
Abstract
The Editors of Tissue Engineering: Part A retract the article entitled, “Cell Fate and Tissue Remodeling in Canine Urethral Repair Using a Bone Marrow Mesenchymal Stem Cell+Endothelial Progenitor Cell Amniotic Patch,” by Wenxin Zhang, Xin Zhang, Yihua Zhang, Xinke Zhang, Tong Zou, Wen Zhao, Yangou Lv, Jinglu Wang, Pengxiu Dai, Hao Cui, Yi Zhang, Dengke Gao, Chenmei Ruan, and Xia Zhang (epub ahead of print September 21, 2020; DOI: http://`.org/10.1089/ten.tea.2020.0129).
After the online publication of the article, the authors have indicated that they “feel that we have not yet studied our work completely and some new great results are discovered. So after carefully thinking, we are going to rearrange this manuscript and try to give more precise model. [sic]” The authors have not explained what those expected results will be, so it remains unclear the direction their work is headed.
The authors also indicated that they plan to submit an updated version of the paper to Tissue Engineering in the future. Upon submission the new manuscript will undergo rigorous peer review, and there is no guarantee of acceptance.
Impact statement
In the complete repair of a 3-cm-long circumferential urethral defect of the canine perineum using bone marrow mesenchymal stem cell (BMSC)+endothelial progenitor cell (EPC) amniotic membrane patch labeled with green and red fluorescent proteins, BMSCs mainly differentiated into urothelial cells, EPCs mainly differentiated into vascular endothelial cells, these two cells together participated in the repair of the urethral mucosal epithelium, and most of the submucosal tissues were formed by the growth of the adjacent recipient cells. Fluorescent protein labeling had no influence on stem cell proliferation and tissue repair ability.
Introduction
Urethral defect is a common disease that is caused by segmental urethral necrosis due to factors such as injury, tumor resection, and stones. The traditional surgical treatment for the disease is used to only repair small or partial urethral defect, but not a long segment of circumferential urethral defect. The tissue-engineered urethra made from scaffold can cause ischemic necrosis and infection after transplantation. Bone marrow mesenchymal stem cells (BMSCs) not only differentiate into multiple mesodermal and nonmesodermal cell lineages, including epithelial cells, but also possess low immunogenicity 1 and immunosuppression by modulating the immune function of the major cell population involved in alloantigen recognition and elimination. 2
Endothelial progenitor cells (EPCs) participate in vascular remodeling and angiogenesis by migrating to the site in need of blood vessels 3 and BMSCs can contribute to EC survival, proliferation, and mobility, and help recruit EPCs for angiogenesis. 4 Previously, a tissue-engineered amniotic membrane (dHAS+BMSCs+EPCs) patch was constructed using decellularized human amniotic membrane scaffold (dHAS)-seeded canine BMSCs and EPCs, and used to repair canine 3-cm-long circumferential urethral defects; good results of urethral patency, smooth lumen, complete mucosal epithelialization, and abundant submucosal vessels were achieved after 2 months. 5
To further clarify the differentiation fate of the stem cells and the process of tissue remodeling in the repair of long circumferential urethral defects using the dHAS+BMSCs+EPCs, BMSCs and EPCs were labeled with green fluorescent protein (GFP) and red fluorescent protein (RFP), respectively, and the labeled cells were used to construct the dHAS+BMSCs+EPCs that was then used to repair a 3-cm-long circumferential urethral defect of the canine perineum. The differentiation fate of BMSCs and EPCs and the process of tissue remodeling were assessed using antibodies against GFP and RFP. The obtained results were satisfactory and provide a foundation for the clinical application of this technique.
Materials and Methods
Labeling of canine BMSCs and EPCs
The methods used to isolate, culture, and identify canine BMSCs and EPCs have been described previously. 5
Fourth-generation canine BMSCs were transfected with the lentiviral vector pCDH-CMV-MCS-EF1-copGFP-T2A-Puro, which expresses GFP. The virus particles were added to alpha-Minimum Essential Medium (α-MEM), a complete medium containing 10% fetal bovine serum (FBS) and 8 μg/mL polybrene at a multiplicity of infection (MOI) of 25. The mixture was slowly added to the cell culture plate, and the cells were then cultured in a 37°C, 5% CO2 incubator for 12 h. The mixture was aspirated and replaced with α-MEM, a complete medium containing 10% FBS. Two days after the transfection, the expression of GFP was observed under an inverted fluorescence microscope (Olympus, Japan). When the transfection rate reached 70% or more, puromycin (2 μg/mL) screening was performed.
