
Abstract
Bone engineering makes it possible to grow unlimited amounts of viable tissue products for basic and applied research, and for clinical applications. A common trend in tissue engineering is the use of decellularized tissue matrices as scaffolding materials, which display structural, mechanical, and biological attributes typical of the native tissue. Due to the limited availability and high cost of human samples, decellularized tissue matrices are typically derived from animal sources. It is unclear, however, whether interspecies differences in tissue parameters will influence the quality of tissue grafts that are engineered using human stem cells. In this study, decellularized cow and human bone scaffolds were compared for engineering bone grafts using human induced pluripotent stem cell-derived mesodermal progenitor cells. After seeding, the cell-scaffold constructs were cultured for 5 weeks in osteogenic medium under dynamic conditions in perfusion bioreactors. The architectural and chemical properties of the scaffolds were studied using microscopic, spectroscopic, and thermogravimetric techniques, while cell behavior and formation of mineralized tissue were assessed using a combination of molecular assays, histological methods, and imaging technologies. The results show that while scaffolds derived from cow and human bone differ somewhat in architecture and composition, both equally support cell viability, tissue growth, and formation of a mineralized bone matrix. Taken together, the results suggest that scaffolds derived from cow bone represent a suitable and convenient alternative to engineer human bone grafts for various biomedical applications.
Impact Statement
Decellularized tissue matrices are popular as scaffolding materials for tissue engineering application. However, it is unclear whether interspecies differences in tissue parameters influence the quality of tissue grafts that are engineered using human stem cells. In this study, decellularized cow and human bone scaffolds were compared for engineering bone grafts using human induced pluripotent stem cell-derived mesodermal progenitor cells and despite minor differences in architecture and mass composition, both scaffolds equally support cell viability and tissue mineralization. Decellularized cow bone scaffolds therefore represent a suitable and more affordable alternative for engineering human bone grafts for basic and applied research.
Introduction
B
Among others, decellularized tissue matrices are commonly used as biomaterials scaffolds for engineering tissues3–14 because they display structural, mechanical, and biological characteristics typical of the native tissue. 15 Due to the limited availability and exorbitant cost of human tissues for research, 16 generation of decellularized scaffolds from human sources cannot be routinely attained. Therefore, for tissue engineering applications researchers have typically utilized scaffolds derived via decellularization of animal tissues.4–7,9–14
We recently demonstrated that viable and functional bone grafts could be engineered from human induced pluripotent stem cells-derived mesenchymal progenitor (iPSC-MP) cells and decellularized cow bone scaffolds. 10 However, several studies have shown that interspecies differences exist in bone parameters such as architecture, density and composition,17–19 raising the question whether decellularized scaffolds derived from tissue of different species are equally suitable for engineering tissue products using human stem cells.
To test this hypothesis, this study compared decellularized cow bone scaffolds to scaffolds derived via decellularization of human cadaveric bone for engineering bone grafts using human stem cells. First, the scaffolds were characterized to examine their architectural and chemical properties using microscopic, spectroscopic, and thermogravimetric techniques. Then, the scaffolds were seeded with human iPSC-MP cells and the cell-scaffold constructs cultured for 5 weeks in osteogenic inducing medium under dynamic conditions in perfusion bioreactors. The samples were thus analyzed to study cell attachment, viability and differentiation, and formation of mineralized tissue using a combination of molecular assays, histological techniques, and imaging technologies.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM)—high glucose, KnockOut DMEM (KO-DMEM), glutaMAX solution 100 × , nonessential amino acids 100 × , β-mercaptoethanol, antibiotic-antimycotic 100 × (Anti-Anti), Dulbecco's phosphate buffered saline (DPBS) solution 1 × , calcium and magnesium free, were purchased from Gibco (Gaithersburg, MD). HyClone fetal bovine serum (FBS) was purchased from Thermo Scientific (Waltham, MA). Basic fibroblast growth factor (bFGF) was purchased from Invitrogen (Carlsbad, CA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Preparation of decellularized bone scaffolds
Decellularized cow bone scaffolds were prepared from bone specimens obtained from 2-week to 4-month old calves (Green Village Packing®). After removing the skin and adjacent muscles, cylindrical plugs (4 mm in diameter) of trabecular bone were drilled from the subchondral region of metacarpal bones and cleaned under high-velocity stream of water. The decellularization process consisted of incubation of the bone plugs in hypotonic buffer, detergent, and enzymatic solution as follows: (1) phosphate buffered saline (PBS) and 0.1% EDTA for 1 h at room temperature (RT), (2) 10 mM Tris and 0.1% EDTA for 12 h at 4°C, (3) 10 mM Tris and 0.5% sodium dodecyl sulfate (SDS) for 24 h at RT, (4) several washes in PBS (15 min each) at RT until all the bubbles were gone, (5) 10 mM Tris and DNAse and RNAse (100 U/mL) for 6 h at 37°C, and (6) twice PBS followed with incubation in 70% ethanol (each for 5 min). Finally, bone plugs were lyophilized, cut, and polished to 4 mm height and 4 mm diameter cylindrical scaffolds.
Decellularized scaffolds derived from cadaveric human bone (59-year-old male) were provided by LifeNet Health®, and prepared following the same protocol used for the preparation of decellularized cow bone scaffolds.
Following decellularization, the dry weight and volume of the scaffolds were measured (using an analytical balance and a digital caliper, respectively) to calculate their density. For the experiments, 26 scaffolds of each group in the density range of 0.333–0.858 mg/mm3 were chosen (Table 1). For the same analysis, scaffolds with similar densities across the density range (low, middle, and high) were selected.
Scanning electron microscopy of decellularized bone scaffolds
Scanning electron microscopy (SEM) images were obtained on a TM-1000 tabletop SEM (Hitachi, Chiyoda, Japan), with a solid-state back scattered electron detector, accelerative voltage of 15 keV and 195 pA or 52 μA emission current, at a working distance of 7 mm. Scaffolds were mounted on SEM studs using a double-sided adhesive carbon tape prior imaging.
Thermogravimetric analysis of decellularized bone scaffolds
Each sample was ground into a fine powder using a mortar and pestle, and 3–5 mg of the powder was loaded on a Q500 thermogravitational analysis machine (TA Instruments, Wakefield, MA). Samples were heated at 10°C/min, from 25°C to 800°C. The mass was recorded twice per second and the mass loss was categorized as follows: removal of water from 50°C to 130°C, decomposition of organic matter (i.e., collagen) from 300°C to 500°C, and decomposition of CO2 (primarily from carbonated mineral—hydroxyapatite) at 700–800°C. The fraction of mineral was determined from the mass remaining at 600°C.
X-ray photoelectron spectroscopy analysis of decellularized bone scaffolds
The chemical composition of the samples was analyzed using X-ray photoelectron spectroscopy (XPS; PHI Quantum II, Al Kα X-ray source; ULVAC-PHI, Inc., Kanagawa, Japan). A presputtering process (pass energy = 1 keV, sputtering time = 30 s) was performed to remove the possible contamination on the surfaces. Survey spectra (pass energy = 224 ev, 0–1100 eV) were recorded from areas of size 150 × 150 μm2. The relative concentration (atom %) of detected elements was calculated from the relative intensities of peaks, after correction for tabulated sensitivity factors in the MultiPak PHI software of the instrument.
