
Abstract
Osteoarthritis (OA) is a highly prevalent disease with limited treatment options. The search for disease-modifying OA therapies would benefit from a more comprehensive knowledge of the genetic variants that contribute to chondrocyte dysfunction and the barriers to cartilage regeneration. One goal of this study was to establish a system for producing engineered cartilage tissue from genetically defined primary human chondrocytes through genome editing and single-cell expansion. This process was utilized to investigate the functional effect of biallelic knockout of the cell cycle inhibitor p21. The use of ribonucleoprotein (RNP) CRISPR/Cas9 complexes targeting two sites in the coding region of p21 resulted in a high frequency (16%) of colonies with homozygous p21 knockout. Chondrogenic pellet cultures from expanded chondrocytes with complete loss of p21 produced more glycosaminoglycans (GAG) and maintained a higher cell number. Single-cell-derived colonies retained the potential for robust matrix production after expansion, allowing for analysis of colony variability from the same population of targeted cells. The effect of enhanced cartilage matrix production in p21 knockout chondrocytes persisted when matrix production from individual colonies was analyzed. Chondrocytes had lower levels of p21 protein with further expansion, and the difference in GAG production with p21 knockout was strongest at early passages. These results support previous findings that implicate p21 as a barrier to cartilage matrix production and regenerative capacity. Furthermore, this work establishes the use of genome-edited human chondrocytes as a promising approach for engineered tissue models containing user-defined gene knockouts and other genetic variants for investigation of OA pathogenesis.
Impact Statement
This work provides two important advances to the field of tissue engineering. One is the demonstration that engineered cartilage tissue can be produced from genetically defined populations of primary human chondrocytes. While CRISPR/Cas-9 genome editing has been extensively used in cell lines that divide indefinitely, this work extends the technique to an engineered tissue model system to support investigation of genetic changes that affect cartilage production. A second contribution is the finding that chondrocytes with p21 knockout synthesized more cartilage matrix tissue than unedited controls. This supports the continued investigation of p21 as a potential barrier to effective cartilage regeneration.
Introduction
Osteoarthritis (OA) is characterized by joint pain and the progressive degradation of articular cartilage and surrounding tissues.1,2 Primary human chondrocytes are a valuable resource for understanding the molecular pathways involved in cartilage dysfunction and OA, but the tools for manipulating chondrocytes have been mostly limited to partial knockdown, overexpression, and pharmacological inhibition. Genetics contribute approximately half of the risk for OA 3 and there is a need to develop methods for the functional characterization of genetic variants that have been identified by genome-wide association studies (GWAS).4–6 Genome editing has been applied to chondrocytes for the generation of a chondrosarcoma cell line with aggrecan knockout 7 and for the disruption of matrix metalloproteinase 13 from a subset of cells within a bulk population of human chondrocytes. 8 For genome editing strategies to be extended to investigation of complete knockout or GWAS variant modeling in chondrocytes, methodologies must be developed to expand and analyze single-cell-derived colonies as a way of overcoming the stochastic nature of editing outcomes. While this approach is compatible with immortalized cell lines and induced pluripotent stem cells (iPSCs), it presents challenges for primary cell types that would otherwise be ideally suited for the research question. One goal of this study was to establish a system by which primary human chondrocytes could be edited and expanded as individual colonies for subsequent cartilage tissue engineering.
The poor healing capacity of articular cartilage has spurred the development of numerous approaches to provide compensatory regeneration for focal cartilage defects, including marrow stimulation by microfracture, 9 autologous chondrocyte implantation, 10 and tissue engineering. 11 Understanding the blocks to endogenous regeneration will improve these treatment paradigms and may also lead to new strategies for the treatment and prevention of OA, as work with a set of recombinant inbred mice has connected the lack of regenerative capacity to OA susceptibility.12–14 Genetic backgrounds conducive to superior healing of ear holes have increased articular cartilage regeneration 12 and are protected from OA.13,14 One mediator of cartilage healing is the cell cycle inhibitor p21, as p21 knockout mice were able to recapitulate the ear closure regenerative phenotype of Murphy-Roths-Large (MRL) “superhealer” mice. 15 Our previous work used short-hairpin RNA knockdown of p21 in iPSC-derived chondrocytes to demonstrate that reducing the level of p21 enhanced the synthesis of cartilage matrix. 16 This finding is consistent with the relationship between p21 levels and differentiation potential of synovial mesenchymal stem/stromal cells (MSCs), 17 as well as the enhanced chondrogenesis with pharmacologic inhibition of p21 in this cell type. 18 The current study generates engineered tissue from edited primary human chondrocytes to investigate the cartilage-forming potential of cells with and without the complete knockout of p21.
Materials and Methods
An overview of the methodological approach is summarized in Figure 1 and involves delivering ribonucleoprotein (RNP) complexes for CRISPR/Cas9-based editing, screening single-cell-derived colonies, and then generating chondrogenic pellet cultures after further expansion.
