
Abstract
Successful human spermatogonial stem cell (hSSC) culture could enable cell therapy for male infertility. Mammalian extracellular matrix (ECM) promotes mitogenesis, migration, and/or differentiation of various stem/progenitor cells, and can plausibly facilitate hSSC survival in culture. Hydrogel forms of human testicular ECM (htECM), porcine testicular ECM (ptECM), porcine small intestinal submucosa ECM (SIS), and porcine urinary bladder ECM (UBM) were used to coat tissue culture plates for hSSC culture. In addition, hSSCs were cultured on Sandos inbred mice (SIM) 6-thioguanine-resistance, ouabain-resistant (STO) mouse embryonic fibroblast feeder cells (control), murine laminin, or human laminin. Undifferentiated embryonic cell transcription factor 1-positive (UTF1+) human spermatogonia were quantified at days 0, 7, and 14 of culture. htECM was the only condition that retained a significantly higher number of UTF1+ cells than control STO feeder cell cultures (22% vs. 3%). Overall, the number of hSSCs declined during the 14 day culture period under all conditions. A multiparameter flow cytometry analysis of cells cultured on htECM and ptECM revealed that stage-specific embryonic antigen 4+ undifferentiated spermatogonia may be lost to differentiation (cKIT+ spermatogonia) and apoptosis (annexin V+ spermatogonia). Proliferation of undifferentiated human spermatogonia (Ki67+) was limited, suggesting that hSSCs may have different growth factor requirements than mouse SSCs. ECM from the homologous species (human) and homologous tissue (testis) was the most effective substrate for hSSCs, and establishes a foundational feeder-free, serum-free condition for future iterative testing of culture conditions toward the long-term goal of stable hSSC cultures.
Impact Statement
This study developed and characterized human testis extracellular matrix (htECM) and porcine testis ECM (ptECM) for testing in human spermatogonial stem cell (hSSC) culture. Results confirmed the hypothesis that ECM from the homologous species (human) and homologous tissue (testis) is optimal for maintaining hSSCs. We describe a simplified feeder-free, serum-free condition for future iterative testing to achieve the long-term goal of stable hSSC cultures. To facilitate analysis and understand the fate of hSSCs in culture, we describe a multiparameter, high-throughput, quantitative flow cytometry approach to rapidly count undifferentiated spermatogonia, differentiated spermatogonia, apoptotic spermatogonia, and proliferative spermatogonia in hSSC cultures.
Introduction
Chemotherapy or radiation treatment for cancer or other conditions can cause permanent infertility.1,2 For men, the extent and persistence of treatment-related azoospermia is determined by a combination of factors, including the disease itself, the drugs, their doses, and the treatment regimen.3–5 Currently available fertility preservation methods for men rely on the isolation and cryopreservation of sperm from the ejaculate or testis that can be used for intrauterine insemination, in vitro fertilization (IVF), and IVF with intracytoplasmic sperm injection. These methods are available to adult and adolescent males but not to prepubertal boys who are not yet producing sperm. However, boys do have spermatogonial stem cells (SSCs) in their testes that might be used to regenerate spermatogenesis.6,7
Brinster and colleagues showed that transplantation of frozen and thawed murine SSC into the seminiferous tubules of an infertile testis leads to complete regeneration of spermatogenesis in the recipient mouse.8,9 This finding, in turn, led to the conceptualization that SSCs might be exploited to preserve and restore the fertility of prepubertal males, wherein SSCs obtained by testicular biopsy and cryopreserved before the onset of cancer treatment can be transplanted back into the patient's testes at a later time to restore complete spermatogenesis.7,10–14 However, SSCs are rare cells in the seminiferous tubule epithelium, and it is likely that a small testicular biopsy obtained from a prepubertal patient would contain only a small number of these cells. 15 The efficiency of SSC transplantation depends on the number of SSCs introduced into the recipient niche.16,17 Therefore, it may be necessary to first expand patient SSC in vitro to achieve robust engraftment and regeneration of spermatogenesis.
Conditions for maintenance and expansion of rodent SSC in long-term culture are well established.18,19 However, these methods are ineffective in supporting proliferation and maintenance of human SSC (hSSC). 20 Methods for long-term propagation of nonhuman primate and hSSC have been described in several recent reports,20–42 but tissue sources were variable; the analytical endpoints were varied (ranging from quantitative real-time polymerase chain reaction to immunocytochemistry to xenotransplantation), and there is no consensus on best methods.
However, review of the published hSSC culture work in Supplementary Table S1 does reveal some trends. Most studies have used some method of sorting or differential planting to enrich hSSCs and/or deplete testicular somatic cells, and included some concentration of glial cell line-derived neurotrophic factor (GDNF). There is a lack of consensus about the cell culture substrate with options ranging from plastic, laminin, Matrigel, or gelatin to various feeder cell preparations.
Mammalian extracellular matrix (ECM) is produced by the resident cells of every tissue and organ, and contains numerous signaling molecules that promote mitogenesis, migration, and/or differentiation of various stem/progenitor cells,43–47 angiogenesis, 48 and immune cell modulation.49–52 Biologic scaffold materials composed of ECM have been widely used to facilitate the repair and reconstruction of diverse tissue types, including esophagus,53,54 skeletal muscle,47,55 dura mater,56,57 tendon,58,59 breast tissue, 60 and others. 61
The use of ECM hydrogels as substrates for in vitro cell culture, or the use of solubilized ECM as a supplement to culture media, can augment the proliferation and/or differentiation of selected cell types and therefore may be desirable for hSSC culture.62–64 The development and use of testicular ECM to culture testicular somatic and germ cells have been reported recently.65–68 While these studies demonstrate the maintenance of the somatic compartment, the use of testis ECMs for maintenance and growth of SSCs in two-dimensional (2D) culture systems is yet to be evaluated.
In this study, a novel approach was used to isolate ECM from human and porcine testicular tissue. The objective was to establish serum-free, feeder-free conditions for maintaining human stem/progenitor spermatogonia in 2D culture on ECM-coated plates. Herein, we tested the hypothesis that ECM from the homologous species (human) and/or homologous tissue (testis) will provide an optimal environment for maintaining or expanding hSSC in culture.