Preparation of cell-scaffold constructs
Human iPSC-MP cells (line 1013A) were expanded as described previously. 20 Briefly, cells at passage (P) 4 were plated in gelatin-coated tissue culture flasks (6.6 × 103 cells per cm2) and expanded in medium consisting of KO-DMEM, 20% (v/v) HyClone FBS, 1 ng/mL bFGF, 2 mM GlutaMAX, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, and 100 U/mL of antibiotic-antimycotic solution.
Before cell seeding, scaffolds with cylindrical shape (4 mm in height and 4 mm in diameter) were sterilized in 70% ethanol overnight and then incubated for 4 h in osteogenic medium consisting of high-glucose DMEM supplemented with 10% (v/v) HyClone FBS, 1 μM dexamethasone, 10 mM β-glycerophosphate, 50 μM ascorbic acid-2-phosphate sesquimagnesium salt hydrate, and 100 U/mL of antibiotic-antimycotic solution.
Cells were seeded at a density of 1.2 × 106 cells per scaffold. Seeding was performed in six-well ultra-low attachment plate (one scaffold per well) using a droplet technique. Briefly, 40 μL aliquots of a single-cell suspension were prepared using the expansion medium and added to the top of paper-blotted scaffolds. The cell-scaffold constructs were incubated for 20 min, and then flipped every 30 min for a total of 4 h to facilitate uniform cell distribution. To avoid drying, aliquots of expansion media (2–10 μL) were added to the scaffolds following each flip. At the end of the seeding procedure, 4 mL of expansion medium were added to each well and samples incubated overnight at 37°C.
The day after the seeding, samples were transferred to 24-well ultra-low attachment plates filled with fresh expansion medium, and the old medium collected to count the number of unattached cells using a disposable Kova® Glasstic® hemocytometers (Kova International, Garden Grove, CA). The seeding efficiency was thus estimated using the following equation:
Following seeding, the samples were cultured in expansion medium for 3 days before being transferred to perfusion bioreactors.
Cultures were screened for mycoplasma using the MycoAlert Mycoplasma Detection kit (Lonza, Basel, CH). Briefly, culture media were collected, centrifuged at 450 g for 5 min, and luminescence measured on aliquots of supernatant using the Synergy Mx microplate reader and Gen5 software (BioTek, Winooski, VT).
Culture in perfusion bioreactor
Three days after seeding, the samples were transferred to bioreactors (each containing six samples) and cultured under direct perfusion in osteogenic medium for 5 weeks. Bioreactor systems were set up as described previously.21,22 Each bioreactor contained six samples that were perfused in parallel. A perfusion flow rate of 3.6 mL/min was set using a digital, low-flow, multichannel Masterflex peristaltic pump (Cole Palmer, Vernon Hills, IL). Media change was performed twice a week for the duration of the study. Aliquots of culture media were collected weekly and stored at −80°C for further analysis. At select times points, cultured samples were collected and processed to study cell differentiation and tissue formation.
LIVE/DEAD assay
Cell viability and distribution were analyzed using the LIVE/DEAD assay (Thermo Scientific) 3 days after seeding. Briefly, samples were cut longitudinally in half, washed in DPBS, and incubated with a solution of calcein AM (2 μM) and ethidium bromide (4 μM) in DPBS for 1 h at 37°C in the dark. Following incubation, the samples were washed in DPBS, and then placed in RPMI (medium without red phenol; Lonza) for imaging. Fluorescence mosaic images of the center of the scaffold were taken with an Olympus IX71 and processed with ImageJ (National Institutes of Health). Confocal images were taken with the Axiovert 200 M microscope (Carl Zeiss AG, Oberkochen, Germany) mounted with LSM 5 Pascal exciter using the LSM 5 Pascal software under defined settings.
Nanostring analysis
The expression of developmental genes was analyzed simultaneously using the NanoString nCounter system (Nanostring Technologies, Seattle, WA). After 5 weeks of culture in perfusion bioreactors, samples were washed in DPBS and RNA isolated using TRIzol (Life Technologies) and RNeasy Mini Kit (Qiagen, Venlo, NE) as described previously. 23 Following extraction, RNA was quantified with the NanoDrop 8000 (Thermo Scientific), and 100 ng used for hybridization (65°C for 18 h). Data were analyzed using the nSolver™ 3.0 software with the following criteria. First, quality control was executed on raw counts using default criteria of the nSolver 3.0 software. Then, background correction was performed by subtracting a constant count value, a mean plus two standard deviations of negative control probes (i.e., probes for which no target is expected to be present), from each probe in a lane. To normalize all platform-associated sources of variation (e.g., automated purification, hybridization conditions, etc.), positive control normalization was performed to the geometric mean of the 50–100 targets with the highest counts. Lanes with positive control scaling factor outside the 0.3–3.0 range were omitted from further gene expression analysis. For housekeeping gene normalization, the following genes were selected: ACTB, ALAS1, and POLR2A. A normalization factor was calculated for each of the lanes based on the mean of counts for the selected housekeeping genes in each lane relative to the mean of counts for these genes across all lanes. Lanes with normalization factor outside the 0.1–10 range were omitted from further gene expression analysis. For data presentation, bar chart showing the expression of investigated genes as normalized counts for each developmental layer or combination of layers was chosen. Supplementary Table S1 lists all investigated genes.
Real-time polymerase chain reaction
Expression of genes involved in osteogenesis, chondrogenesis, and adipogenesis was analyzed using real-time polymerase chain reaction (PCR) after 5 weeks of culture in perfusion bioreactors. RNA was extracted as described in the Nanostring analysis section and reverse transcribed with random hexamers using the GoScript™ Reverse Transcription System (Promega, Madison, WI) according to the manufacturer's protocol. The expression of runt-related transcription factor 2 (RUNX2; Hs00231692_m1), collagen type I alpha (COL1A1; Hs00164004_m1), liver/bone/kidney alkaline phosphatase (ALPL; Hs01029144_m1), osteopontin (OPN; Hs00959010_m1), platelet-derived growth factor receptor beta (PDGFRB; Hs01019589_m1), SRY-Box 9 (SOX9; Hs00165814_m1), peroxisome proliferator-activated receptor gamma (PPARG; Hs01115513_m1), and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Hs02758991_g1) was analyzed using the StepOnePlus PCR System cycler (Applied Biosystems, Waltham, MA) in a 20 μL volume reaction using the TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assays (Applied Biosystems). The expression levels of investigated genes are shown as normalized to the expression level of GAPDH.
Alkaline phosphate activity
Alkaline phosphatase activity (ALP) was analyzed 3 days after seeding and 5 weeks after culture in perfusion bioreactors. ALP activity was determined using a modification of the method described in the literature. 24 Briefly, samples were lysed in 90 μL of Triton (0.1%, Sigma-Aldrich) and through three freeze (liquid nitrogen)/thaw (water bath at 37°C) cycles. Then after, 10 μL aliquots of cell lysates were added to a transparent, flat-bottom, 96-well plate and mixed with 50 μL of 2-amino-2-methyl-propanol buffer (pH 10.3) and 50 μL of 15.2 mM paranitrophenylphosphate (PNPP) in a 2 mM solution of magnesium chloride at 37°C. The reaction was stopped by adding 200 μL of a 1 N solution of sodium hydroxide, and the amount of produced paranitrophenol determined spectrophotometrically at 410 nm using the plate reader SYNERGYMx (BioTek) equipped with Gen 5 1.09 software. Results are expressed as “pmoles of PNPP cleaved per sample per min.”