Fourth-generation canine bone marrow EPCs were transfected with the lentiviral vector pLVX-mcherry-IRES-Puro expressing RFP. The virus particles were added to M199 complete medium containing 10% FBS and 4 μg/mL polybrene at a MOI of 50. The mixture was slowly added to the cell culture plate, and the cells were cultured in a 37°C, 5% CO2 incubator for 12 h. The mixture was then aspirated and replaced with fresh M199 complete medium containing 10% FBS. Two days after the transfection, the expression of RFP was observed under an inverted fluorescence microscope. When the transfection rate reached 70% or more, puromycin (2 μg/mL) screening was performed.
Coculture and detection of proliferative activity of the fluorescent protein-labeled BMSCs and EPCs
To observe the cogrowth of BMSCs and EPCs and the effect of fluorescent protein labeling on cell viability, the labeled BMSCs and EPCs were cocultured, and their proliferative activity was measured. The labeled second-generation BMSCs and EPCs were made into a 1:1 mixed cell suspension, and was placed in a 60-mm cell culture dish. The dish was shaken in a “+” pattern to evenly distribute the cells and placed in a 37°C, 5% CO2 incubator. The growth and distribution of the cells were observed using an inverted fluorescence microscope.
At the same time, the mixed cell suspension was inoculated into a 96-well plate at 1 × 103 cells/well, and the CCK-8 Cell Proliferation and Activity Assay Kit (BL-782748; Zeta Life, Inc.) was used to evaluate the cells' proliferative activity after coculture for 1, 3, and 5 days. Sixth-generation (equivalent passage) unlabeled mixed BMSCs and EPCs were used as a control. Before detection, the medium was replaced with fresh medium; then, 10 μL of CCK-8 solution was added to each well, the plate was incubated for 3 h in a 37°C 5% CO2 incubator, and the optical density (OD) at 450 nm was measured using a Microplate Reader. Four replicates were processed for each group at each time point; the results are presented as the mean ± standard deviation.
Construction and detection of tissue-engineered dHAS patches with labeled BMSCs and/or EPCs
The preparation of dHAS and the construction of tissue-engineered dHAS patches have been described previously. 5 The sterilized and rehydrated dHAS was put into a 60-mm dish containing the BMSC culture medium and/or EPC culture medium, and 1 × 106 fourth-generation labeled BMSCs and/or labeled EPCs were seeded onto it. Then, the cell-seeded sheet was cultured for 2 days to obtain tissue-engineered dHAS+BMSCs+EPCs, dHAS+BMSCs, and dHAS+EPCs patches, respectively.
Detection of the engineered dHAS patches
The growth of cells on the dHAS was observed with a microscope. A small piece of an engineered dHAS patch was used to prepare paraffin sections; the sections were stained with Hematoxylin–Eosin (HE), and the morphology of the cells growing on the dHAS was examined.
To detect the proliferative activity of the cells on the dHAS patch, 12 pieces of 0.5 × 0.5 cm dHAS were respectively placed in a plate hole of a 96-well plate and inoculated with a 1:1 cell suspension of fourth-generation labeled BMSCs and EPCs at a density of 1 × 103 cells/well. After 1, 3, and 5 days of culture, cell proliferation activity was evaluated using the CCK-8 Cell Proliferation and Activity Assay Kit; the same preparation of cells cultured directly in 12 plate holes without dHAS of the 96-well plate served as a control. The medium was replaced with fresh medium before detection; then, 10 μL CCK-8 solution was added to each well, the plate was incubated for 3 h in a 37°C, 5% CO2 incubator, and the OD at 450 nm was measured using a Microplate Reader. Four replicates were processed for each group at each time point; the results are presented as the mean ± standard deviation.
Preparation and repair of the canine urethral defect model
A total of 20 healthy adult male mongrel dogs each weighing 6–9 kg were used in this study. The animals were provided by the Experimental Animal Center of Northwest A&F University, and this study was approved by the Animal Ethics Committee of Northwest A&F University. The dogs were randomly divided into five groups of four dogs each: three experimental groups (dHAS+BMSCs+EPCs group, dHAS+BMSCs group, and dHAS+EPCs group) and two control groups (dHAS group and sham-operated group). The insertion of an 8F catheter, preparation, and repair of urethral defect model in dog were as reported previously (Fig. 1). 5

Surgical modeling and patch graft repair.