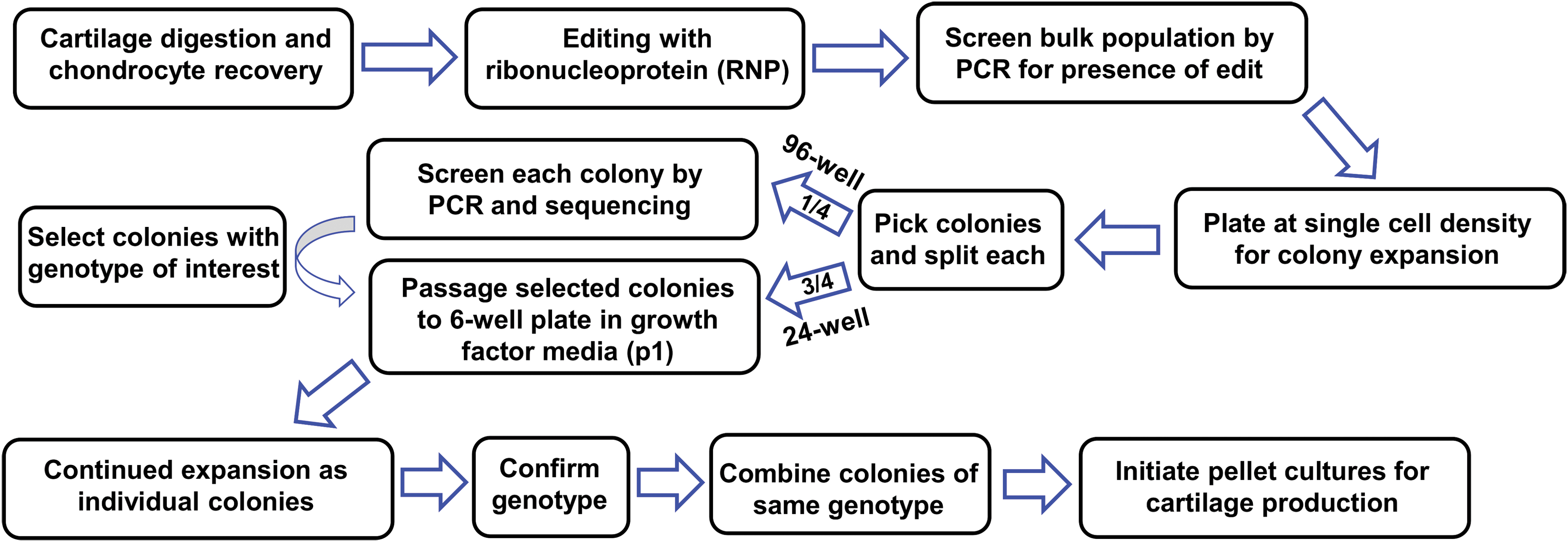
Workflow of system for engineering cartilage tissue from genome-edited human primary chondrocytes. Color images are available online.
Isolation and culture of human articular chondrocytes
Normal human cartilage was obtained from ankle joints without OA from three male donors aged 49 (donor 1), 47 (donor 2), and 50 (donor 3) years through the Gift of Hope Organ and Tissue Donor Network (Itasca, IL) via Rush Medical College (Chicago, IL). Primary human articular chondrocytes were isolated by sequential pronase and collagenase digestion as previously described. 19 Chondrocytes were plated at a density of 1.5 million cells per 6 cm2 dish (∼70,000 cells/cm2) in chondrocyte media consisting of Dulbecco's modified Eagle's medium (DMEM)/Nutrient Mixture F-12 HEPES (11330-032; Gibco) supplemented with 10% fetal bovine serum (FBS; Cat. No. 1500-500; VWR), 1 × Penicillin–Streptomycin (15140122; Gibco), 2.5 μg/mL Amphotericin B (A2942; Sigma), and 4 μg/mL Gentamicin (1575-060; Gibco). Following recovery for 8–10 days, cells were trypsinized (T3924; Sigma), washed twice with phosphate-buffered saline (PBS), and centrifuged at 100 g for 8 min in preparation for transfection.
Preparation of genome editing components
RNP complex containing the Cas9 enzyme and sequence-targeting guide RNAs were prepared according to manufacturer's recommendation (Integrated DNA Technologies). In brief, the tracrRNA (1072533; Integrated DNA Technologies) and crRNA (Human p21 guide AD: CCAAGCTCTACCTTCCCACG, Human p21 guide AC: TTCTGACGGACATCCCCAGC, Negative Control crRNA No. 1) were each resuspended in Tris-EDTA buffer to 200 μM stock concentration. Equimolar concentrations of crRNA and tracrRNA were combined and heated at 95°C for 5 min and then cooled to produce the 100 μM crRNA:tracrRNA duplex. Separate RNP complex for each guide was prepared by combining the crRNA:tracrRNA duplex with 10 μg/μL Alt-R® Cas9 Nuclease (1081058; Integrated DNA Technologies) and PBS at a ratio of 1.2:1.7:2.1 at room temperature for 15 min.