Osteocalcin release
The content of osteocalcin in the medium was analyzed weekly till the end of the culture period (5 weeks) using the Gla-type Osteocalcin EIA kit (Takara, Mountain View, CA) according to the manufacturer's protocol. Absorbance was measured at 450 nm using the plate reader SYNERGYMx (BioTek) equipped with Gen 5 1.09 software. Results are expressed as cumulative osteocalcin release into culture media in “ng/mL.”
Histology
Formation of new tissue was analyzed histologically after 5 weeks of culture in perfusion bioreactors. Nondemineralized samples were embedded in resin and processed using a modified version of a method described in the literature.23,25 Briefly, samples were fixed, dehydrated, and immersed into methylmethacrylate. Polymerized samples were then cut to ∼300 μm-thick sections using an IsoMet™ low-speed saw (Buehler, Lake Bluff, IL), ground to ∼100-μm thickness, polished using MicroPolish™ Alumina powder with 0.3 and 0.05 μm grain size (Buehler), and stained with Stevenel's blue solution. Histological slides were then digitalized using the ScanScope® GL scanner (Aperio, Vista, CA) at 20 × magnification at standard resolution. Images were visualized using the Aperio ImageScope v12.1.0.0529 software and exported to JPG format using the Extract Region tool.
Immunohistochemistry
Deposition of bone matrix proteins was analyzed via immunohistochemistry (IHC) 3 days after seeding and 5 weeks after culture in perfusion bioreactors. Briefly, samples were cut longitudinally in halves, washed in DPBS for 5 min at RT, and fixed in 4% (v/v) paraformaldehyde in DPBS (Santa Cruz Biotechnology, Dallas, TX) for 2 days at 4°C. After fixation, samples were washed in PBS for 5 min at RT, and decalcified in Immunocal (Decal Chemical Corp., Tallman, NY) for 2 days at 4°C. Samples were finally dehydrated through graded concentrations of ethanol before embedding in paraffin. Following embedding, serial longitudinal sections (5 μm-thick) were cut in the middle of the sample and mounted on glass slides for immunohistochemical staining.
Briefly, sections were deparaffinized by heating at 60°C for 30 min, incubated in CitriSolv for 5 min (twice), rehydrated through graded concentrations of ethanol, incubated in deionized water for 2 min (3 times), and washed in DPBS for 5 min. The sections were then incubated in citrate buffer (pH 6) at 90°C for 30 min to retrieve the antigens, washed in deionized H2O for 5 min, and incubated with 3% H2O2 in methanol for 30 min to block the endogenous peroxidase activity. Following a wash in DPBS for 5 min, sections were incubated with 1% normal horse serum in DPBS to block the nonspecific binding and stained overnight at 4°C in a humidified chamber with primary antibodies (all purchased from Millipore and diluted 1:500 in DPBS) against bone sialoprotein (rabbit polyclonal anti-BSP II, No. AB1854), osteopontin (rabbit polyclonal anti-osteopontin, No. AB1870), and osteocalcin (rabbit polyclonal anti-osteocalcin, No. AB10911). Specific antigen detection was performed using the biotinylated secondary antibody and biotin/avidin complex (Universal Vectastain ABC kit Elite; Vector Laboratories, Burlingame, CA) diluted in DPBS according to manufacturer's instructions and via incubation with 3,3′-diaminobenzidine peroxidase substrate for 5 min (Vector DAB kit; Vector Laboratories). All sections were counterstained with hematoxylin for 30 s (Richard-Allan Scientific, San Diego, CA), dehydrated with a graded series of ethanol washes (50%, 70%, 95%, and 100% twice—each for 2 min), incubated with citrisolv (twice 5 min), dipped into xylene, and coverslipped (Fisher Scientific, Hampton, NH) using a Permount mounting media (Fisher Scientific). Negative controls were performed following the same procedure but omitting either the primary or secondary antibody incubation.
Routine hematoxylin and eosin (HE) staining was performed using Gill's HE Y dye on the first serial section before IHC staining. Stained slides were then digitalized following the same procedure described in the Histology section.
Micro-computed tomography
Mineralization of newly formed tissues was studied via micro-computed tomography (μCT). Scans were performed on the same samples before cell seeding and 5 weeks after culture in perfusion bioreactors. Briefly, samples were aligned along their axial direction and stabilized in a 15-mL centrifuge tube that was clamped within the specimen holder of a vivaCT 40 system (SCANCO Medical AG, Brüttisellen, Switzerland) with the following settings: voltage 55 kV, current 0.109 mA. The 4-mm length samples were scanned at 21-μm isotropic voxel size. A global threshold technique was applied to differentiate the mineralized tissue. Results are expressed as bone volume (BV)/total volume (TV), trabecular number (Tb.N.), trabecular thickness (Tb.Th.), and trabecular separation (Tb.Sp.).
Image processing and generation
Figures were produced in Adobe Illustrator (Adobe Systems Incorporated, San Jose, CA). Background color of histology and IHC figures was corrected to pure white using Adobe Photoshop (Adobe Systems Incorporated).
Statistical analysis
Statistical analysis was conducted using the GraphPad Prism 6 version 6.0 (GraphPad Software, Inc., La Jolla, CA). Unpaired t-test was used for single comparison between groups. All results are shown as means and standard deviations. A difference between the mean values for each group was considered statistically significant when the p-value was <0.05.
Results
Scaffold characterization
Topographical images of decellularized cow and human bone scaffolds were obtained using a scanning electron microscope (Fig. 1A). Decellularized cow and human bone scaffolds display similar porous, trabecular structure although decellularized human bone scaffolds display higher porosity, characterized by the presence of pores with larger size. High magnification SEM images also reveal that both scaffold types display a similar topographic pattern, with bundles of fibers oriented parallel to the surface of the scaffolds in different spatial directions.