The postoperative analgesia and care were the same as reported previously. 5 The wound was checked daily to ensure that it was clean and dry, and the urination of the dogs was observed. The suture was removed after the wound healed. The Elizabeth circle and the catheter were withdrawn 2 weeks after the surgery.
Ascending urethrogram
To detect the effect of the labeled cells on the repair of canine urethral defects, ascending urethrogram of the urethras of the experimental dogs was performed before surgery as well as at 4 and 8 weeks after surgery. The method of ascending urethrogram was as reported previously. 5
Urethral necropsy
To observe the effect of the labeled cells on canine urethral defect repair and to collect the tissue samples, two dogs in each group were euthanized 1 and 2 months postoperatively, and their urethras were cut longitudinally to observe whether the urethra was smooth and level, whether the mucosal epithelialization was complete, and whether stenosis or scars had formed. The related images were photographed. In each group of animals, the newly generated urethra was sampled. The sampling included an anterior segment (proximal end), an anterior middle segment, a middle segment, a posterior middle segment, and a posterior segment (distal end); the samples were fixed with 4% paraformaldehyde.
Monitoring of cell differentiation fate and tissue remodeling during urethral repair using tissue-engineered dHAS patches
Paraffin sections and frozen sections were prepared from the samples collected 1 and 2 months after surgery. Paraffin sections obtained from each group were stained with HE, and the structure of the newly generated urethra was observed and photographed under a microscope. Frozen sections obtained from the dHAS+BMSCs group were treated with mouse anti-GFP monoclonal antibody (Abcam Shanghai Trading Co., Ltd.) and goat anti-mouse IgG H&L (Alexa Fluor® 488) polyclonal antibody (Abcam Shanghai Trading Co., Ltd.), and those obtained from the dHAS+EPC group were treated with rabbit anti-mcherry polyclonal antibody (Abcam Shanghai Trading Co., Ltd.) and goat anti-rabbit IgG H&L (Cy3®) polyclonal antibody (Cowin Biotech Co., Ltd.).
The samples from the dHAS+BMSCs+EPC group were treated with all four of the above antibodies, and the samples obtained from the two control groups were also treated with all four of the above antibodies as a control. The samples from each group were also treated with DAPI for nuclear staining. The stained sections were placed under an inverted fluorescence microscope, and the distribution of green and/or red fluorescence was observed and photographed.
Results
Labeling of canine BMSCs and EPCs
Two days after transfection of BMSCs with the pCDH-CMV-MCS-EF1-copGFP-T2A-Puro lentiviral vector, the GFP expression rate reached 80%; 3 days after screening with 2 μg/mL puromycin, the transfected cells reached 80% confluence and the GFP expression rate was close to 100% (Fig. 2A, B).

Fluorescent protein labeling of EPCs and BMSCs, coculture of the labeled cells, and tissue-engineered dHAS patches.
Two days after transfection of EPCs with the pLVX-mcherry-IRES-Puro lentiviral vector, the RFP expression rate reached 80%; 3 days after screening with 2 μg/mL puromycin, the transfected cells reached 80% confluence and the RFP expression rate was close to 100% (Fig. 2C, D).
Coculture of labeled BMSCs and EPCs and detection of proliferative activity
The labeled BMSCs and EPCs were mixed in a 1:1 ratio, placed in a 60-mm culture dish, and cultured for 1 day at 37°C in a 5% CO2 cell culture incubator. Observation under a fluorescence microscope showed that fusiform cells and cells shaped like paving stones were evenly distributed on the bottom of the culture dish and grew well; after 2 days of culture, the cells had reached 80% confluence (Fig. 2).
The cell proliferation activity was measured using a Microplate Reader. The ODs of the labeled BMSCs and EPCs on days 1, 3, and 5 were 0.309 ± 0.006, 0.676 ± 0.018 and 1.469 ± 0.012, respectively. The ODs of the unlabeled BMSCs and EPCs on days 1, 3, and 5 were 0.314 ± 0.011, 0.517 ± 0.028, and 1.312 ± 0.015, respectively. There was no significant difference in proliferative activity in the two groups of cells (n = 4, p = 0.601).