Transfection of primary human chondrocytes with RNP complex
RNP complex was delivered using the Amaxa Nucleofector™ system (Lonza). Following trypsinization and washing with PBS, ∼200,000 cells were resuspended in a mixture containing 20 μL of P3 Primary Cell Nucleofector solution (V4XP-3032; Lonza), 5 μL of RNP complex for each guide (both guides AC and AD, or negative control), and 1 μL of 100 μM Alt-R Cas9 Electroporation Enhancer (1075916; Integrated DNA Technologies). The mixture was gently pipetted up and down and transferred to the 16-well Nucleocuvette™ strip (V4XP-3032; Lonza) and transfected using program ER-100 on a 4D-Nucleofector Core unit (Lonza). Transfected cells were incubated at room temperature for 5–10 min and then transferred to 24-well plate containing prewarmed antibiotic free chondrocyte media supplemented with 20% FBS for recovery. An aliquot of the cells was placed in a 96-well to be used for confirmation of successful genome editing by polymerase chain reaction (PCR). Following confirmation of editing in the bulk cell population of the 96-well plate (2–4 days), the cells from the 24-well plate were passaged and seeded at low cell density (200 cells per 6 cm2 dish) for 8–12 days of expansion as single-cell-derived colonies. Notably, colonies formed as seen in previous work 20 despite the difference that the current approach did not perform fibronectin-based selection of a chondrocyte subpopulation. 20 Individual colonies were picked with a pipette tip using a microscope (EVOS FL; Thermo Fisher) placed in a laminar flow hood and split into 96-well and 24-well plates for analysis of genome editing and continued expansion, respectively.
Screening single-cell-derived colonies for editing outcomes by PCR and sequencing
Cells in 96-well plates were washed once with PBS before addition of QuickExtract™ DNA Extraction Solution (Lucigen). The volume was adjusted based on the estimated cell number, with a minimum of 35 μL and maximum of 100 μL for a nearly confluent well. After a 15-min incubation at 37°C, cells were pipetted up and down, transferred to tubes (490003-722; VWR), and vortexed for 1 min. The cell suspension was then heated at 65°C for 6 min, vortexed briefly, and placed at 98°C for 2 min. Extracted DNA solution was kept at −20°C until PCR. PCR amplification was performed by adding 5 μL template DNA, 1 μM forward (TTAGCTTGCCCTTCAGTTGC) and reverse (5′-GGTCTTTGCTGCCTACTTGC-3′) primers, and EconoTaq PLUS GREEN 2 × Master Mix (Lucigen) in a 25 μL reaction. PCR conditions included an initial denaturation at 94°C for 2 min, 35 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 50 s, followed by a final extension at 72°C for 10 min. For sequencing of PCR products, the DNA from the PCR reaction was purified using a cleanup kit (T1030; New England Biolabs) and sequenced using SimpleSeq™ tubes (Eurofins Genomics) and the following primer: ACCAGCTGGAAGGAGTGAGA. Chromatograms were visualized using BioEdit software.
Cell expansion in monolayer
Selected colonies with confirmed edits (or negative controls) from the 24-well plate were trypsinized and plated as passage 1 cells at a density of ∼3,000 cells/cm2 in media supplemented with 1 ng/mL TGF-β1 (PHG9214; Life Technologies) and 5 ng/mL bFGF (PHG0264; Life Technologies). Pellets were made at the end of passage 3 (donor 1), the end of passage 1 (donor 2), or at the end of passage 1 through passage 4 (donor 3). The fold increase and number of population doublings were calculated based on counts obtained using a Countess™ II Automated Cell Counter.
Pellet culture for evaluation of chondrogenic potential
Pellets were made from pooled colonies after expansion (donors 1 and 3) or from individual single-cell-derived colonies (donor 2). In brief, 250,000 cells were placed in 15 mL tubes and washed with serum free DMEM-high glucose (DMEM-HG, 11995-065; Sigma). DMEM was replaced with differentiation media consisting of DMEM-HG supplemented with 1% ITS+Premix (354352; Corning), 1 × Penicillin–Streptomycin (Gibco), 100 nM dexamethasone (D4902; Sigma), 50 μg/mL proline (P5607; Sigma), 50 μg/mL
Analysis of total DNA and glycosaminoglycan content
Pellets were digested with a solution consisting of 125 μg/mL papain, 0.1 M sodium phosphate, 5 mM EDTA, and 5 mM cysteine hydrochloride at pH 6.5. Digestion was performed for 24 h at 65°C in an oven or with agitation in a ThermoMixer C (Eppendorf) at 600 rpm. The QuantiFluor® double-stranded DNA (dsDNA) System was used to measure dsDNA content according to manufacturer's instructions. Glycosaminoglycans (GAG) content of the pellet was evaluated using the DMMB assay as previously described 21 with 1,9 dimethylmethylene blue dye at pH 3 and a chondroitin-4-sulfate standard curve.