Serum-free media supplemented with GDNF and basic fibroblast growth factor (bFGF) were used to culture cells on various ECM substrates, including human and porcine testis ECM (htECM and ptECM), porcine small intestinal submucosa (SIS-ECM), porcine urinary bladder matrix (UBM-ECM), and human laminin, an isolated component of ECM. Sandos inbred mice (SIM) 6-thioguanine-resistance, ouabain-resistant (STO) feeder cells and mouse laminin that have been used to culture mouse SSCs were used as controls. Viable stem/progenitor spermatogonia were quantified at days 0, 7, and 14 of culture through the expression of undifferentiated embryonic cell transcription factor 1 (UTF1) and stage-specific embryonic antigen 4 (SSEA4).
Materials and Methods
Procurement and processing of human testis tissue
Deidentified human testes were obtained through the Center for Organ Recovery and Education and the University of Pittsburgh Health Sciences Tissue Bank under University of Pittsburgh CORID no. 686. Tissue was obtained from postpubertal male organ donors (Supplementary Table S2) and transported on ice in Lactated Ringer's solution after procurement. Seminiferous tubules were removed from the tunica albuginea. A single cell suspension of human testicular parenchyma was prepared by sequential enzymatic digestion with 2 mg/mL collagenase (Worthington Biochemical Corporation), followed by 0.25% trypsin (Invitrogen) plus 7 mg/mL DNase I (Sigma-Aldrich) in Hank's Balanced Salt Solution (HBSS; Invitrogen), as described previously. 69
Cells were cryopreserved at a final concentration of 20 × 106 cells/mL in cryoprotectant medium containing 15% fetal bovine serum (FBS) and 10% dimethyl sulfoxide (DMSO; Sigma) in minimum essential media (MEMα; Invitrogen). Cryovials were frozen at a rate of −1°C/min in controlled-rate freezing containers (Nalgene-Nuc International) in a −80°C freezer and transferred into liquid nitrogen the following day. For experiments, frozen testis cells were thawed rapidly, washed, and suspended in Dulbecco's phosphate-buffered saline (DPBS) with 1% FBS.
Magnetic activated cell sorting and differential plating
Frozen and thawed cells were incubated with rat anti-CD49f-PE antibody (10 μL/106cells; BD Biosciences) in DPBS-based sorting medium supplemented with 1% FBS (Supplementary Table S3). Cells were then washed and incubated with antiphycoerythrin microbeads (2 μL/106 cells; Miltenyi Biotec) for 45 min, then sorted on a magnetic column; the positive fraction was collected in serum-free medium (Supplementary Table S4) supplemented with 20 ng/mL GDNF and 1 ng/mL bFGF, washed and cultured on collagen I-coated plates (1 μg/cm2; Corning) for 48 h to remove adherent somatic cells. Cells that were floating after 24 h were removed and replated on fresh collagen I-coated wells. Cells that were still floating after an additional 24 h were collected and plated in the experimental culture conditions described below.
Production of porcine SIS-ECM and UBM-ECM
ECM was isolated from porcine small intestine and urinary bladder as previously described.70,71 In brief, tissue was harvested from market weight pigs, and the tunica submucosa, muscularis mucosa, and stratum compactum were mechanically isolated from the intestines. The basement membrane and lamina propria were mechanically isolated from the bladders. Tissues were further decellularized and disinfected by agitation in 0.1% peracetic acid (PAA) with 4% ethanol before rinsing, lyophilization, and milling into powder. ECM was solubilized by digestion with 1 mg/mL pepsin at pH 2.5 for 24 h.
Production of htECM and ptECM
Several detergents were evaluated to identify a preferred decellularization protocol for native human testis (htNative). Frozen testes were thawed, decapsulated, and sliced to 4 mm, rinsed in type I water, which was replaced every 20–60 min until water remained colorless, and agitated in a 0.02% trypsin and 0.05% EGTA solution at 37
Decellularization efficiency was determined by histology, including staining with hematoxylin and eosin (H&E) and 4′,6-diamidino-2-phenylindole (DAPI). PicoGreen assay (Quant-IT PicoGreen dsDNA reagent; Invitrogen) was used to quantify remnant double-stranded DNA (dsDNA), and gel electrophoresis with a 1.5% agarose gel was used to determine base pair size of any remnant DNA. Once validated as an effective method for decellularization of htECM, the same protocol was used on native porcine testis (ptNative) to produce ptECM. htECM and ptECM were lyophilized, milled into powder, and digested in 0.1% pepsin for 24 h before use in cell culture.
Biochemical composition of htNative, htECM, ptNative, and ptECM
Retention of sulfated glycosaminoglycans (sGAGs) after decellularization was qualitatively determined by histology through Alcian Blue staining. sGAG concentrations in the pepsin-digested samples were quantitatively measured with the Blyscan Sulfated Glycosaminoglycan Assay Kit (Blyscan #B1000; Biocolor Ltd., Carrickfergus, Co Antrim, United Kingdom). Three independent biological testis samples of each of the four groups (htNative, htECM, ptNative, and ptECM) were pooled for urea–heparin extraction to assay total protein content (BCA Protein Assay Kit; Pierce, Rockford, IL) and bFGF content (Human FGF basic Quantikine ELISA Kit; R&D Systems DFB50). All kits were used according to manufacturer's instructions.
Substrate preparation and cell culture
For feeder cell-based culture, previously mitomycin-treated and cryopreserved STO feeder cells (ATCC SCRC 1049) were rapidly thawed and washed in STO media (Supplementary Table S5), and plated on 24-well culture plates coated with 0.1% gelatin. Cells were allowed to attach to the substrate for 24–48 h.
For culture on laminin, 24-well plates were coated with mouse laminin at 2 μg/cm2 as previously reported. 72 The concentration of Human Laminin from Fibroblasts (Sigma) used for coating plates was optimized by culturing human spermatogonia on plates coated with human laminin at the concentration used for mouse laminin (2 μg/cm2) and a lower concentration of 0.5 μg/cm2. Both concentrations led to a similar recovery of UTF1+ cells. Hence, the lower concentration of 0.5 μg/cm2 was selected as the working concentration of human laminin for the subsequent experiments. Plates were incubated at 37°C for 2–4 h according to manufacturer's instructions.