Examination of the tissue-engineered dHAS patches containing labeled BMSCs and/or EPCs
Examination under a phase-contrast microscope showed that after the labeled BMSCs and/or EPCs were placed on dHAS and incubated in a 37°C, 5% CO2 cell incubator for 2–4 h, the cells adhered to the surface of the dHAS and were mostly round in shape; after 2 days of culture, the cells appeared to be growing and in good condition. dHAS cultured for 2 days after inoculation of the cells were used to prepare tissue sections. HE staining revealed that the cells adhered to the dHAS at moderate density and grew in a good condition. Observation under a fluorescence microscope showed that the labeled BMSCs, EPCs, and BMSCs+EPCs were evenly distributed on the dHAS and grew well (Fig. 2).
According to the results of the cell proliferation assay, the ODs of the labeled BMSCs and EPCs after 1, 3, and 5 days of culture on dHAS were 0.314 ± 0.011, 0.517 ± 0.028, and 1.312 ± 0.015, respectively; when the cells were cultured directly in a 96-well plate for 1, 3, and 5 days, the cell proliferation assay yielded OD values of 0.314 ± 0.011, 0.571 ± 0.032, and 1.385 ± 0.033, respectively. These values show a significant upward trend, indicating good cell proliferation. There was no significant difference in the results of the cell proliferation assay in the two groups (n = 4, p = 0.825).
The repair effect of BMSCs and/or EPC dHAS patches to canine urethral defects
Observation of urination
All the experimental dogs in this study urinated through the catheter within 2 weeks after surgery. After withdrawal of the catheter, the dogs in the dHAS+BMSCs+EPC group and the dHAS+BMSC group showed urinary patency; the urination time of the dogs in the dHAS+EPC group was gradually extended; for the dogs in the dHAS group and the sham-operated group, urination gradually became difficult and urinary dripping even occurred at the later stage, but urinary retention did not occur.
Ascending urethrogram
Ascending urethrogram showed similar results at 4 weeks (Fig. 3A1–E1) and 8 weeks (Fig. 3A2–E2) postoperatively, but progressive urethral contracture was significant in the dHAS+BMSC group and the dHAS group. The urethrography area of operative region was analyzed and the percentage of the postoperative area to the preoperative area revealed the unclogging degree of newborn urethra (Table 1). The unclogging degree at 8 weeks was significantly lower than that at 4 weeks in the dHAS+BMSC group (Table 1), which showed that the newborn urethra showed progressive stenosis or contracture over time.
The Newborn Urethra Unclogging Degrees (Postoperative Area/Preoperative Area %) in Repair Urethrogram: Comparison Between 4 and 8 Weeks, n = 4
Adopting follow-up test software, a and b showed statistical difference (p < 0.05) and significant difference (p < 0.01) between 4 and 8 weeks, respectively.
BMSCs, bone marrow mesenchymal stem cells; dHAS, decellularized human amniotic membrane scaffolds; EPCs, endothelial progenitor cells.
The unclogging degree at 8 weeks was extremely higher in the dHAS+BMSCs+EPC group than in the other four groups (p < 0.001), significant or extremely higher in the dHAS+BMSC group than in the dHAS+EPCs (p < 0.01), dHAS (p < 0.01), and sham-operated group (p < 0.001), extremely higher in the dHAS+EPC and dHAS groups than in the sham-operated group (p < 0.001) (Table 2).
The Newborn Urethra Unclogging Degrees (Postoperative Area/Preoperative Area %) in Repair Urethrogram: Comparison Among the Groups One by One, n = 4
Using follow-up test software, *, ** and *** showed statistical difference (p < 0.05), significant difference (p < 0.01) and extremely significant difference (p < 0.001) between groups, respectively.
NS, no significant difference.