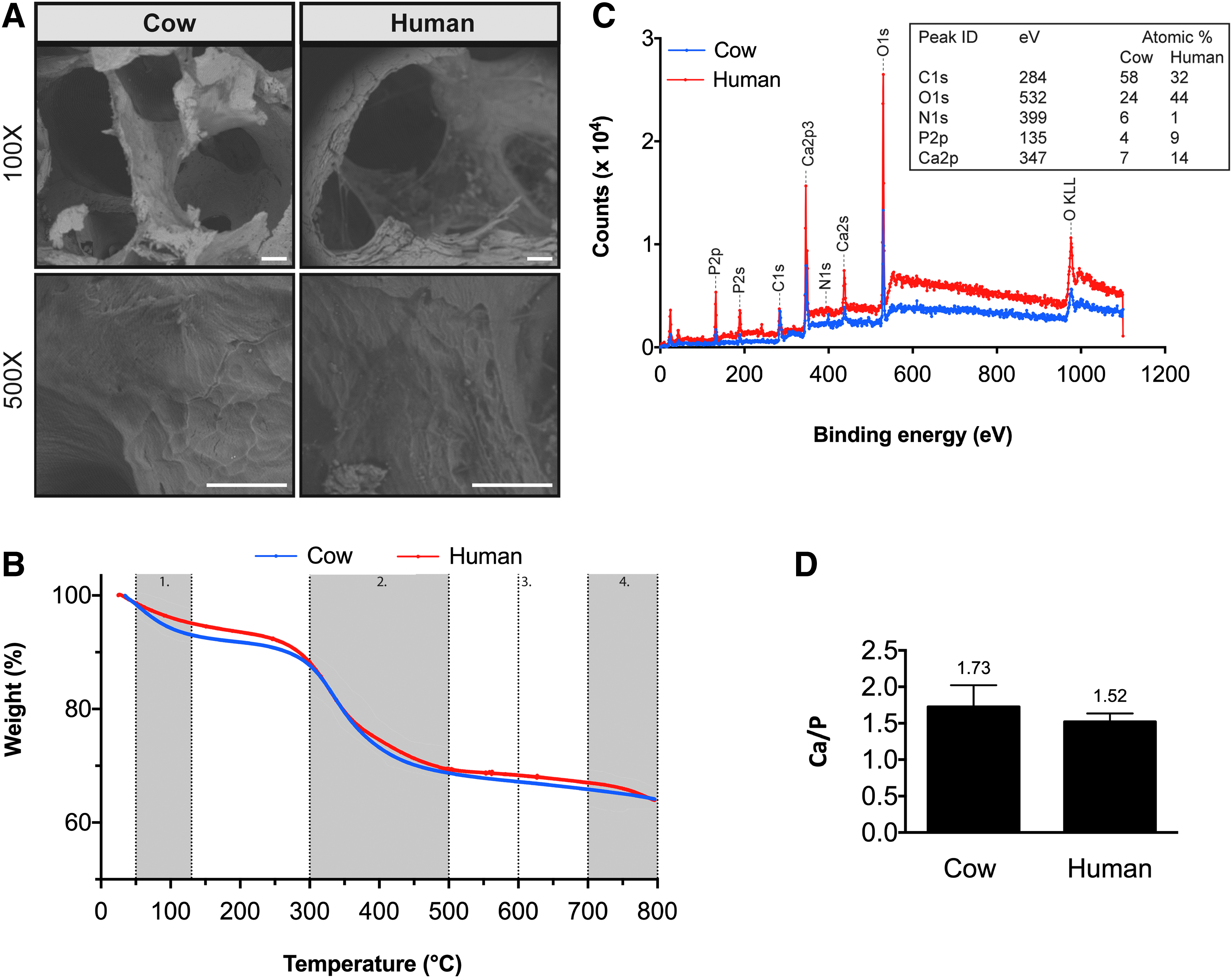
Characterization of decellularized cow and human bone scaffolds.
Mass composition of decellularized cow and human bone scaffolds were studied using thermogravimetric analysis (Fig. 1B). Decellularized cow and human bone scaffolds display a weight loss of 12.40% ± 1.10% and 10.93% ± 0.88%, respectively between 50°C and 130°C that can be attributed to the loss of water. The second weight loss depression for decellularized cow and human bone scaffolds appears between 300°C and 500°C due to decomposition of organic matter and is 20.76% ± 1.33% and 21.01% ± 2.24%, respectively. Combustion of remaining weight of decellularized cow and human bone scaffolds at 600°C determines the mineral fraction and scaffolds display a weight loss of 63.87% ± 0.15% and 64.67% ± 2.08%, respectively. Decellularized cow and human bone scaffolds display a weight loss of 2.96% ± 0.15% and 3.52% ± 0.84%, respectively between 700°C and 800°C that can be attributed to decomposition of CO2 (primarily from carbonated mineral—hydroxyapatite). There are no significant differences in weight loss at all temperatures between decellularized cow and human bone scaffolds. Altogether, these results show decellularized cow and human bone scaffolds have very similar mass composition.
Elemental analysis using XPS shows that both scaffold types display similar spectra of detected elements, characterized by the presence of peaks for carbon, nitrogen, oxygen, phosphate, and calcium (Fig. 1C). Further, the analysis shows that the calcium to phosphorus (Ca/P) ratio for decellularized cow and human bone scaffolds is 1.73 and 1.52, respectively (Fig. 1D).
Cell seeding, distribution, and viability
The efficiency of seeding was estimated by counting the number of nonattached cells 1 day after seeding. Figure 2A shows that the seeding efficiency was higher than 90% for both scaffold types. Interestingly, decellularized human bone scaffolds show significantly higher (p < 0.05) number of attached cells compared with decellularized cow bone scaffolds, resulting in higher seeding efficiency estimated for these scaffolds.
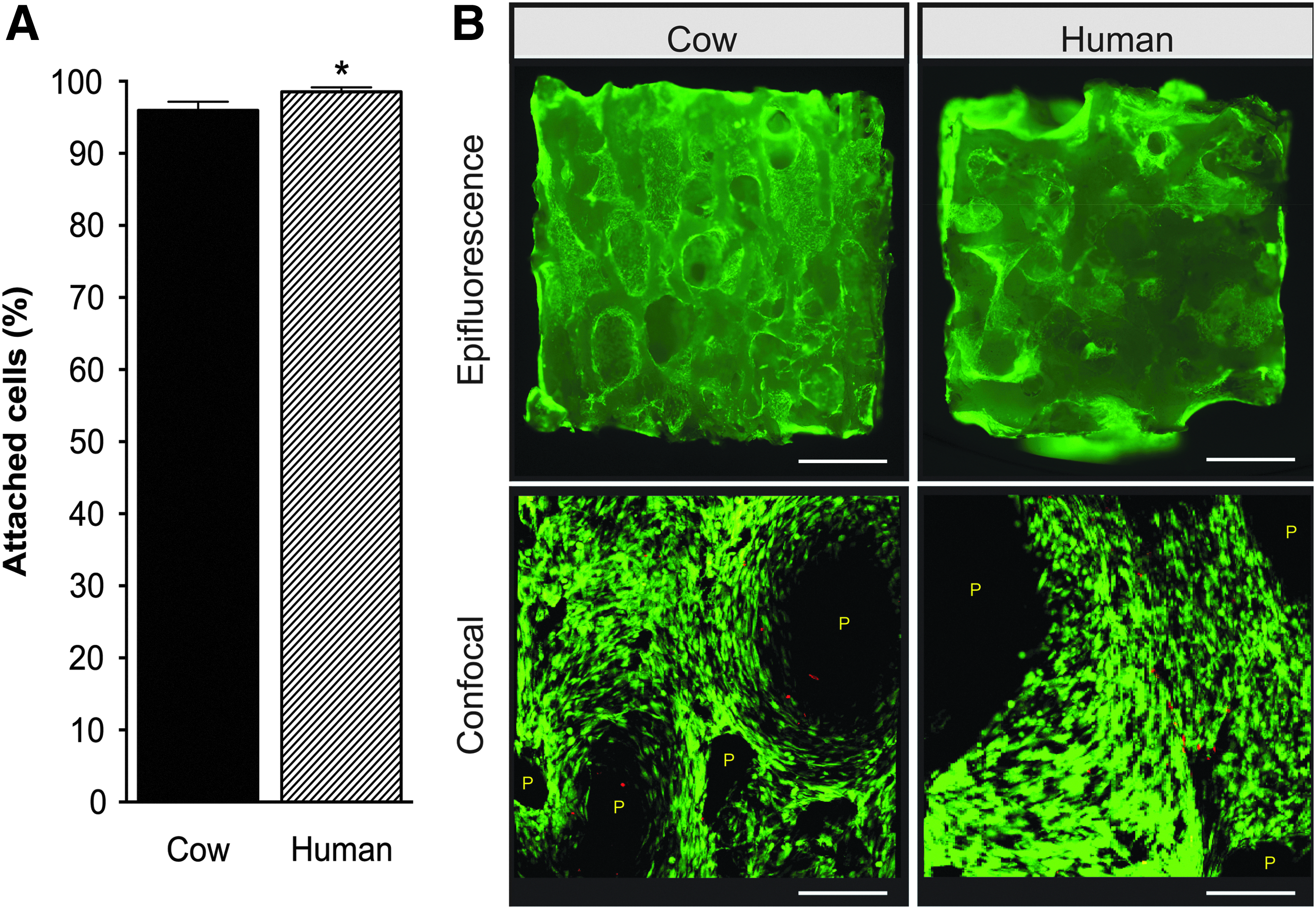
Cell seeding and viability on decellularized cow and human bone scaffolds.