For culture on tissue-derived ECM, ECM digests were neutralized to pH 7.6 with 0.1 N NaOH and diluted in PBS to a final concentration of 300 μg/mL, and then added to 24-well plates and incubated for 2–4 h at room temperature. At the time of initiation of culture, media from wells with STO feeder cells and residual ECM from the ECM-coated wells were aspirated. Any adherent molecular species within the solubilized ECM remained as a substrate for the SSC, but no visible disc of ECM was present. The wells were then rinsed with HBSS before introduction of cells for culture. Floating and weakly adherent cells from collagen I-coated plates used for differential plating were removed, washed by centrifugation at 600 g, resuspended in mouse serum-free medium summarized in Supplementary Table S3.21,73
Medium was supplemented with GDNF (20 ng/mL) and bFGF (1 ng/mL), and plated on substrates at a concentration of 50–70 × 103 cells/cm2. Fresh medium was supplemented into culture wells every 3–4 days, and cells were passaged 2:1 at day 7 using 0.05% trypsin-EDTA (Life Technologies).
Immunolabeling
Cells were spotted on Superfrost slides (Fisherbrand) at a concentration of 5 × 106 cells/mL, fixed with chilled 100% acetone, and allowed to dry. Fixed cells were rehydrated with DPBS, blocked for 1 h with a blocking buffer containing 3% bovine serum albumin (MP Biochemicals) and 5% normal donkey serum (Jackson ImmunoResearch). Mouse monoclonal anti-UTF1 antibody (1:200; Millipore) was added to cells and incubated for 90 min. Isotype-matched normal IgG monoclonal antibody (BD Pharmingen) was used as negative control.
Slides were washed with DPBS containing 0.1% Tween20 (Sigma) and incubated with donkey antimouse secondary antibody conjugated with AlexaFluor488 (1:200; ThermoFisher) for 45 min. Slides were washed, and coverslips were mounted using Vectashield mounting medium containing DAPI (Vector Laboratories). Cells were observed under the Nikon Eclipse 90i microscope and were analyzed using the NIS Elements Advanced Research Software.
Flow cytometry
Cells were washed with azide-free and serum-free PBS, and incubated with viability dye, GhostDye Red780 (Tonbo Biosciences), according to manufacturer's instructions. Cells were washed and incubated in buffer containing 2% FBS and 0.09% sodium azide in PBS with AlexaFluor488 mouse anti-SSEA4 antibody (2.5 μL/106 cells; BD Biosciences) and APC mouse antihuman CD117 (5 μL/106 cells cKIT) antibody (BD Biosciences).
Cells were then washed and stained with BV421 Annexin V (5 μL/106 cells; BD Biosciences) in annexin V binding buffer composed of 100 mM Hepes (ThermoFisher), 140 mM NaCl, and 25 mM CaCl2. After annexin V staining, cells were fixed in a 1:1 solution of annexin V buffer and Cytofix/Cytoperm solution (BD Biosciences), and washed in Perm/Wash solution (BD Biosciences) containing annexin V buffer, mixed 1:1. Cells were then stained with BUV396 mouse anti-KI-67 antibody in perm/wash/annexin V buffer supplemented with Brilliant Stain Buffer (BD Biosciences) according to manufacturer's instructions, washed and analyzed using the BD FACSAria II flow cytometer. Isotype-matched IgG monoclonal antibodies were used as negative control. Compensation controls were prepared using OneComp eBeads compensation beads (ThermoFisher).
Statistical analysis
One-way analysis of variance (ANOVA) was used to compare the recovery of UTF1+ cells in the various cell culture conditions at days 7 and 14 using the GraphPad Prism software. One-way ANOVA and two-tailed t-tests were used for the statistical analysis of flow cytometry results. p Values <0.05 were considered statistically significant.
Results
Decellularization protocol establishment
Detergents commonly used to decellularize tissue were compared for their ability to decellularize human testicular tissue (Fig. 1A). After exposure to the various decellularization protocols, remnant dsDNA in the ECM was quantified with PicoGreen reagent and compared with dsDNA amounts in native tissue (Fig. 1B). ECM made with any of the detergents contained significantly less dsDNA (p < 0.001) than found in the native tissue (29,900 ± 2250 ng of dsDNA per mg of dry weight tissue). Remnant dsDNA in ECM made with 4% SDC (9790 ± 1610 ng/mg) was comparable with that of the 3% TX-100 (7860 ± 544 ng/mg), and both contained significantly more dsDNA (p < 0.0001) than ECM made with 0.25% SDC/TX-100 (1010 ± 124 ng/mg). ECM made with 0.075% SDS contained the least dsDNA (p < 0.001 compared with the 0.25% SDC/TX-100 mixture) having 208 ± 23 ng/mg. The SDS protocol was used for both htECM and ptECM for the remainder of the study.