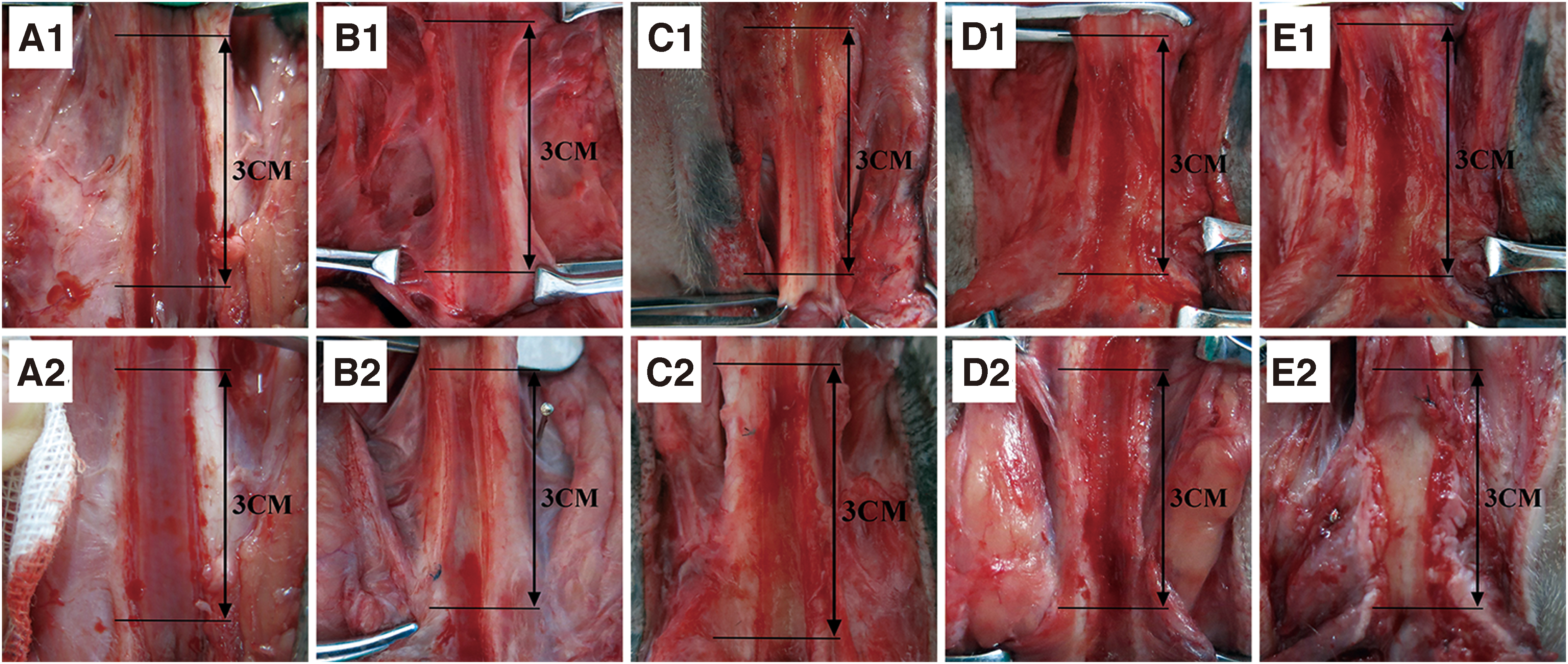
Urethral necropsy
The results of urethral necropsy were similar at 1 month (Fig. 4A1–E1) and 2 months (Fig. 4A2–E2), postoperatively. In the dHAS+BMSCs+EPC group, the cavity of the newly generated urethra was unobstructed, the surface of the urethra was smooth, and the color was lustrous, similar to the normal urethra (Fig. 4A1, A2). In the dHAS+BMSC group, the cavity of the newly generated urethra was relatively unobstructed, the surface was relatively smooth, and the color was light and uneven (Fig. 4B1, B2). In the dHAS+EPC group, the cavity of the newly generated urethra had slight stenosis, and the surface of the anterior middle and middle segments was rough and showed scar formation (Fig. 4C1, C2). In the dHAS group, the cavity of the newly generated urethra was significantly stenotic, and the surface was unsmooth with a large number of scars and contractures (Fig. 4D1, D2). In the sham-operated group, the surface of the newly generated urethra was extremely unsmooth, with severe scarring and contracture (Fig. 4E1, E2).
The results of postoperative urination, urethrography, and necropsy showed that the repair effect of the dHAS patch containing labeled BMSCs and/or EPCs on the canine urethral defect was similar to that of previously constructed amniotic patches containing unlabeled BMSCs and/or EPCs, 5 indicating that labeling of the stem cells had no significant influence on their differentiation and repair ability.
