no increase in apoptosis. Propidium iodide staining also showed no significant difference between iPSC exposed to pSi loaded with both inhibitors (3.1 ± 1.6%) and cells alone (4.1 ± 2.5%), suggesting no increase in cell necrosis.

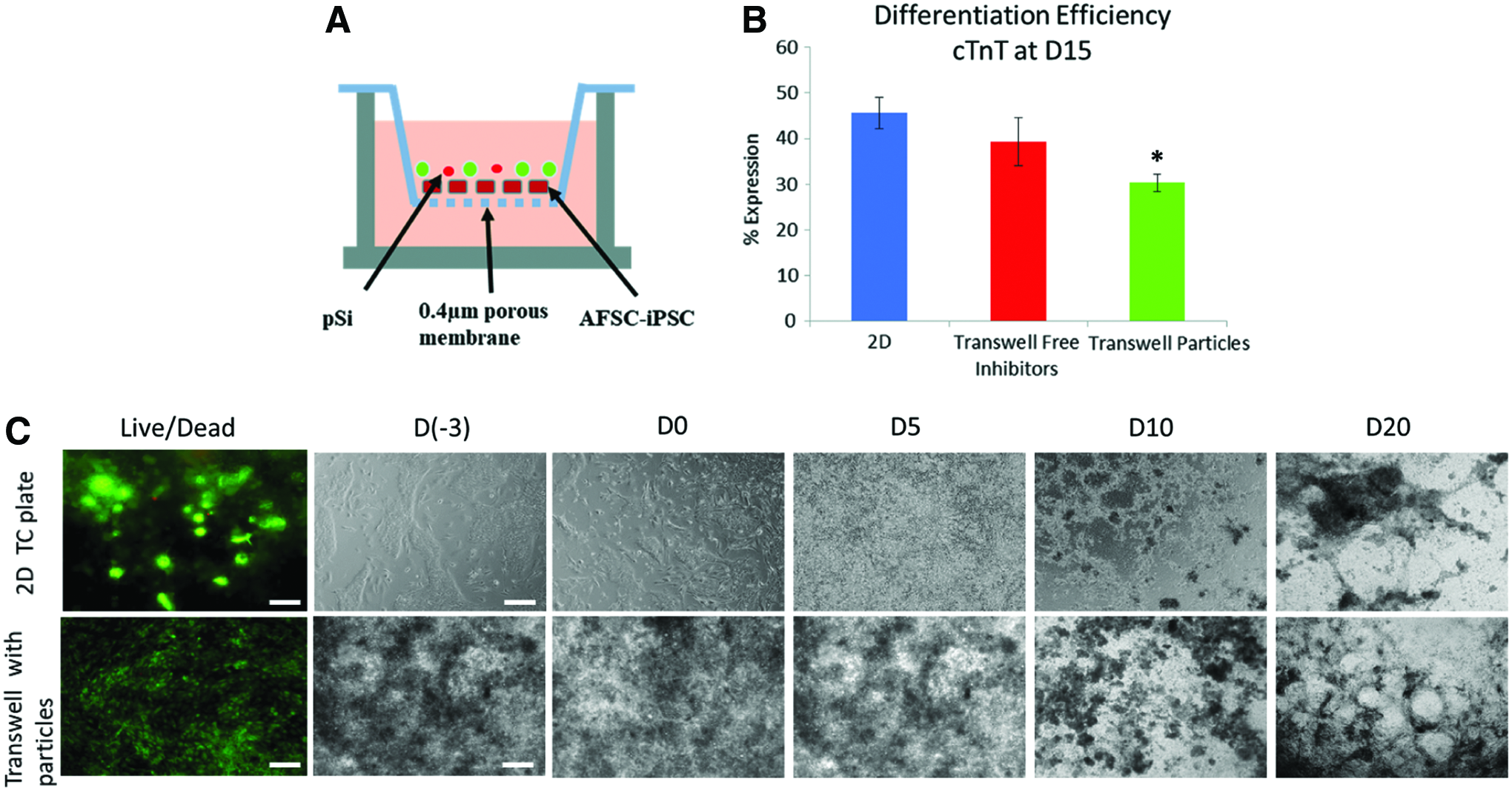
AFSC-iPSC cardiac differentiation by pSi.