Cell viability and distribution were analyzed using the LIVE/DEAD assay 3 days after seeding. Figure 2B shows that decellularized cow and human bone scaffolds support cell viability equally, with cells homogenously distributed across the scaffold surface showing comparable morphological features.
Gene expression
Gene expression was studied via Nanostring and real-time PCR 5 weeks after culture in perfusion bioreactors. Figure 3 shows that the cells display overall a similar profile of expression of developmental genes when cultured on decellularized cow and human bone scaffolds.

Expression of developmental genes. Nanostring analysis showing the expression of genes involved in germ layer specification 5 weeks after culture of cell-scaffold constructs in perfusion bioreactors. Results are expressed as mean ± SD (n = 3). Asterisk (*) denotes significant difference between groups based on t-test (p < 0.05).
Out of 83 tested genes, 15 genes are highly expressed (displaying counts above 1000) in cells cultured on decellularized cow and human bone scaffolds. Specifically, four genes involved in ectodermal differentiation (CRABP2, PDGFRA, SNAI2, and SOX9), six genes involved in mesodermal differentiation (ANPEP, INHBA, ITGAV, KDR, STAT3, and TNFRSF1A), one gene involved in endodermal differentiation (CTNNB1), and four genes involved in ecto-meso-endoderm differentiation (CD44, CDH2, ITGB1, and THY1) are highly expressed.
On the other hand, nine genes are differentially expressed when comparing cells cultured on decellularized cow and human bone scaffolds. Cells cultured on decellularized cow bone scaffolds display significantly higher (p < 0.05) expression of INHBA, LEF1, MAPT, MCAM, NES, PDX1, and PECAM, while cells cultured on decellularized human bone scaffolds display significantly higher (p < 0.05) expression of CD44 and SRF.
The expression of the chondrogenic gene SOX9 and adipogenic gene PPARG show no differences when cells are cultured on the two scaffold types (Supplementary Fig. S1). On the other hand, genes involved in osteogenic differentiation are differentially expressed when comparing cells cultured on decellularized cow and human bone scaffolds, with cells cultured on decellularized cow bone scaffolds displaying significantly higher (p < 0.05) expression of RUNX2, COL1A1, ALPL, BSP, OPN, and PDGFRB (Fig. 4).
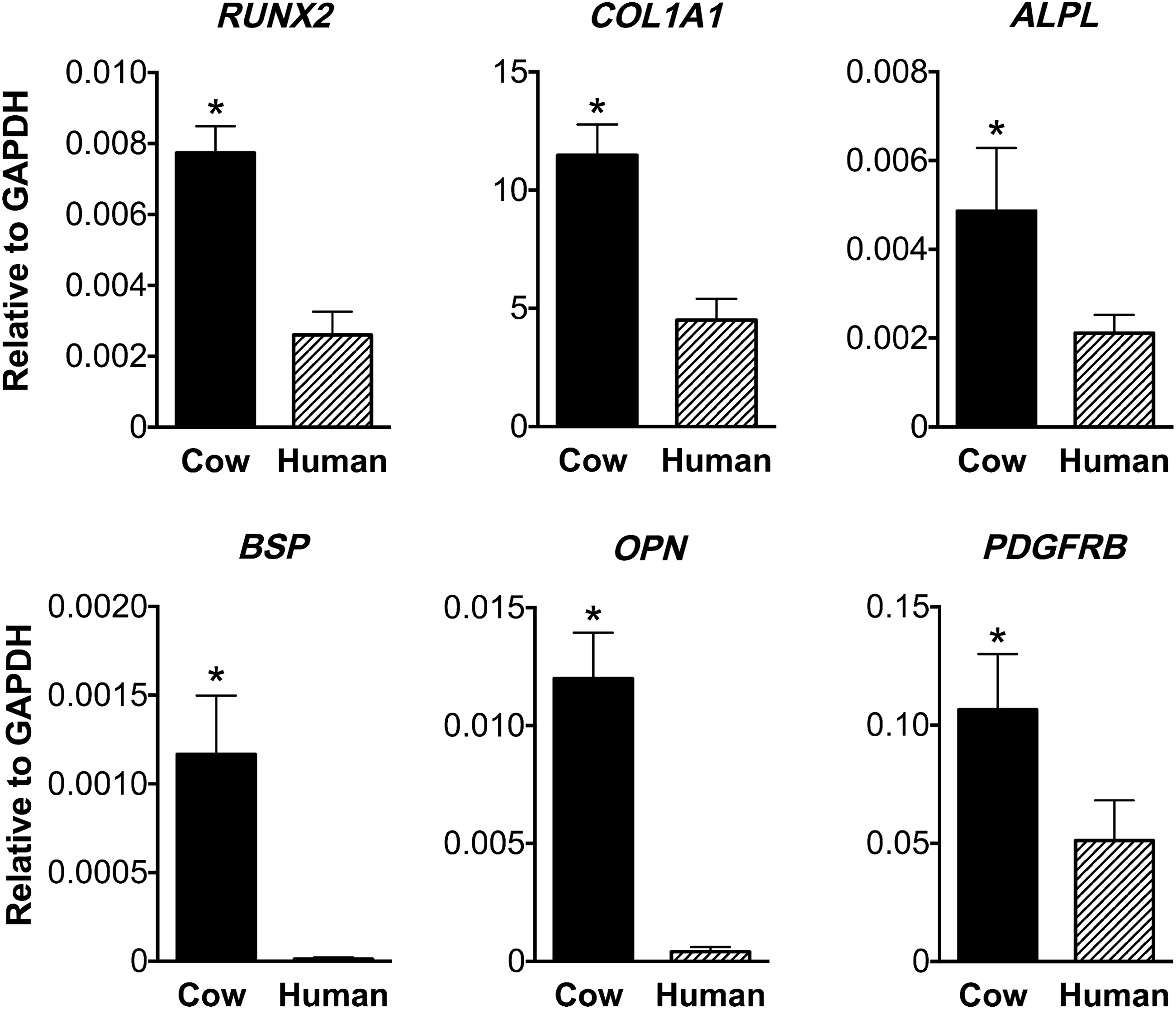
Expression of osteogenic genes. Real-time PCR analysis showing the expression of genes involved in osteogenic differentiation 5 weeks after culture of cell-scaffold constructs in perfusion bioreactors. Results are expressed as mean ± SD (n = 3). Expression values are shown as relative to the expression of the housekeeping gene GAPDH. Asterisk (*) denotes significant difference between groups based on t-test (p < 0.05). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PCR, polymerase chain reaction.