Tracing the differentiation of BMSCs and EPCs and tissue remodeling
dHAS+BMSCs+EPC group
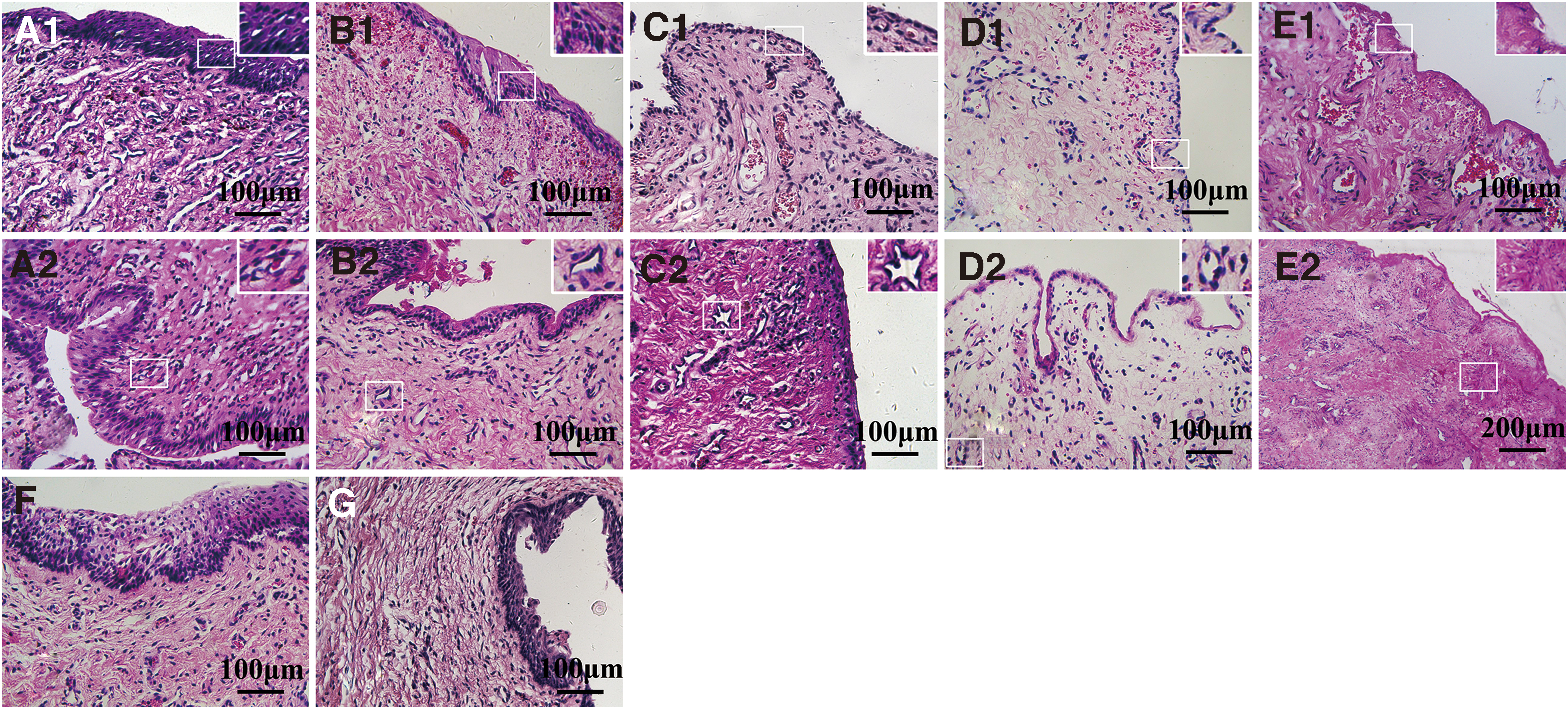
Microscopic observation revealed that the newly generated urethra was covered by a mucosal epithelium composed of five to six layers of columnar cells, similar to the epithelial structure of the normal urethra; the submucosa was richly vascularized (Fig. 5A1, A2). In the area of newly generated urethral mucosal epithelium, red fluorescence and green fluorescence coexisted; more cells emitting green fluorescence than cells emitting red fluorescence were present.
However, in the submucosa, there were few cells emitting green fluorescence and many cells emitting red fluorescence; the cells emitting red fluorescence formed a large number of vascular structures (Fig. 6A–E). This indicated that most BMSCs and a small number of EPCs had differentiated into epithelial cells and participated in the formation of the newly generated urethral mucosal epithelium; most EPCs had differentiated into vascular endothelial cells and participated in the formation of blood vessels, and most of the submucosal tissues were formed by the growth of the adjacent recipient cells.
dHAS+BMSCs group
Microscopic observation revealed that most of the newly generated urethra was covered by a mucosal epithelium composed of three to four layers of columnar cells besides the anterior and posterior segments of the mucosal epithelium covered by five to six layers of columnar cells, and the submucosal blood vessels were abundantly distributed (Fig. 5B1, B2). The green fluorescence was concentrated in the mucosal epithelium of the newly generated urethra, showing little submucosal distribution and weak intensity (Fig. 6F–J). This indicates that the BMSCs mostly differentiated into epithelial cells during the repair of the urethral defect and participated in the formation of urethral mucosal epithelium; the submucosal tissues were mostly formed by the growth of the surrounding recipient cells.