Histology and immunohistochemistry
Pellets were fixed in 4% paraformaldehyde for 24 h at 4°C and processed for paraffin embedding. For histology, slides were stained with Fast Green (0.02% in dH20, 25 min; 7252; Sigma) and Safranin-O (1.5% in dH20, 30 min; S8884; Sigma). For immunohistochemistry, antigen retrieval was performed using pepsin (Digest-All™ 3; 003009; Thermo Fisher) for 5 min at room temperature. Samples were incubated overnight at 4°C using a primary antibody targeting type II collagen (II-II6B3, no dilution; Developmental Studies Hybridoma Bank), followed by an anti-mouse secondary antibody (715-065-151, 1:1000; Jackson ImmunoResearch) and visualization using VECTASTAIN® Elite ABC-HRP (PK-6100; Vector Laboratories) and DAB Peroxidase (SK-4100; Vector Laboratories). For both histology and IHC, slides were counterstained with hematoxylin and mounted with Permount™ for imaging.
Protein isolation and western blotting
Protein isolation and western blotting were performed as described. 22 In brief, TRIzol™ (Thermo Fisher) was used on both monolayer cells and pellets. For monolayer, TRIzol was added to cells and vortexed for 1 min. Pellets were placed in bead beater tubes (10158-610; VWR) with TRIzol and homogenized at 6500 rpm × 30 s for three cycles (Precellys® 24; Bertin Corp.). Protein concentration was determined using the Micro BCA Protein Assay Kit (Thermo Fisher) and either 12 μg (monolayer cells) or 8 μg (pellets) was used for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The membrane was cut after blocking and the lower half was incubated in p21 antibody (2947, 1:2000; Cell Signaling Technology), while the upper half was incubated in anti-G3PDH Human Polyclonal Antibody (GAPDH; 2275PC, 1:2000; Trevigen), both overnight at 4°C. Following incubation in secondary antibody solution (1:2000; Cell Signaling Technology) for 1 h, the membranes were washed and placed in the Radiance Plus chemiluminescent substrate and imaged with the Azure c600 gel imaging system (Azure Biosystems).
Gene expression analysis
RNA isolation and reverse transcription was performed as described. 22 In brief, TRIzol (Thermo Fisher) was used in combination with column-based purification, followed by use of the qScript XLT Supermix (VWR). Quantitative PCR was performed using SYBR Green Master Mix (Catalog No. 172-5271; Bio-Rad) and the following primers written as 5′–3′: Peptidylprolyl Isomerase A (PPIA, F: GTCAACCCCACCGTGTTCTT, R: CTGCTGTCTTTGGGACCTTGT); Type II collagen (COL2, F: GGCAATAGCAGGTTCACGTACA, R: CGATAACAGTCTTGCCCCACTT); Type I collagen (COL1, F: CAGCCGCTTCACCTACAGC, R: TTTTGTATTCAATCACTGTCTTGCC); Aggrecan (ACAN, F: TCGAGGACAGCGAGGCC, R: TCGAGGGTGTAGCGTGTAGAGA), Versican (VCAN, F: TGGAATGATGTTCCCTGCAA, R: AAGGTCTTGGCATTTTCTACAACAG).
Statistical analysis
Statistical analysis and plotting were performed using Prism 7 (GraphPad, La Jolla, CA). Data are plotted as individual points and error bars indicate mean ± standard error of the mean. Normality was assessed by Shapiro–Wilk test and data were analyzed by unpaired t-test or two-way analysis of variance (ANOVA) with Tukey's post hoc analysis.
Results
Genome editing in primary human chondrocytes
We developed an efficient system for generating engineered tissue from primary human chondrocytes with the homozygous deletion of a target gene. Transfection efficiencies were estimated at 40–60% based on trial experiments using a green fluorescence protein (GFP) plasmid (Supplementary Fig. S1). The genomic DNA of p21 was targeted with guide RNAs that generate double-stranded breaks at 15 base pairs and 237 base pairs into the coding region (Fig. 2A). Thus, cutting at both locations would generate a 222 base pair deletion. The bulk population of edited cells was assessed by PCR amplification of the targeted region, which showed that including the p21 guide RNAs generated a novel amplicon that is ∼222 base pairs smaller than the parent product (Fig. 2B, left). Repeating this analysis on single-cell-derived colonies allows for the discrimination of the editing outcome in each colony: fully intact (parent product only), heterozygous targeted (both parent and smaller product), and homozygous targeted (smaller product only) (Fig. 2B, right). The efficiency of editing was estimated by analyzing the percentage of screened colonies with each editing outcome. Analysis of 80 colonies from donors 2 and 3 showed that 53 (66%) colonies had a heterozygous deletion and 13 (16%) had a homozygous deletion (Fig. 2C). To gain further insight into the exact editing outcomes, we performed Sanger sequencing on a subset of the PCR products (Fig. 2D). The homozygous colonies confirmed a single read with the expected deletion, with the example shown having the same 221 base pair deletion on both alleles (Fig. 2D). Notably, even the colonies scored as “wild type” by PCR due to the lack of a large deletion showed multiple reads beginning at the expected guide cut site, which is consistent with the error-prone nonhomologous end joining (NHEJ) repair pathway (Fig. 2D).