Production of bone-specific proteins
Osteogenic differentiation was also assessed by measuring the activity of ALP and the content of osteocalcin released in the culture medium. The results show that cells cultured on decellularized cow and human bone scaffolds display significantly higher (p < 0.05) ALP activity 5 weeks after culture in perfusion bioreactors. Interestingly, cells cultured on decellularized human bone scaffolds display significantly higher ALP activity at day 3 compared to those cultured on decellularized cow bone scaffolds. The cumulative curve of osteocalcin release show that, during the entire culture period, cells cultured on decellularized human bone scaffolds release significantly higher osteocalcin (p < 0.05) compared with those cultured on decellularized cow bone scaffolds (Fig. 5).
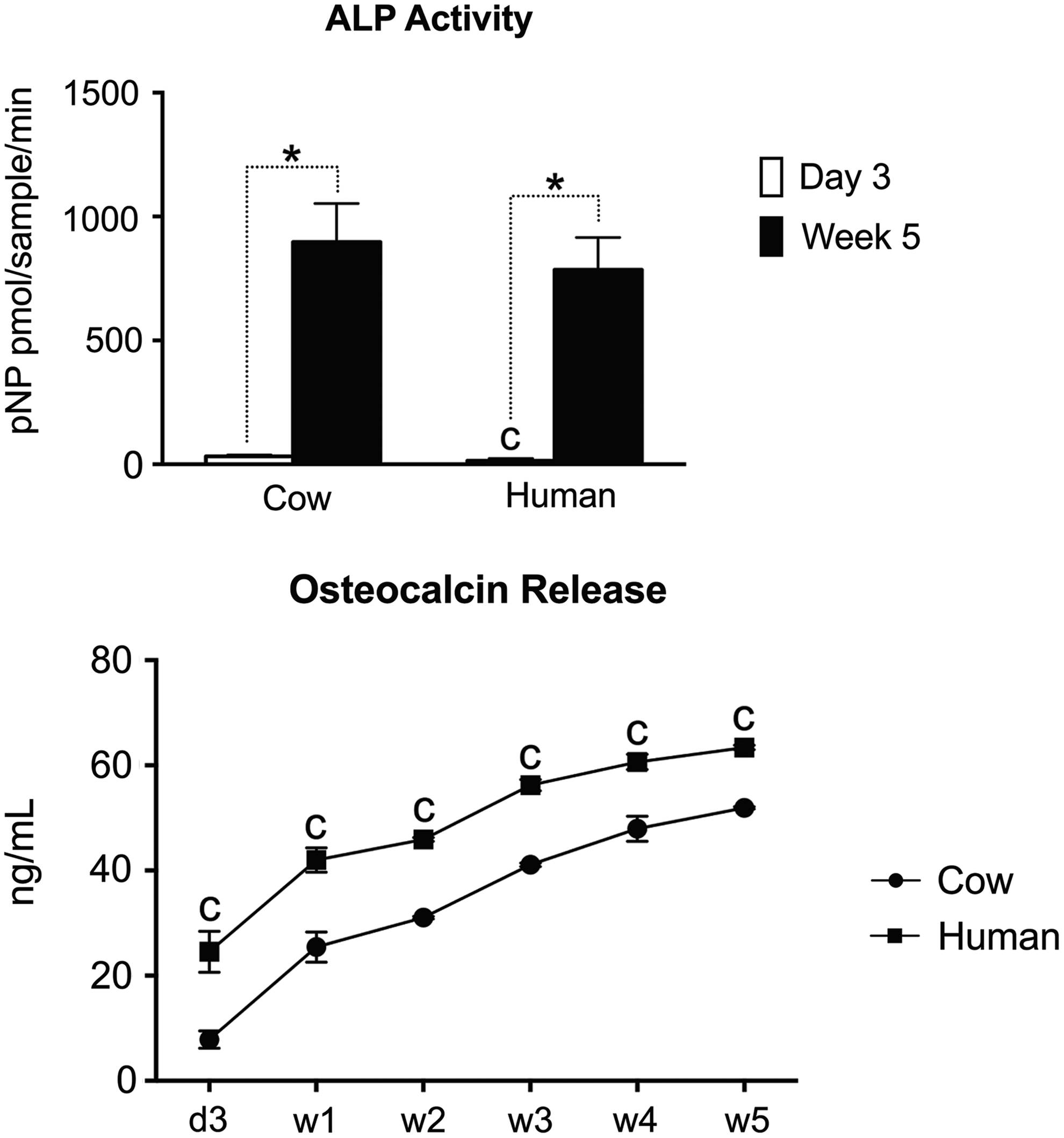
Alkaline phosphatase activity and osteocalcin release. Alkaline phosphatase activity 3 days after seeding and 5 weeks after culture of the cell-scaffold constructs in perfusion bioreactors. Cumulative release of osteocalcin after 1, 2, 3, 4, and 5 weeks of culture in perfusion bioreactors. Results are expressed as mean ± SD (n = 3). Asterisk (*) denotes significant difference between time points for the same condition, c denotes significant difference between cow and human groups for the same time point based on t-test (p < 0.05).
Tissue formation and mineralization
Tissue formation was analyzed histologically 5 weeks after culture in perfusion bioreactors. Resin-embedded nondemineralized samples display a similar extent of tissue formation, irrespective of the investigated scaffold type, with the cells distributed uniformly throughout the scaffolds and embedded in a thick extracellular matrix (Fig. 6). Immunohistochemical analysis of paraffin-embedded samples reveals that the extracellular matrix of the newly formed tissues stains positive for bone-specific extracellular matrix proteins such as bone sialoprotein, osteopontin, and osteocalcin (Fig. 7). The distribution of the staining appears similar when comparing the two scaffold types. However, samples of decellularized human bone scaffolds appear to stain stronger. Negative IHC controls are provided in Supplementary Figure S2 and show no color production when the primary or secondary antibodies are omitted.

Tissue formation on decellularized cow and human bone scaffolds. Histological analysis of samples after 5 weeks of culture in perfusion bioreactors. Samples are stained with Stevenel's blue. Cell nuclei are stained in blue. Areas highlighted in yellow are shown at higher magnification. Scale bars: 1 mm (upper) and 100 μm (lower). Cross-sectional images are representative of three samples.