Cardiac gene expression in pSi differentiation comparable to dissolved inhibitors
Neither the 2D monolayer culture nor transwell culture showed any significant difference in viability after 3 days in culture. The morphology of differentiating cells in transwells at day 0 showed denser cell localization and less cell spreading compared with 2D monolayer differentiation. During the first 5 days of differentiation, the 2D monolayer differentiation had a phase-dark layer of fibroblastic cells below a layer of spherical phase-bright cells, whereas a phase-bright cell layer was absent in transwell culture. At timepoints greater than 10 days, both groups exhibited a clustered morphology with elongated web-like sheets. Spontaneous beating occurred in all groups at ∼18–25 days of differentiation (Supplementary Video S1; Supplementary Data are available online at www.liebertpub.com/tea). Localized beating colonies were observed in all groups and were not synchronously paced.
cTnT expression showed 30.3 ± 1.9% differentiation efficiency in pSi differentiated cells at day 15. Differentiation efficiency was greater in both 2D and transwell dissolved inhibitor groups with 45.6 ± 3.5% and 39.3 ± 5.3%, respectively (Fig. 3B).
pSi released inhibitor differentiation expressed markers of cardiac differentiation
Cardiac differentiation was monitored through gene expression of brachyury, early-stage cardiac markers (nkx2.5 and isl-1), and later-stage cardiac markers (cTnT, β-MHC, Cx43). Pluripotent stem cell marker OCT4 was also monitored throughout the differentiation to quantify remaining pluripotency in the cell population.
Brachyury expression was measured 24 h after the introduction of CHIR99021 in both conditions. The relative expression compared with day (−3) of differentiation showed that all groups had little to no brachyury expression at the start of differentiation, but after 24 h all groups showed significant upregulation. The 2D monolayer differentiation and transwell dissolved inhibitors had a fold change increase of 236.3 ± 6.7, whereas the transwell with inhibitor loaded particles showed a significant fold change increase of 99.3 ± 27.1 (Fig. 4B).

Cardiac marker expressions during differentiation.
When compared with day (−3), OCT4 expression was shown to be downregulated over the course of differentiation in all groups (Fig. 4C). At day 10 and 20, OCT4-fold expression in all conditions was below 0.06 ± 0.01 and 0.007 ± 0.006, respectively.
Early-stage cardiac markers, Nkx2.5 and Isl-1, were upregulated throughout the differentiation compared with day 0 (Fig. 4D, E). At each of the timepoints, Nkx2.5 expression was lower in the transwell groups compared with the 2D monolayer group. When compared with the other timepoints within groups, day 10 was shown to have significant upregulation of Nkx2.5 (Fig. 4D).
Cardiac troponin T was shown to be upregulated as early as day 5 in the transwell group with free inhibitors when compared with day 0 (Fig. 4F). However, significant upregulation of cTnT was only shown in the transwell with dissolved inhibitors group at day 20 of differentiation. β-MHC was upregulated at day 10 in the 2D monolayer group, whereas later timepoints showed β-MHC to be significantly upregulated across all groups (Fig. 4G). At day 30, the transwell group with pSi had the highest expression of β-MHC. Gap junction protein connexin 43 expression was not significantly different compared with day 0 at each timepoint in all groups (Fig. 4H).
Early cardiomyocyte characteristics and arrangement
Immunofluorescence staining showed expression of late-stage cardiac markers β-MHC and connexin 43 in pSi differentiation at day 30 (Fig. 5). Western blot analysis supports the translation of upregulated late-stage cardiac proteins in the pSi differentiation group comparable to the 2D differentiation group (Fig. 6B).