Screening and efficiency of homozygous p21 KO.
Single-cell-derived colony expansion
Individual colonies that had been targeted by the negative control guide RNA (p21 intact) and those screened and sequenced as p21 knockout were expanded in either 10% FBS chondrocyte medium or with the addition of 1 ng/mL TGF-β1 and 5 ng/mL bFGF. The growth factors resulted in a larger fold increase during passage 1 regardless of genotype (Supplementary Fig. S2A). As engineered tissue production requires a substantial number of cells and previous studies have shown that culture with TGF-β1 and bFGF is conducive to subsequent chondrogenesis,20,23 further studies were completed using the growth factors for expansion. Under these conditions, all colonies from p21 intact and p21 knockout groups were capable of more than 22 population doublings from the time of single-cell plating through the end of the third passage (Supplementary Fig. S2B), which is equivalent to over 4 million cells per starting colony. Gene expression analysis of chondrocytes at the time of single-cell plating and after colony expansion shows the emergence of a “de-differentiated” chondrocyte phenotype with reduced expression of COL2 and ACAN, as well as stable COL1 and increased VCAN (Supplementary Fig. S3). This phenotype was reversible, as expression levels of COL2 and ACAN after pellet culture were above baseline and expression levels of COL1 and VCAN were below baseline (Supplementary Fig. S3).
Effect of homozygous p21 knockout on pellet chondrogenesis (donor 1)
To determine the effect of p21 knockout, we first combined the expanded colonies of each genotype and confirmed the complete loss of measurable p21 protein by western blot (Fig. 3A). Chondrogenic pellet cultures with 250,000 cells each were established from both genotypes and those made from p21 knockout chondrocytes had a higher weight at the end of the 28-day culture period (Fig. 3B). Digestion of the pellets for analysis of biochemical content showed that p21 knockout pellets also had significantly higher GAG content (Fig. 3C). Part of this increased matrix production is likely due to the higher number of cells in pellets from p21 knockout chondrocytes as assessed by dsDNA content (Fig. 3D). Paraffin sections were analyzed to show the presence of enhanced GAG (Fig. 3E) and type II collagen (Fig. 3F) in the p21 knockout pellets compared with controls.
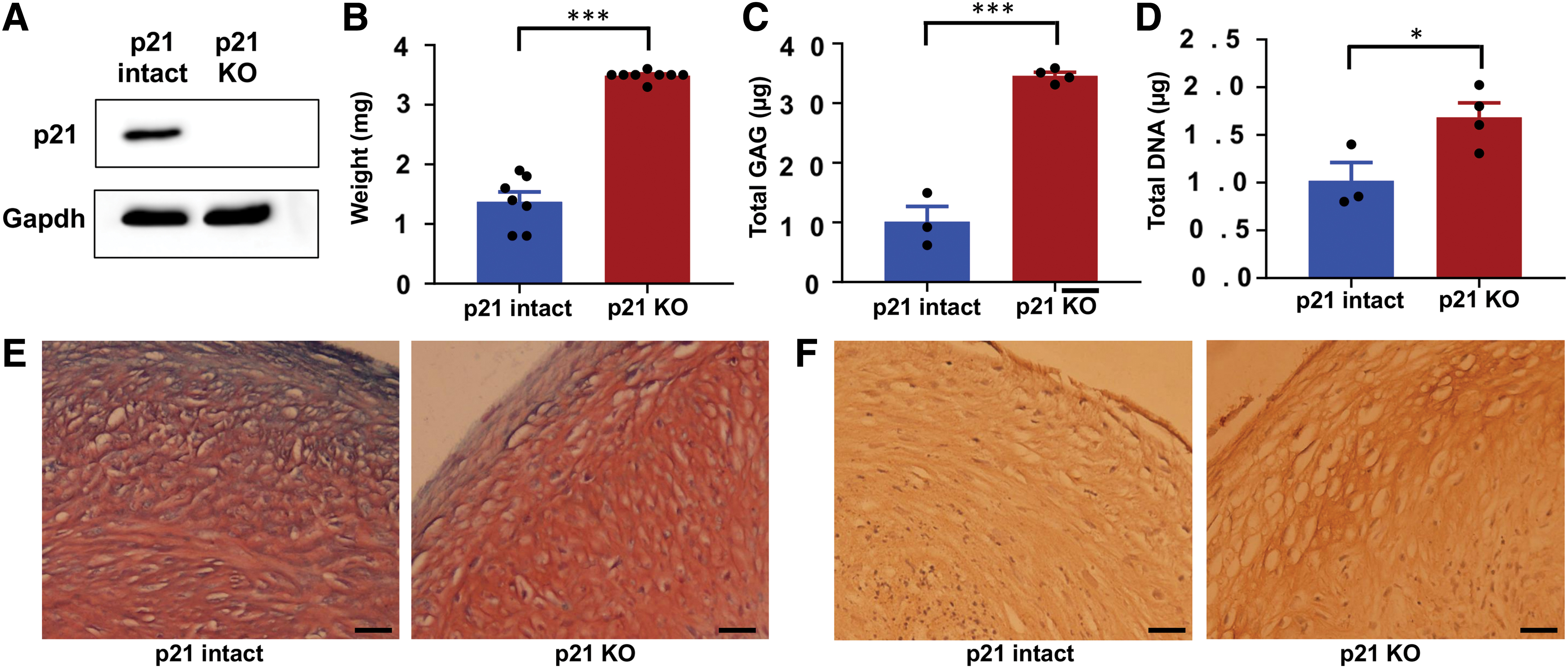
Cartilage matrix production with p21 KO (donor 1).