Tissue formation on decellularized cow and human bone scaffolds. HE and IHC staining of samples 3 days after cell seeding and 5 weeks after culture in bioreactors. HE stains cell nuclei in blue and extracellular matrix in pink. IHC stains positively for BSP, OPN, and OC in brown. Nuclei were counterstained with hematoxylin (blue). Scale bar: 1 mm. Cross-sectional images are representative of three samples. BSP, bone sialoprotein; HE, hematoxylin and eosin; IHC, immunohistochemistry; OC, osteocalcin; OPN, osteopontin.
Tissue mineralization was analyzed via μCT 5 weeks after culture in perfusion bioreactors, and results were compared to those obtained from empty scaffolds prior seeding. Three-dimensional reconstructions of scaffolds and samples 5 weeks after culture in perfusion bioreactors are shown in Figure 8A. Following culture, the mineral content significantly increases (p < 0.05) for both experimental groups as evidenced by increased bone volume to total volume (BV/TV) ratio, increased trabecular number (Tb.N.), and decreased trabecular separation (Th.Sp.) (Fig. 8B). On the other hand, the trabecular thickness increases only for decellularized cow bone scaffolds, while decellularized human bone scaffolds show significantly higher (p < 0.05) trabecular thickness before and after culture compared to decellularized cow bone scaffolds.
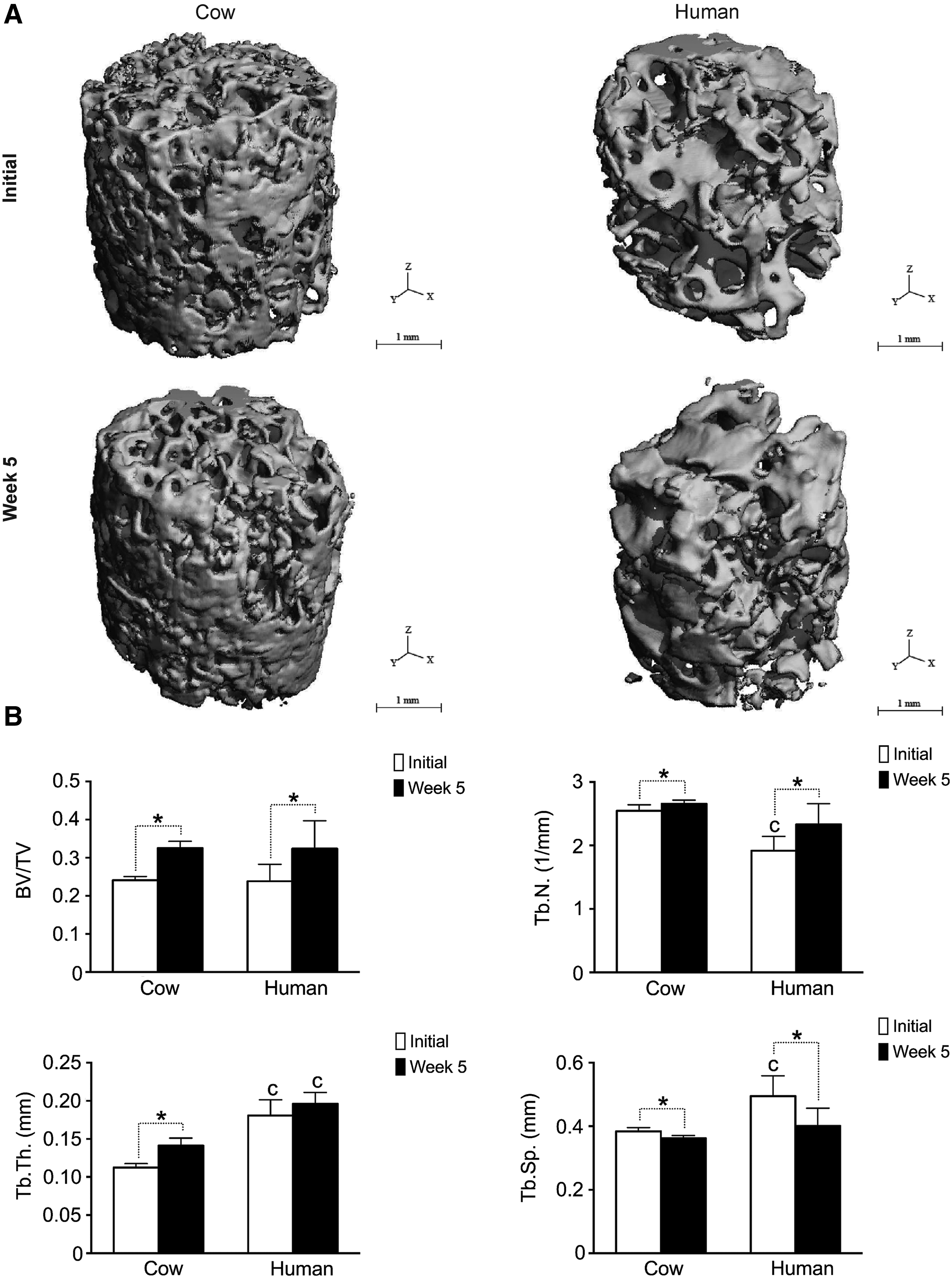
Tissue mineralization.
Discussion
When engineering bone grafts in the laboratory, osteocompetent cells are interfaced with biomaterial scaffolds, which act as frameworks for the cells and also provide the cells with intrinsic signals that can affect their biology. Understanding the role that biomaterial scaffolds play in supporting osteogenic differentiation and bone tissue formation is therefore critical to engineer tissue products with superior experimental and clinical potential.
Over the past decade, there has been a growing interest in developing scaffolds via decellularization of tissues and organs. Due to their unique biomimetic properties, these scaffolds are commonly used in combination with cells for tissue engineering applications3–14 or alone as tissue replacement products. 26 We recently engineered viable bone grafts by combining human iPSC-MP cells with decellularized cow bone scaffolds. 10 These grafts represent valid experimental models to conduct advanced basic and applied research, and might 1 day be used in the clinics to treat patients suffering from skeletal defects.
Especially for research applications, decellularized tissue scaffolds are typically derived from livestock animals.7,9–14 In fact, human cadaveric tissues are not accessible in large amounts and may be extremely expensive 16 precluding use for routine preparation of decellularized tissue scaffolds. However, interspecies differences exist in bone composition, density and quality, and it is unknown whether these differences can affect cell behavior and influence the quality of bone grafts engineered using human stem cells. Therefore, to test this hypothesis, this study compared decellularized cow and human bone scaffolds, combined with human iPSC-MP cells (line 1013A), to assess their ability to support differentiation, tissue growth, and formation of a bone matrix.