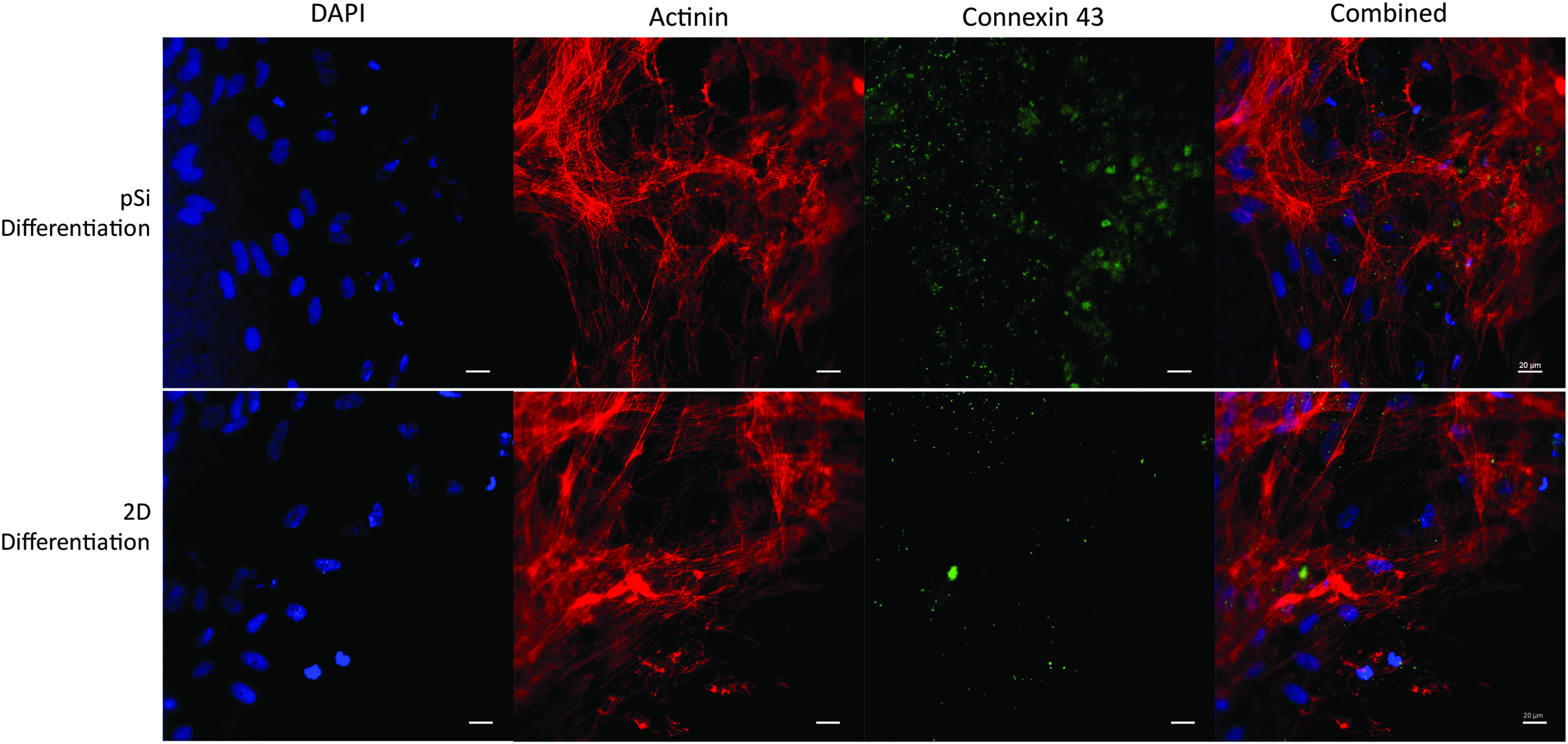
Confocal immunofluorescence of differentiated AFSC-iPSC. Top: Cells differentiated with pSi particles, Bottom: Cells differentiated in 2D with dissolved inhibitors. Actinin (red), Cx43 (green), DAPI (blue) (Scale 20 μm). 2D, two-dimensional; DAPI, 4′,6-diamidino-2-phenylindole. Color images available online at www.liebertpub.com/tea
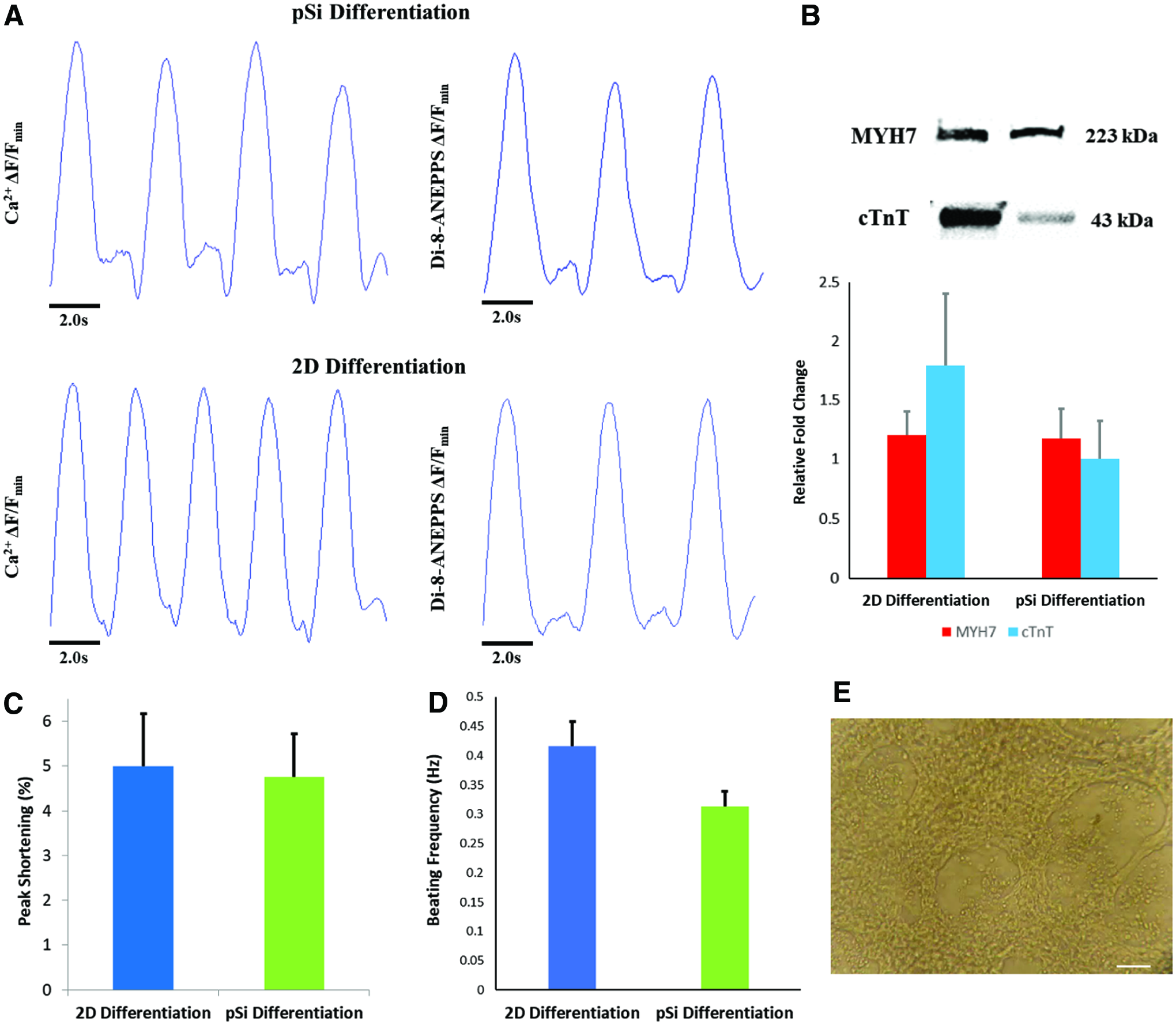
Traces of spontaneously beating cardiac cells.
Electrophysiological activity and peak shortening of pSi differentiated cells
Both control and experimental groups had spontaneously beating cells at days 18–21. Calcium handling measurements showed that pSi differentiated cells spontaneously beat at a frequency of 0.31 ± 0.03 Hz (Fig. 6A). Differentiation with dissolved inhibitors resulted in cells with a beating frequency of 0.48 ± 0.05 Hz (Fig. 6A). Contractile cell peak length shortening was not significantly different between dissolved inhibitor and pSi differentiation groups at 5.0 ± 1.2% and 4.8 ± 1.0%, respectively.
Discussion
In this study, we have shown that GSK3/Wnt inhibitors released from pSi can be used to differentiate iPSC into cardiac cells. Previous studies showed highly efficient cardiac differentiation by temporal exposure to dissolved GSK3/Wnt inhibitors, where direct exposure to the GSK3 inhibitor (CHIR99021) occurs during the first 24 h and subsequent exposure to the Wnt inhibitor (IWP2) occurs between days 3 and 5.4,18 Here, we showed that controlling the release of inhibitors by pSi encapsulation could produce spontaneously contracting cardiac cells, minimizing handling interactions.
iPSC did not exhibit cellular apoptosis or necrosis when exposed to pSi. Previous studies have shown that internalization of pSi can induce cell programmed death.19,20 Therefore, the preservation of iPSC cellular activity was likely due to minimal particle internalization. Both pSi formulations were internalized in iPSC but did not show a significant difference in apoptosis and necrosis compared with controls. This was likely due to the high metabolic nature of iPSC and lack of cellular waste exchange for 72 h will incite apoptosis.