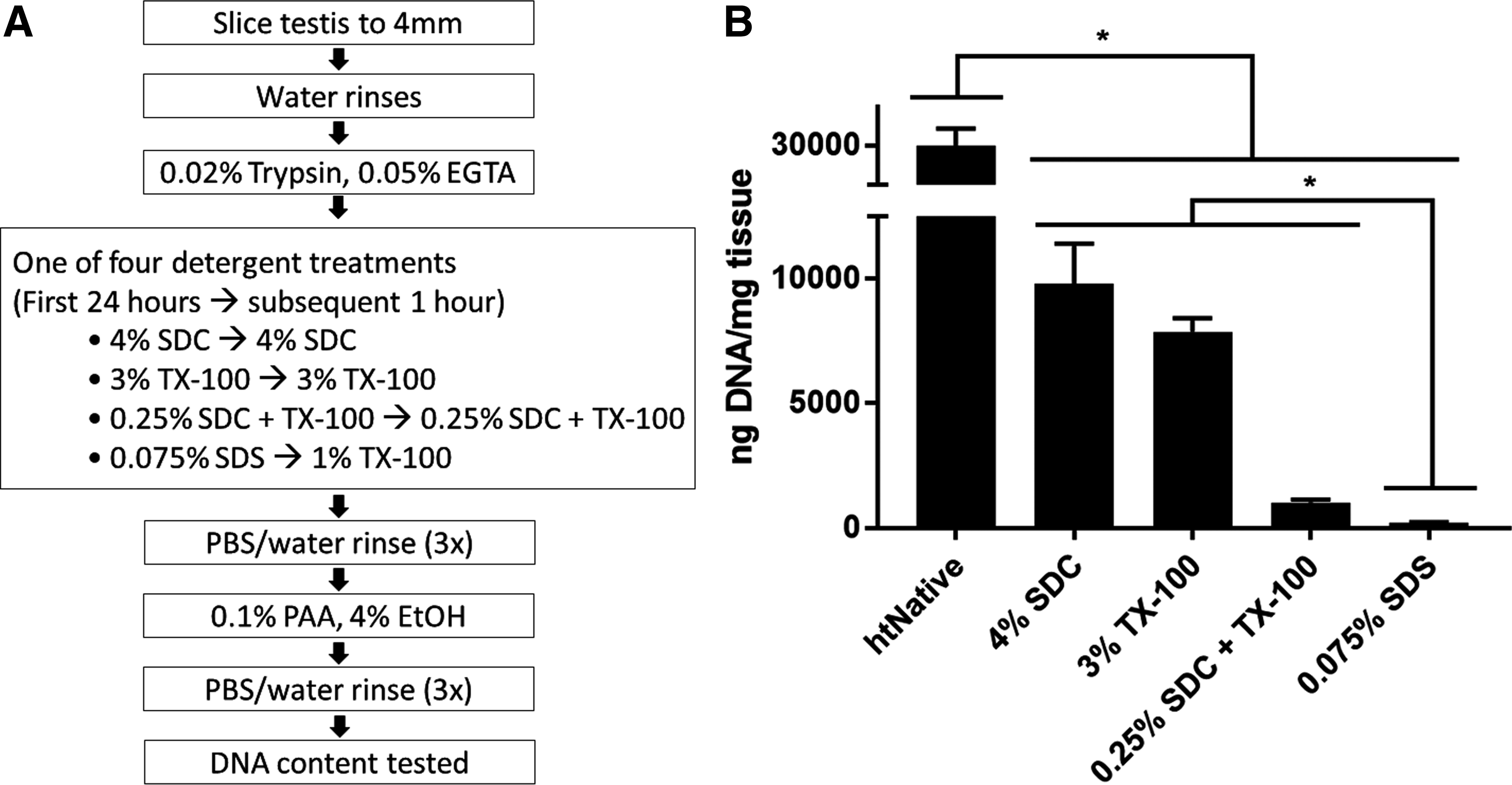
Overview of human testis decellularization strategy.
Decellularization efficacy
The degree to which htECM and ptECM were free of cells and cell remnants was assessed using previously established guidelines. 74 For htECM, no intact nuclei were visible by H&E or DAPI staining (Fig. 2A, C). For ptECM, no intact nuclei were visible by H&E, but rare clusters of nuclei were observed with DAPI staining (Fig. 2B, D).

Efficacy of decellularization protocol. Three biological replicates of human and porcine testis tissue were evaluated for DNA content before and after decellularization. Images in
dsDNA was quantified from three biological replicates before and after decellularization. Concentration of dsDNA in native tissue between the two species was similar; htNative #1: 31,100 ± 1070 ng/mg, htNative #2: 28,100 ± 325 ng/mg, htNative #3: 26,000 ± 4720 ng/mg, ptNative #1: 46,500 ± 242 ng/mg, ptNative #2: 44,000 ± 217 ng/mg, ptNative #3: 38,800 ± 827 ng/mg. htECM contained three orders of magnitude less dsDNA than htNative, and ptECM contained one order of magnitude less dsDNA than ptNative; htECM #1: 26 ± 1 ng/mg, htECM #2: 169 ± 14 ng/mg, htECM #3: 45 ± 8 ng/mg, ptECM #1: 4320 ± 75 ng/mg, ptECM #2: 3340 ± 28 ng/mg, ptECM #3: 3660 ± 48 ng/mg (Fig. 2E, F).
Gel electrophoresis showed that each native sample contained high amounts of genomic DNA. Two htECM samples contained no visible DNA, although the third showed a smear >200 base pairs (Fig. 2G). All three ptECM samples showed a faint smear of DNA at all sizes (Fig. 2H).
Biochemical composition
The amount of sGAGs, total protein, and bFGF in the native tissue and ECM for both species was quantified. Alcian Blue staining revealed that many of the sGAGs that were present in the htNativetissue were preserved in the htECM throughout the decellularization process (Fig. 3A). However, the porcine native tissue did not stain strongly for sGAG, and no sGAG staining was visible in the ptECM histology (Fig. 3B). These qualitative results were verified by the sGAG quantification. sGAG content in htECM was quantified in technical triplicate for both the human native and htECM samples of the three aforementioned biologically distinct donors (Fig. 3C).