Before cell culture, the scaffolds were characterized to explore any relevant difference in structure and composition. To minimize variations, in this study the bone plugs were drilled from corresponding anatomical locations in cows and humans. The results show that, irrespective of their origin, the scaffolds display similar porous, trabecular structure both at microstructural and submicrostructural level, with decellularized human bone scaffolds showing generally larger pores. Comparison studies have shown that interspecies differences exist in cortical bone parameters such as architecture, composition, and density. For example, human cortical bone displays a laminar structure while cow cortical bone contains both a laminar and a plexiform (i.e., laminar bone with a dense system of vascularization) structure. 17 In addition, comparison studies have shown that the mineral density and volume fraction of the mineral phase are lower in human cortical bone compared to cow cortical bone, while no differences exist in the volume fraction of the organic phase. 18 In particular, the Ca/P ratio, which is generally considered an indicator of bone quality,19,27,28 is recognized to be different in different species. Accordingly, in this study the Ca/P ratio was lower in scaffolds derived from human bone (1.52) compared with those derived from cow bone (1.73). Also, the Ca/P ratio tends to decrease with age and in osteoporotic patients, 27 and the difference observed in this study perhaps reflects the age difference of the human and cow subjects from which the scaffolds were derived. Importantly, Ca/P ratio was measured after the decellularization process, which may have slightly altered the content of these elements. Altogether, the results demonstrate that, after the decellularization process, decellularized cow and human bone scaffolds are very similar in structure and mass composition. However, differences in the organization of extracellular matrix fibers and other biomolecules might exist, which could affect cell behavior and influence the bone engineering potential of decellularized bone scaffolds derived from different species.
To explore this possibility, the scaffolds were seeded with human iPSC-MP cells and constructs cultured for 5 weeks in perfusion bioreactors. The cells were seeded on the scaffolds using a previously established technique, 11 which results in high seeding efficiency and uniform cell distribution throughout the scaffold.10,20,23 In this study, the seeding efficiency was higher than 90% for both scaffold types, although decellularized human bone scaffolds showed higher number of attached cells, possibly due to differential expression of cell adhesion proteins present on these scaffolds. Understanding the phenomenon behind improved cell adhesion on scaffolds derived from human bone is worth further investigation, since cell adhesion is recognized to regulate cell cycle, differentiation, migration, and survival. 29
Despite the minor difference observed in cell attachment, cells cultured on the two scaffold types displayed overall a very similar profile of expression of developmental genes 5 weeks after culture in perfusion bioreactors, indicating no major effect played by the scaffolds derived from cow and human bone on the developmental potential of the human iPSC-MP cells used in this study. Few genes were differentially expressed when comparing cells cultured on decellularized cow and human bone scaffolds, specifically genes encoding proteins of cytoskeleton (MAPT, NES), proteins involved in cell adhesion (CD44, MCAM, PECAM) and transcription factors involved in several signaling pathways (INHBA, LEF1, PDX1, SRF). When studying the expression of genes involved in mesodermal differentiation toward the chondrogenic (SOX9) and adipogenic (PPAR-γ) lineages no significant differences were observed for cells cultured on scaffolds derived from cow and human bone.
On the other hand, genes involved in osteogenic differentiation and bone development such as RUNX2, COL1A1, ALPL, BSP, OPN, and PDGFRB30–33 were differentially expressed when comparing the two scaffold types, with cells cultured on decellularized cow bone scaffolds showing significantly higher expression. It is not clear whether the observed differences truly reflect an effect played by the scaffold type on the differentiation process. The reason for this difference may result from the fact that the decellularized human bone scaffolds display higher porosity and larger pore size. Scaffold porosity is recognized to affect cell behavior34–37 and can dictate the bone engineering potential of biomaterial scaffolds. While the presence of large pore size can facilitate neovascularization and tissue formation in vivo, 37 the opposite seems to be true when engineering bone grafts in vitro. 35 Small pore sizes and lower porosity may suppress cell proliferation, force cell aggregation, and thus stimulate osteogenesis under select culture conditions. On the other hand, a higher porosity appears to facilitate cell proliferation, because it facilitates transport of oxygen and nutrients. 34 Few studies reported an increased expression of osteogenic genes with decreasing pore size of biomaterials,38–40 which is in line with results observed in this study. Alternatively, these differences may be only representative of the time point analyzed in this study. Studying the expression levels of osteogenic genes at earlier time points may help to answer these questions and shed light on the osteogenic differentiation process of human iPSC-MP cells in combination with these scaffold materials. The increased expression of osteogenic genes observed when cells are cultured on decellularized cow bone scaffolds may be of benefit for the construction of qualified in vitro model of development and disease using these scaffolds.
While the ALP activity increased significantly upon culture in bioreactors, no significant differences were observed when comparing the two groups at the end of the culture period. On the other hand, cells cultured on decellularized human bone scaffolds released a higher amount of osteocalcin. As osteocalcin is produced by osteoblasts, it is often used as a marker for the bone formation process.41,42 The difference observed between the two experimental groups indicate that the scaffolds somehow influence the osteogenic differentiation process, and this may be related to differences in the physical parameters of the scaffolds such as porosity and pore size.20,43 Similar to the expression of osteogenic genes, one would expect osteocalcin to be released in higher amounts when cells are cultured on decellularized cow bone scaffolds. However, decellularized human bone scaffolds display larger pores and overall higher porosity, which are recognized to favor cell proliferation. 34 Therefore, the higher release of osteocalcin observed in this study might be associated with a higher number of cells in the constructs, which result from the higher cell adhesion following seeding and increased cell proliferation during culture in bioreactors. It remains to be determined whether the higher amount of released osteocalcin boosts the regenerative properties of these grafts in vivo.
Despite the difference observed in the expression of osteogenic genes and production of bone-specific markers, histological and μCT analysis of the samples revealed similar formation of mineralized tissue for both experimental groups, as evidenced by progressive cell penetration throughout the scaffolds, synthesis of an extracellular matrix with characteristics typical of the bone tissue, and increased mineral content. Even if the content of inorganic material is the same, differences in the amount and ratio of bone matrix proteins may affect the quality and regenerative properties of tissue-engineered bone grafts. In vivo studies in animal models of skeletal defects are needed to test this hypothesis, and understand the relation between gene expression, matrix composition, and tissue engineering potential of the bone grafts engineered in this study. Although important questions remain on the effect of the investigated scaffolds on the profile of expression or production of bone-specific markers, taken together the results of this study indicate that decellularized cow bone scaffolds represent a suitable and more affordable alternative to decellularized human bone scaffolds for engineering bone grafts using human stem cells.
Conclusion
The results of this study demonstrate that, despite minor differences in architecture and mass composition, decellularized cow and human bone scaffolds equally support cell viability and formation of mineralized tissue when combined with human iPSC-MP cells. Decellularized cow bone scaffolds therefore represent a suitable and more affordable alternative for engineering human bone grafts for basic and applied research.
Footnotes
Acknowledgments
We thank Dr. Dieter Egli for the human iPSC line 1013A; Prof. Gordana Vunjak-Novakovic for help with processing of decellularized cow bone scaffolds and for the perfusion bioreactors; Dr. Steve Chang and Dr. Darja Marolt for help with procurement of human cadaveric bone; Hector Martinez for mycoplasma testing; Kennedy Agwamba for helpful suggestions concerning the Nanostring analysis; Dr. Rick Mosma for proofreading the article. Funding was provided by the New York Stem Cell Foundation Research Institute (G.M.d.P.), The Ralph and Ricky Lauren Family Foundation (G.M.d.P.), and by the EU FP7 NMP Biodesign programme (H.E.).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.