dHAS+EPC group
Microscopic observation revealed that the number of cell layers in the newly generated urethral epithelium was small; most of the middle segment had a monolayered cell structure, a small amount of scar tissue was present, and the submucosa was richly vascularized (Fig. 5C1, C2). Red fluorescence was less prominent in the mucosal epithelium of the newly generated urethra and more prominent in the submucosa, mainly forming the vascular structure (Fig. 6K–O). This indicates that a small number of EPCs had differentiated into epithelial cells and participated in the formation of mucosal epithelium during the repair of the urethral defect, while most EPCs differentiated into vascular endothelial cells and participated in neovascularization. The submucosal tissues were mostly formed by the growth of the surrounding recipient cells.
dHAS group
The anterior and posterior segments of the newly generated urethra were covered by multiple layers of columnar endothelial cells of various lengths; most of the other areas were covered by a single layer of endothelial cells, and scar tissue was obvious (Fig. 5D1, D2). Immunofluorescence staining was negative for both red and green fluorescence.
The sham-operated group
The anterior and posterior segments of the newly generated urethra were covered by multiple layers of columnar endothelial cells of various lengths; most of the other areas were not covered by epithelial cells, and granulation tissue hyperplasia and scarring were severe (Fig. 5E1, E2). Immunofluorescence staining was negative for both red and green fluorescence.
Based on careful histological observation, the structure of the newly generated urethra in each group did not appear to change significantly at 1 or 2 months postoperatively, but the fluorescence intensity of the experimental groups became relatively weak, and the scar tissue present in the sham-operated groups tended to age.
Discussion
Differentiation fate of BMSCs and EPCs and tissue remodeling during the repair of urethral defects
Shukla et al. used constructed tissue-engineered labeled BMSCs and small intestinal submucosal scaffolds to expand a porcine bladder, demonstrating that most of the cells differentiated into urothelial cells. 6 Lijun et al. implanted CM-DiI-labeled BMSCs on a collagen membrane and used the engineered tissue to repair rat uterine horn defects; they found that the BMSCs were localized in the basal layer of the endometrium and could secrete growth factors, promote the proliferation and migration of peripheral endometrial cells, and participate in the repair of uterine defects. 7 Lu et al. injected human amniotic membrane MSCs into the uterine muscle of rats with intrauterine adhesions, demonstrating that MSCs can promote endometrial regeneration. 8 Matsumura et al. inoculated bone marrow-derived stem cells into biodegradable scaffolds and obtained fully endothelialized tissue-engineered vascular grafts after 8 weeks. 9 Duttenhoefer et al. obtained a tubular structure displaying endothelial markers after coculturing human EPCs and MSCs on a polyurethane scaffold for 7 days. 10 Markus et al. performed transmembrane coculture of BMSCs and EPCs and found that EPCs could promote the differentiation of BMSCs into an epithelioid cell phenotype. 11
In the present study, tissue-engineered dHAS patches were constructed using fluorescent protein-labeled BMSCs and/or EPCs and used to repair a 3-cm-long urethral defect, and immunofluorescence detection was performed on samples collected 1 and 2 months postoperatively. The results showed that BMSCs were mainly distributed in the mucosal layer of the newly generated urethra, with little submucosal distribution and that they had differentiated into mucosal epithelial cells; EPCs were mainly distributed in the submucosal layer of the newly generated urethra, where they formed vascular structures, and a few of them were distributed in the mucosal layer and had differentiated into mucosal epithelial cells. Especially in the dHAS+BMSCs+EPC group, both types of cells differentiated into mucosal epithelial cells and formed the mucosal epithelial layer of the new urethra (Fig. 6), which demonstrates the differentiation fates and mutual collaboration of BMSCs and EPCs in the repair of urethral defects, and further confirm and expand the results of previous studies.