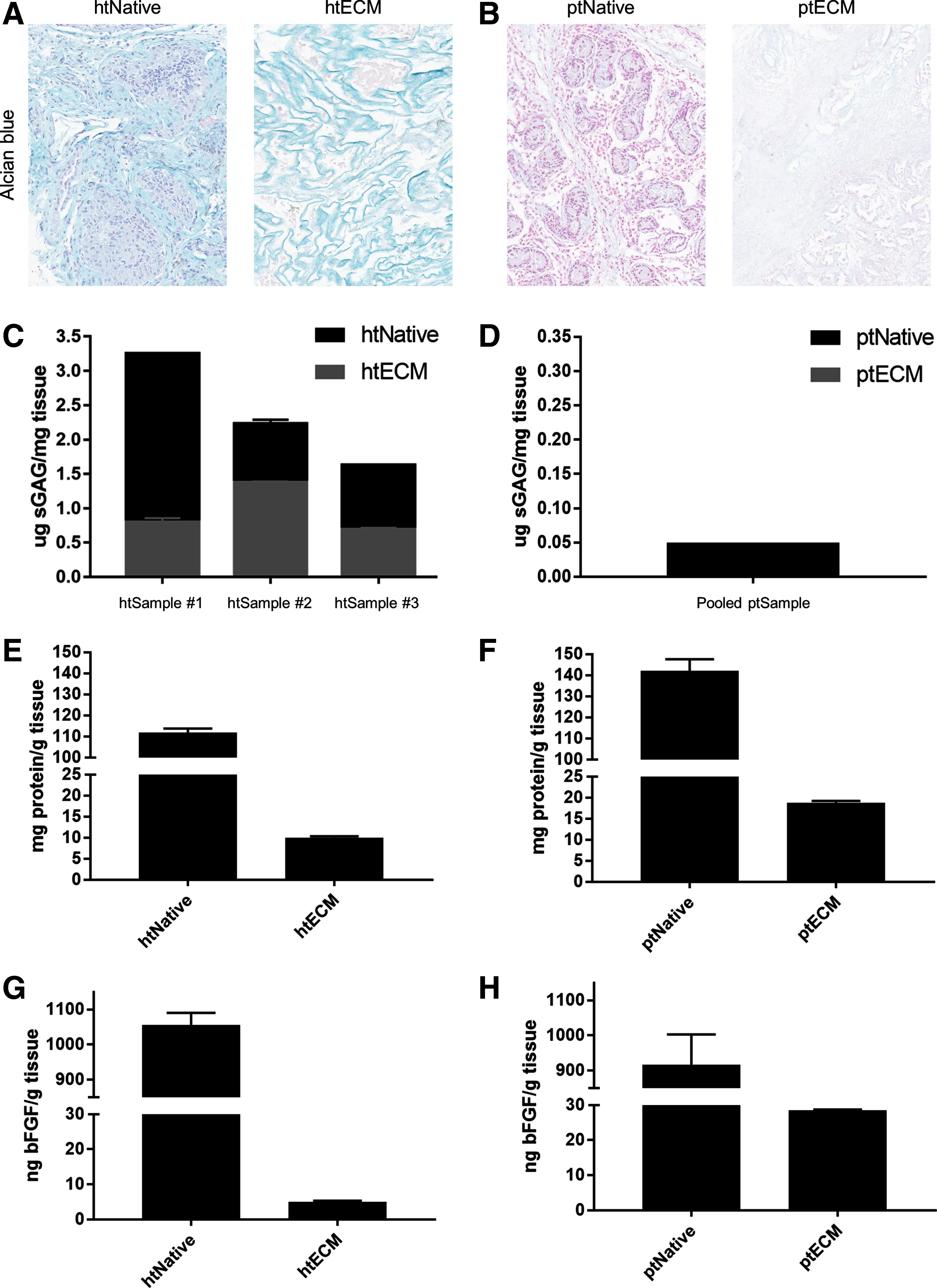
Biochemical composition of ECM.
Due to low tissue supply, the porcine native and ptECM samples were pooled into one sample for sGAG quantification (Fig. 3D). htNative #1 contained 3.25 ± 0.01 μg of sGAG per milligram of tissue (mean ± SD), and htECM #1 contained 0.80 ± 0.06 μg/mg. htNative #2 contained 2.23 ± 0.06 μg/mg of sGAG per milligram of tissue and htECM #2 contained 1.38 ± 0.01 μg/mg. htNative #3 contained 1.63 ± 0.01 μg/mg of sGAG per milligram of tissue and htECM #3 contained 0.69 ± 0.02 μg/mg (Fig. 3C). The pooled ptNative contained 0.05 μg of sGAG per milligram of tissue, and no sGAGs were detected in the pooled ptECM.
To quantify total protein before and after decellularization, a urea–heparin extraction was performed on pooled samples. htNative yielded 111.8 ± 1.9 mg of protein per gram of tissue and htECM contained 10.0 ± 0.3 mg/g (Fig. 3E). htPorcine yielded 142.9 ± 5.5 mg of protein per gram of tissue and ptECM contained 18.7 ± 0.5 mg/g (Fig. 3F). Total bFGF was measured by ELISA for the urea–heparin extracts of each material. htNative contained 1050 ± 35 ng of bFGF per gram of tissue, and htECM contained 5.0 ± 0.3 ng/g (Fig. 3G). ptNative contained 917 ± 86 ng of bFGF per gram of tissue, and ptECM contained 28.5 ± 0.2 ng/g (Fig. 3H).
Rheological properties of htECM hydrogel
The rheological properties of htECM hydrogel were determined for concentrations of 10 and 20 mg/mL, and were shown to be concentration dependent (Supplementary Fig. S1). A time sweep test showed that both concentrations of hydrogel had storage (G′) and loss (G″) moduli approximately one order of magnitude apart from each other, indicating the material to be a true hydrogel. Rheological testing indicated the viscosity of the pregel, forming gel, and formed gel of the 20 mg/mL material to be higher than that of the 10 mg/mL material. Interestingly, the time to reach 50% gelation for both materials was ∼39 s, but time to full gelation was ∼35 min for the 20 mg/mL gel and ∼49 min for the 10 mg/mL gel.
UTF1+ cell enrichment
Human spermatogonia were enriched from frozen and thawed testicular cell suspensions using magnetic-activated cell sorting based on integrin alpha-6 (ITGA6) expression, as previously described. 75 Quantification of UTF1 expression by immunolabeling showed a nearly threefold enrichment of UTF1+ cells in the ITGA6+ fraction (13.78% ± 1.06%) compared with the unsorted cells (5.05% ± 1.07%) (p = 0.004) (Fig. 4A).

Enrichment and culture of undifferentiated spermatogonia on ECM substrates.
The positive fraction of the sort contained a significant proportion of somatic cells that tended to overgrow the culture and therefore dilute the population of spermatogonia by day 14 (data not shown). Hence, we performed differential plating of the ITGA6+ positive fraction to further enrich UTF1+ cells and remove adherent somatic cells (Fig. 4B). Somatic cells in the positive fraction adhered to the collagen-coated plates, while germ cells remained floating in the medium. Approximately 33.68% ± 6.18% of the floating cells expressed UTF1 and were used to initiate culture (Fig. 4B).