Differences in chondrogenic potential by colony (donor 2)
Combining the cells of expanded colonies before pellet culture eliminates the potential to investigate the variability in how a particular edit (in this case p21 knockout) affects chondrogenesis. Thus, edited chondrocytes from donor 2 were expanded through end of the first passage and made into pellet cultures from 10 p21 intact and 5 p21 knockout colonies. Biochemical data from independent pellets for each colony were typically clustered for both GAG (Fig. 4A) and DNA (Fig. 4B), with larger variation between different colonies of the same genotype. For clarity, data were also averaged to a single value per colony for both GAG (Fig. 4C) and DNA (Fig. 4D). The variability between colonies from the same bulk population of cells is consistent with previous work analyzing the chondrogenic potential of single-cell-derived chondrocyte colonies.23,24 However, the effect of p21 knockout persisted through the colony variability, as both DNA content and GAG content were increased in the set of colonies with homozygous loss of p21 compared with control.
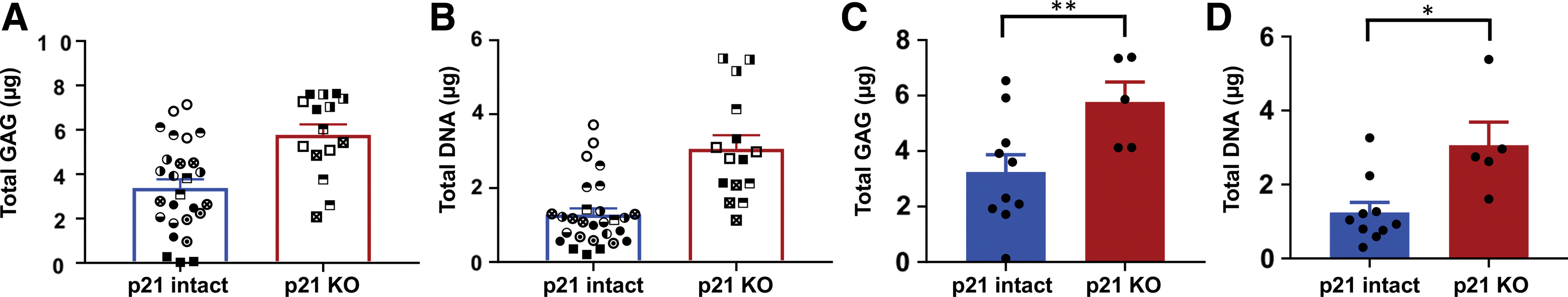
Pellets from individually expanded colonies (donor 2). For
The effect of p21 knockout on chondrogenesis of expanded chondrocytes (donor 3)
Reducing the level of p21 through hypoxia, growth factors, or pharmacological inhibition has been shown to maintain proliferative and differentiation potential during the expansion of MSCs.18,25–27 To determine the way in which p21 knockout and continued expansion affect chondrogenesis, we established pellet cultures at the end of passages 1 through 4. As expected, given the genetic nature of the edit, the complete knockout of p21 was confirmed to be stable both during expansion and throughout pellet culture (Fig. 5A). Protein levels in the p21 intact samples were quantified and the production of p21 was similar at the end of pellet culture compared to monolayer cells at that passage (Fig. 5B). The level of p21 protein decreased with extensive passaging (Fig. 5B), perhaps due to repression by the presence of bFGF or due to preferential expansion of p21 low chondrocytes within the population.