Inhibitors were able to direct cardiac differentiation after release from pSi. The release profiles from pSi were not binary and, therefore, had a sustained release past day 1 and 5 for GSK3 and Wnt inhibition, respectively. Temporal regulation of these inhibitors has been previously shown to be crucial for efficient cardiac differentiation, but a sustained release produces cardiac cells with less efficiency. 21 Our data support these findings as we showed an average pSi differentiation efficiency of 30.3%. Calcium handling and membrane potentials show contractility of pSi differentiated cells similar to that of direct inhibitor differentiation. A lower rate of spontaneous contraction in pSi differentiated cells may have resulted from sampling of differing populations of contractile cells or delayed measuring of transient intensities, allowing the cultures to decrease in temperature. Future studies showing the beating frequency at different timepoints after the start of spontaneous beating will help suggest any electrophysical development over time.
Although plating density was the same in all groups, the differentiating cellular morphologies were different. Previous studies have shown that single iPSC can sense micro and nanoscale topographies altering their pluripotency.22,23 The rounded and clustered appearance of cells within the transwells was likely due to differences in substrate topography and may have promoted maintenance of pluripotency in a population of cells. After 10 days from the start of differentiation, the morphology between the two groups became more similar, with beating colonies forming in localized areas. In terms of cardiac maturation, the morphology of differentiated cardiac cells did not show a clear pattern of cellular alignment. Although immunofluorescent imaging and western blot analysis confirmed β-MHC and connexin 43 were present in differentiated cells, they ultimately lacked sarcomeric pattering and organization. Continued culture and mechanical/chemical conditioning may be required for maturation.24,25
Genetic expression of cardiac differentiation markers in pSi differentiation showed comparable results to direct inhibitor differentiation. The release of CHIR99021 from uncoated pSi was sufficient in transitioning pluripotent iPSC into the mesendoderm linage, and further Wnt inhibition by IWP2 loaded pSi upregulated late-stage cardiac markers. Peak expressions of β-MHC and connexin 43 were later in pSi differentiated cells compared with the control. Although the cumulative dosage of pSi released inhibitors was similar to that of direct addition of inhibitors, 6 μM CHIR99021 at day 0 and 2.5 μM IWP2 at day 3, the non-binary and sustained release may have delayed cardiac maturation. Alterations to the pSi release kinetics could be altered to further increase cardiac efficacy. The downregulation of OCT4 within all groups of differentiation is important from a translational perspective, signifying reduced risk of pluripotent cells that are able to form teratomas. 26
A major limitation of this study is the significantly lower differentiation efficiency compared with that of fibroblast-derived iPSC. Previous studies have shown differentiation efficiency variability based on cell line.27,28 This may result from innate differences in gene expression or epigenetic traits. AFSC-derived iPSC have been previously shown to have a lower differentiation efficiency compared with fibroblast-derived iPSC lines. 16 Therefore, pSi AFSC-iPSC differentiation was not expected to increase differentiation efficiency compared with the dissolved inhibitor control. A high-throughput screening of differing GSK3/Wnt inhibitors should also be investigated in future studies to possibly enhance differentiation capabilities. This could elucidate more efficient signaling inhibitors and, in turn, would require more dosage optimization if used in this pSi delivery platform. This platform used on a different cell line may produce greater differentiation efficiencies with a less heterogenous population of resulting cells.
Although the burst release of CHIR99021 closely mimicked direct addition of the small molecule at day 0, there were still minimal concentrations over the course of cardiac differentiation. The release of IWP2, on the other hand, mimicked a more delayed and sustained release. Optimization of release parameters can also help to direct a more efficient cardiac differentiation. This could be done by altering the degradation kinetics of the particles in terms of polymer ratio and coating thickness. This would ensure the proper dosage of inhibitors per area of cell. Another limitation to this study is the diffusion of waste products. Transwell culture allowed for the isolation of iPSC and pSi, but elimination of cellular waste was limited to simple diffusion. Further development of this platform would benefit from a bioreactor ensuring sufficient exchange of nutrients.
Conclusion
This study shows that small-molecule inhibitors temporarily released from pSi can direct cardiac differentiation of iPSC. The dual release of GSK3 and Wnt inhibitors from pSi is a simple and effective way to generate spontaneously beating cardiac cells. This self-directing cardiac differentiation platform has potential in future cardiac tissue engineering applications.
Footnotes
Acknowledgments
This work was supported in part by grants from the American Heart Association (14BGIA18750004 to J.G.J.), the National Science Foundation (CBET-1547838 to J.G.J.), the National Institutes of Health (1R01HL130436-01 to J.G.J.), and Texas Children's Hospital.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.