SSC culture on ECM substrates
ITGA6+ cells after differential plating were introduced into culture wells coated with mouse laminin, human laminin, htECM, ptECM, SIS-ECM, UBM-ECM, or STO feeder cells (control) at a concentration of 50–70 × 103 cells/cm2 (17–24 × 103 UTF1+ cells/cm2). We used immunocytochemistry to determine the number of UTF1+ undifferentiated spermatogonia under each condition, which were compared with STO feeder cell controls on days 0, 7, and 14 (Fig. 4C). UTF1+ cells were present in all culture conditions at 14 days.
Relative to the number of UTF1+ cells originally placed in culture (day 0), the number of UTF1+ cells remaining in culture was significantly reduced by days 7 and 14 of culture (p = 0.0001). However, there were differences between treatment groups. STO feeders retained 32.14% ± 11.72% and 3.3% ± 1.36% of UTF1+ cells on days 7 and 14 of culture, respectively. Day 7 and 14 UTF1+ cell retention rates were as follows: murine laminin (53.89% ± 22.67% and 12.09% ± 2.38%), human laminin (52.94% ± 12.47% and 6.29% ± 3.20%), htECM (54.18% ± 12.94% and 21.90% ± 5.50%), ptECM (37.95% ± 4.82% and 16.22% ± 4.00%), SIS (63.88% ± 11.10% and 10.80% ± 3.25%), and UBM (46.78% ± 21.61% and 9.90% ± 1.41%).
While there was no significant difference in the number of UTF1+ cells between conditions at day 7, htECM retained the greatest number of UTF1+ cells by day 14 of culture. In addition, htECM was the only condition that retained a significantly greater number of UTF1+ cells than the control STO feeder cell-based condition at day 14 (p = 0.0039). Flow cytometry results shown below provide additional insights on the cellular mechanisms that explain the loss of undifferentiated human spermatogonia on htECM and ptECM substrates during 14 days of culture.
Flow cytometry analysis of SSCs
We devised a multiparameter flow cytometry strategy to determine the cellular mechanisms that explain the demise of undifferentiated spermatogonia during the 14 day culture period. Cultured cells were stained for SSEA4+ undifferentiated spermatogonia, cKIT (CD117)+ differentiated spermatogonia, annexin V+ apoptotic spermatogonia, and Ki67+ proliferating spermatogonia (Fig. 5A–F). SSEA4 is a robust and validated cell surface marker of human undifferentiated spermatogonia. 76 UTF1 staining on fixed cells for flow cytometry did not work effectively in our hands. Therefore, SSEA4 was substituted for UTF1 as it has been effectively used to mark undifferentiated spermatogonia as reported by others.31,76
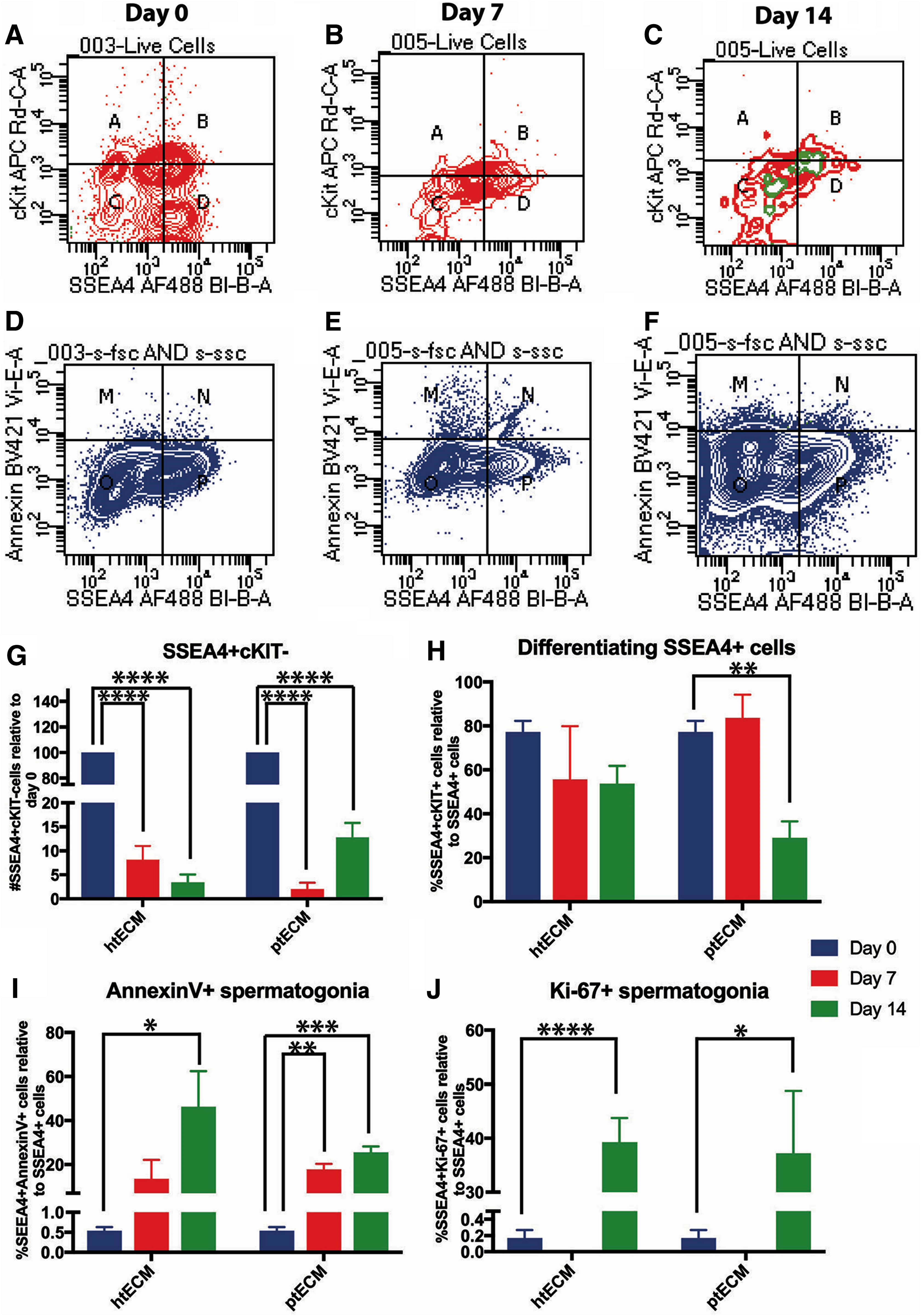
Evaluation of hSSC cultures on htECM and ptECM through flow cytometry:
At day 0, 16.4% ± 5.4% of live, ITGA6+, differentially plated cells were SSEA4+cKIT−, whereas 52.97% ± 5.54% of the cells were SSEA4+cKIT+ and 20.23% ± 7.98% of the cells were SSEA-cKIT+ (Fig. 5A, quadrants A, B, and D). Similar to the results with UTF1 ICC, the number of SSEA4+/cKIT− undifferentiated spermatogonia declined significantly by days 7 and 14 of culture on htECM as well as ptECM (p = 0.0001), with only 3.4% ± 1.66% and 12.84% ± 2.96% of the originally plated SSEA4+/cKIT− cells remaining by culture day 14, respectively (Fig. 5A–C, G).