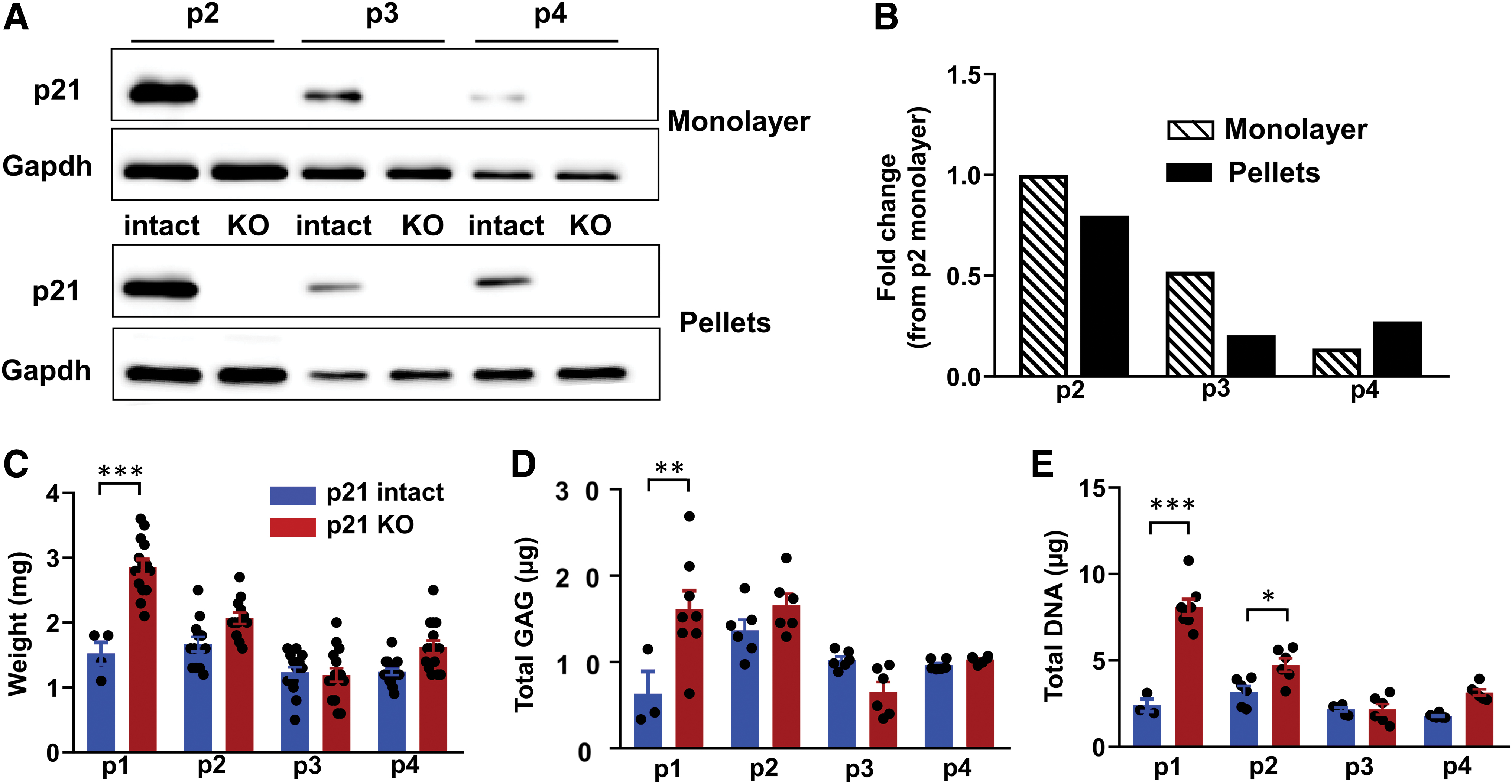
Effect of continued expansion in monolayer (donor 3).
Analysis of the pellets at the end of each passage demonstrated a significant effect of both passage number and genotype by two-way ANOVA for pellet weight (Fig. 5C), GAG content (Fig. 5D), and DNA content (Fig. 5E). Comparisons between genotypes at each passage indicated that the p21 knockout had the strongest effect on pellets made at passage 1, with a significant effect on pellet weight, DNA content, and GAG content. In this donor, the chondrogenic potential of cells at later passages was not affected by the knockout of p21 (Fig. 5C–E).
Discussion
The ability to establish genetically defined populations of primary human articular chondrocytes has profound implications for the study of cartilage biology and OA. 28 However, a major barrier to genome editing in chondrocytes is the challenge of expanding edited colonies without losing the potential to synthesize robust cartilage matrix. This study establishes an efficient approach for engineering cartilage tissue from primary human chondrocytes and validates the method by targeting the cell cycle inhibitor p21. Consistent with the hypothesized role of p21 in restraining regenerative capacity, cartilage tissue generated from chondrocytes with complete p21 knockout had more cells and a higher GAG content.
This work is complementary to other genome editing approaches that have been used to study cartilage biology. Yang et al. expressed Cas9 in a rat chondrosarcoma cell line to allow for the knockout of genes through guide RNA transfection. 7 The authors then generated stable cell lines with complete knockout of aggrecan to demonstrate the effect of this critical cartilage component on cell morphology and chondrosarcoma development. 7 Seidl et al. targeted a bulk population of primary human chondrocytes with the RNP version of CRISPR/Cas9 to initiate a reduction in functional MMP-13. 8 While single-cell-derived colonies were not selected in this study, generating NHEJ in the coding region of MMP-13 was sufficient to reduce the level of MMP-13 in the bulk cell population. Because MMP-13 is a secreted factor that compromises function at the tissue level, the reduced MMP-13 resulted in a higher quality engineered tissue. 8 Another genome editing strategy is to modify pluripotent cells before subsequent differentiation to the chondrogenic phenotype. While using iPSC-derived cartilage for modeling OA is attractive in terms of storing edited progenitors and the opportunity for tissue scale-up, 29 genome modification occurs during the pluripotent stage and thus the effect of the change may be enacted during the complex differentiation toward the chondrocyte phenotype.30–33 Each of these previous approaches (generating stably modified cell lines, bulk targeting of chondrocytes, iPSC editing, and differentiation) has merit for particular applications, and we propose that the current paradigm will be particularly useful in experiments that require genetically defined populations of primary articular chondrocytes.