In contrast to the total number of SSEA4+ undifferentiated spermatogonia, a greater proportion of differentiating SSEA4+/cKIT+ spermatogonia were retained on days 7 and 14 of culture, with only the ptECM number being significantly lower by day 14 (p = 0.01; Fig. 5A–C, H). These results suggest either that SSEA4+/cKIT+ differentiating spermatogonia survive better in culture, or that SSEA4+/cKIT+ spermatogonia that are lost to apoptosis are replaced by differentiation of SSEA4+/cKIT− spermatogonia. Our data shown in Supplementary Figure S2 indicate a trend toward cKIT+ cells being lost to apoptosis during the 14 day culture period (p = 0.056 for day 14 htECM), which may favor the interpretation that SSEA4+/cKIT− spermatogonia are lost to differentiation and contribute to the maintenance of the population of SSEA4+/cKIT+ spermatogonia.
Apoptosis clearly played a role in the demise of SSEA4+ total spermatogonia in culture as the number of SSEA4+/annexin V+ apoptotic spermatogonia increased significantly during the 14 days of culture on both htECM and ptECM (p < 0.05; Fig. 5D–F, I). Ki67 data provided in Figure 5A–C (green) and J indicate little or no proliferation of SSEA4+ cells on days 0 and 7 of culture (0–0.17%), but a significant increase on day 14 of culture on both htECM (39.28% ± 4.42%; p < 0.0001) and ptECM (37.19% ± 11.60%; p < 0.05). Isotype negative controls for the flow cytometry experiments are shown in Supplementary Figure S3.
Together, these data indicate that SSEA4+/cKIT− undifferentiated human spermatogonia are lost to differentiation and apoptosis during the 14 day culture period, and this loss was not compensated by proliferation of SSEA4+ cells. There were no significant differences in the proportions of SSEA4+ cells undergoing differentiation, apoptosis, or proliferation between the two testicular ECMs.
Discussion
This study identified methods to isolate ECM from human testis (htECM) and porcine testis (ptECM), and compared the ability of a solubilized form of this ECM to serve as an adsorbed culture plate substrate and support the viability of hSSC versus mouse laminin, human laminin, small intestinal submucosa (SIS-ECM), urinary bladder (UBM-ECM), and STO mouse embryonic fibroblast feeder cells.
The htECM material was shown to meet established decellularization criteria 74 while retaining sGAG and growth factors, although the same decellularization protocol produced ptECM with higher dsDNA content. This finding may be explained in part by differences in macromolecular content/density, cellularity, and biochemical distinctions between the testis microenvironment of the two species. Identical decellularization methods provided a head-to-head comparison of the two ECM environments, although an alternative protocol may produce ptECM with lower DNA content.
Identification of a preferred testis decellularization process involved the use of several commonly used detergents and subsequently comparing the cellularity, and the amount and integrity of remnant DNA within the testis ECM. This detergent-based approach was also described by Baert et al. 66 who compared 1% TX-100 with 1% SDS for both 24 and 48 h and found SDS at 24 h to be superior, due probably to its “harsh ionic nature.” This study investigated the use of the same detergents but at different concentrations; 3% for the milder nonionic TX-100 and 0.075% for SDS. We also evaluated the use of 4% SDC and a solution containing 0.25% of SDC and TX-100 each. Our results likewise indicated SDS to be a superior decellularization agent but required one order of magnitude less detergent than Baert et al.'s protocol.
Furthermore, we included a 2-h wash step subsequent to the SDS exposure, which has been shown to enhance the removal of residual SDS as it adversely affects the cytocompatibility of ECM scaffold materials. 77 We speculate that minimizing the SDS concentration during decellularization, and subsequently removing as much residual SDS as possible, helped preserve the biochemical composition and integrity of the resident structural and functional molecules.
Rheological studies of pepsin-solubilized htECM showed that upon polymerization at 37°C at both 10 and 20 mg/mL concentrations there was approximately a 10-fold difference between the storage (G′) and loss (G") moduli, indicating that the biomaterial intrinsically behaves as a true hydrogel.78,79
The retention of UTF1+ cells on day 14 of culture was greatest on the htECM (21.9% ± 5.5%) and ptECM (16.2% ± 4.0%) substrates, but only htECM retained significantly more UTF1+ human spermatogonia than the STO feeder cell controls (3.3% ± 1.36%). These observations may justify moving to an ECM-based, feeder-free, serum-free system for downstream experiments to establish conditions for long-term maintenance and expansion of hSSC in culture. ptECM cultures retained the next highest number of UTF1+ spermatogonia after 14 days (16% ± 4% UTF1+ cells relative to day 0), but this was not statistically different than the STO feeder condition.
The ECM of the testis represents the natural in vivo niche for hSSC. The decellularization method reported herein preserves the biochemical composition of the testis ECM environment, and these endogenous cues may contribute to hSSC growth/survival in vitro. The loss of sGAG, total protein, and bFGF after decellularization is not surprising as the bulk of these components lie in the cellular compartment of the tissue. The findings in this study are consistent with those of other studies.80–85 However, it is possible that the ECM proteins were also depleted by the use of SDS in the protocol. The bioactivity of ECM bioscaffolds has been attributed to structural proteins,86–88 glycosaminoglycans, 89 and the ligand landscape, including embedded growth factors and cytokines,90–93 the enzymatic creation of matricryptic peptides,94–97 and most recently, the inclusion of micro-RNA-containing matrix-bound nanovesicles. 52
The ECM is continuously degraded, remodeled, and synthesized by the resident cells of each tissue; therefore, ECM from different source tissues is biochemically distinct and can have tissue-specific effects on cells. It therefore makes intuitive sense that ECM from the homologous tissue (testis) and homologous species (human) might be most suitable for culturing hSSC although this is not always the case (Supplementary Table S6, Refs.62,82,98–103).
Such tissue specificity has been shown in studies involving ECM derived from skin and skeletal muscle, 103 liver, 62 and myocardium. 104 However, other studies show either no difference or a preference for heterologous tissue ECM (Supplementary Table S6). For example, neurites grew equally well in ECM hydrogels derived from CNS, spinal cord, or urinary bladder tissues, 101 and in a clinical trial treating volumetric muscle loss no difference was found between ECM derived from small intestine, urinary bladder, or dermis. 47 ECM tissue specificity has not previously been evaluated for expansion of hSSC in 2D culture.
While there have been several reports on the establishment of hSSC culture methods, the methods for validating cultures have been variable, and there is no consensus on best methods.20–42 One of the challenges for validating hSSC cultures is the absence of a functional assay to test the spermatogenic potential of cultured cells. Human to nude mouse xenotransplantation has been used in some studies to quantify transplantable SSCs in culture, but this approach is time consuming (2 months to analysis) and not compatible with iterative testing of culture conditions in early stages.20,27,39,69,75,105 Therefore, most studies rely on the expression of endogenous markers to characterize spermatogonia, including GDNF family receptor alpha-1 (GFRα1), ITGA6, G-protein-coupled receptor 125 (GPR125), thymocyte differentiation antigen 1 (THY1 or CD90), ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), Sal-like 4 (SALL4), promyelocytic leukemia zinc finger (ZBTB16), undifferentiated embryonic cell transcription factor 1 (UTF1), and SSEA4. However, markers such as GPR125, UCHL1, SSEA4, ITGA6, and THY1 are also expressed by other cells in the testis.33,36,37,106
In contrast, the expression of UTF1 in adult mammals is restricted to the testes, 107 specifically the population of undifferentiated spermatogonia on the basement membrane of seminiferous tubules.36,75,108 UTF1 is expressed by cells that also express the undifferentiated spermatogonial markers SALL4 and UCHL1 (PGP9.5) but does not overlap with the differentiated spermatogonial marker, cKIT. 75
In this study, spermatogonia were enriched by sorting testicular cells based on ITGA6 expression, as previously described. 75 However, the ITGA6+ fraction retained a substantial proportion of somatic cells that tended to overgrow the culture by day 14. To deplete the somatic cell population, the ITGA6+ fraction was differentially plated on collagen I, and the nonbinding fraction was selected for cell culture, as has been reported previously.32,40 Results showed greater retention of UTF1+ cells on htECM substrate at 2 weeks compared with cultures on STO feeder cells. However, UTF1+ cell numbers gradually declined over time on all substrates.
We developed a multiparameter flow cytometry approach to understand the cellular mechanisms that explain the loss of undifferentiated human spermatogonia on htECM and ptECM substrates. Our data indicated that the population of SSEA4+/cKIT− undifferentiated spermatogonia may be lost to differentiation (SSEA4+/cKIT+) and apoptosis (SSEA4+/annexin V+). These observations will help focus future culture experiments to identify conditions that inhibit differentiation pathways and/or reduce apoptosis. We observed almost no proliferation of undifferentiated human spermatogonia (SSEA4+/Ki67+) on days 0 or 7 of culture, but a significant increase in proliferation on day 14. The increased proliferation of SSEA4+ human spermatogonia on day 14 could result from selection of a subpopulation of spermatogonia that were compatible with the culture conditions.
However, the data provided in Figure 5C suggest that the increased proliferation is associated with the cKIT+ differentiating portion of SSEA4+ cells, so may simply reflect a higher rate of proliferation in differentiating spermatogonia compared with undifferentiated spermatogonia. Alternatively, the increased proliferation of SSEA4+ human spermatogonia on day 14 of culture could be an emergency stress response in which apoptosis in some cells causes compensatory proliferation in the neighboring cells to maintain homeostasis. 109 Overall, these data suggest that GDNF and bFGF growth factors that are required for survival and expansion of mouse SSCs in culture are not sufficient to support the survival or expansion of hSSCs in culture, at least at the concentrations tested in this study.
Results of this study indicate that the homologous species and homologous tissue ECM represented the best substrate for maintaining hSSCs in culture. However, the overall significant decline of undifferentiated spermatogonia by day 14 indicates that more work is needed to achieve the long-term goal of expanding hSSC numbers in culture. Unlike the mouse, where SSC cultures could be validated with a functional transplantation assay that regenerates complete spermatogenesis, assessment of hSSC cultures has been largely limited to descriptive markers. We found that UTF1 and SSEA4 are robust markers of human spermatogonia that could be used in ICC and/or FACS analyses, which is consistent with previous reports.31,76 The flow cytometry approach enables high-throughput, quantitative, simultaneous assessment of multiple endpoints, which will help guide future experiments.
The serum-free, feeder-free system described here should provide a simplified platform for iterative testing of various culture conditions.
Footnotes
Acknowledgments
This work was supported by grants from the US–Israel Binational Science Foundation (2011111) to K.E.O.; the Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD092084) to K.E.O. and S.F.B.; and the National Institutes of Health training grants (TL1TR001858) to M.H.M. and (T32 EB001026) to K.T.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.