We selected p21 as a first target for this approach due to the intriguing body of literature suggesting that p21 is a potent repressor of tissue regeneration. The MRL “superhealer” mouse strain was originally identified by the scarless closure of ear holes. 34 MRL mice also showed improved regeneration in many other tissue contexts, including full-thickness articular cartilage defects. 35 Following the lead of altered cell cycle distribution in cells from MRL mice, Bedelbaeva et al. showed that the regenerative capacity was linked to lower inducible levels of p21 and that p21 knockout mice showed the ear hole closure phenotype. 15 Subsequent lineage tracing studies in p21 knockout mice demonstrated that regenerated cartilage is produced by previously existing chondrocytes that reenter the cell cycle and produce new tissue. 36 The enhanced regenerative capacity of cartilage tissue with loss of p21 appears to extend to articular cartilage as well, with a recent study showing that p21 knockout mice have superior healing in response to focal cartilage injury. 37 Interestingly, p21 knockout mice are more susceptible to an instability model of posttraumatic OA, illustrating the complex nature of disease pathogenesis, and raising the possibility that changes to other aspects of OA may counter the potentially beneficial effects of enhanced cartilage regenerative potential.38,39
Given the complexity of in vivo regenerative capacity, in vitro models can be useful as a way to focus on tissue-intrinsic changes with loss of p21. We had previously shown that the stable (but incomplete) small hairpin RNA (shRNA) knockdown of p21 in murine iPSC-derived chondrocytes increased the chondrogenic potential of this cell type. 16 The current use of permanent (and complete) knockout in primary human chondrocytes gave similar results in terms of pellet cultures with increased weight, cell number, and GAGs. However, the interaction of p21 loss and cell expansion on chondrogenic potential was different with the two cell types. In the previous study, p21 knockdown cells generated similar quality engineered cartilage pellets at the end of two or five passages, which was different than control cells and therefore extended the expansion window before the loss of chondrogenic potential. 16 In the current study, p21 knockout had the largest effect on cells at early passages, and p21 knockout cells at the end of passage 4 were similar to controls.
One limitation of the single-cell colony approach is that heterogeneity of the outcome between individual colonies of the same genotype may make it challenging to detect the effect of the genome editing. The potential variability is illustrated by Fellows et al. in their description of the number of population doublings before senescence in chondrocyte colonies from normal and OA patients. 20 Colony variability in expanded chondrocytes can also lead to differing capabilities for cartilage matrix production.23,24 To explore the effect of colony variability in our system, we made pellet cultures from individually expanded colonies of each genotype (donor 2, Fig. 4). While there was differing chondrogenic capacity in these colonies, the genotype effect of p21 knockout was still present as evidenced by a significant increase in GAG content and DNA. This reinforced the data from donors 1 and 3, in which pellets were made from the combined cells of numerous colonies of each genotype at the end of expansion. These findings suggest that other applications of this technology will need to consider colony variability during experimental design, especially when the genetic modification is expected to generate a relatively subtle effect on matrix production. Other limitations of this study include the use of a small number of male donors. Future work will need to determine the consistency of the effects of particular edits across numerous donors of both sexes. Furthermore, functional assays such as mechanical testing would provide additional data to supplement the analysis of tissue composition that is presented in the current work.
A major contribution of this work is to establish a framework for genome editing in primary human chondrocytes. One particularly exciting use of this technology will be to perform functional follow-up of OA risk variants. Follow-up from GWAS has required techniques such as allelic expression imbalance and reporter assays to determine the function of the specific genetic changes.40,41 The current approach extends the possibilities of functional validation by generating isogenic populations of primary chondrocytes—where the only difference from control cells is the user-defined change. While some variants may lead to altered matrix synthesis, other variants may require the engineered tissues to be challenged in vitro with inflammatory cytokines or other methods to detect an aberrant response due to genetic background. These sophisticated models will be critical for assessing the increasing number of genetic variants that have been identified as risk factors for OA.
Footnotes
Acknowledgment
The authors thank the UNC Animal Histopathology core.
Disclosure Statement
Start-up funds provided by the Thurston Arthritis Research Center and the Department of Biomedical Engineering. No competing financial interests exist.
Funding Information
This work was conducted while B.O.D. was an Arthritis and Aging Research Grant recipient from the Arthritis National Research Foundation (ANRF) and the American Federation for Aging Research as well as second-year funding from ANRF.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.