The results of the present study suggest that when stem cells are transplanted into the body a “dominant differentiation phenomenon” may occur that may possibly be affected by the environment. In the environment of a urethral defect, BMSCs mainly differentiated into urothelial cells; EPCs mainly differentiated into vascular endothelial cells, and some of them differentiated into urothelial cells. In the environment of cardiac injury, MSCs differentiated into cardiomyocytes. 12 In the tendon injury model, MSCs can participate in the formation of tendon tissue. 13 MSCs can also differentiate into smooth muscle cells under in vitro induction conditions, but they did not participate significantly in the formation of smooth muscle bundles after implantation in a small intestinal submucosa scaffold and transplantation into the body to repair the bladder. 14 In the present study, no significant formation of smooth muscle bundles was found in either the dHAS+BMSC group or the dHAS+BMSCs+EPC group; this point awaits further investigation.
Stem cell labeling, tracing, and differentiation detection
Fluorescent protein labeling and tracing represents one of the most sensitive and important techniques in stem cell transplantation therapy and tissue engineering research. 15
However, in cell fate detection (especially histological detection) during differentiation remodeling after transplantation of stem cells and engineered tissues, the autofluorescence of biological tissues often produces a strong background that seriously affects the intensity and sharpness of the labeled signal. As shown in the quantitative results of Tamara et al., although the fluorescence derived from labeling may be brighter than the tissue autofluorescence, the specificity of fluorescence imaging is often affected by the autofluorescence of the surrounding tissues. 16
Indirect immunofluorescence detection of stem cell marker proteins using GFP and RFP can amplify specific fluorescence signals, reduce the autofluorescence background of biological tissues, and improve the specificity and sharpness of immunofluorescence imaging. Sales et al. labeled MSCs and EPCs with a retroviral vector carrying a gene encoding a fluorescent protein, constructed engineered vascular patches using these labeled MSCs and EPCs and PGA membranes, and transplanted the engineered vascular patches into sheep pulmonary arteries. At 7 and 14 days after implantation, samples were collected, tissue sections were prepared, and the labeled cells were detected using antibodies against GFP and RFP and the appropriate fluorescent secondary antibodies. The two types of cells were found on the surface of the engineered vascular patches, with sufficient fluorescence resolution to permit cell counting. 17
In the present study, BMSCs and EPCs were labeled with a lentiviral vector expressing a fluorescent protein, and a 3-cm-long urethral defect was repaired by means of a tissue-engineered dHAS patch constructed using the labeled BMSCs and EPCs. At 1 and 2 months after surgery, the labeled cells were detected using antibodies against GFP and RFP and the corresponding fluorescent secondary antibodies, and clear, specific fluorescence images were obtained in both assays (Fig. 6). These findings fully demonstrated that labeling of stem cells using a lentiviral vector carrying a gene encoding a fluorescent protein followed by indirect immunofluorescence detection of the labeled cells using an antibody against the fluorescent protein can be used in stem cell labeling and tracing and in the detection of stem cell differentiation in tissue engineering studies.
The present study compared the proliferative activity of labeled BMSCs and EPCs with that of unlabeled BMSCs and EPCs. The results showed that the expression of fluorescent proteins in cells transfected with lentiviral vectors had no effect on the proliferation of the stem cells, strongly supporting the study results of Junjie et al. 12 Comparison of the results of the present study (Figs. 3–6) with our previous findings 5 reveals that fluorescent protein labeling has no influence on stem cell differentiation or tissue remodeling.

Ascending urethrogram.
Urethral necropsy.
Tissue structure of the middle segment of the newly generated urethra.

Immunofluorescence detection of BMSCs and EPCs.
Conclusions
Fluorescent labeling using lentiviral vectors has no influence on stem cell proliferation or differentiation and can be used in combination with indirect immunofluorescence detection to trace stem cell differentiation and the tissue remodeling process in tissue engineering studies. BMSCs and EPCs have synergistic and complementary effects in the repair of urethral defects. BMSCs mainly form urethral mucosal epithelium, and EPCs mainly form blood vessels in the submucosa; altogether, these cells can improve the mucosal epithelium; and most of the submucosal tissues were formed by the growth of the adjacent recipient cells. The dHAS+BMSCs+EPC patch can be used to completely repair canine 3-cm-long urethral defects and expected to be used in clinical applications. Although we have obtained satisfactory results, it is necessary to further study urethral smooth muscle formation and confirm the long-term effects of the dHAS+BMSCs+EPC patch repair as a final guarantee of the actual clinical applicability of this technique.
Author Contributions
Z.W. and Z.X.: Test operation, collection, and/or assembly of data. Z.Y.: Conception and design, Financial support and article writing. X.Z., Z.T., Z.W., and L.Y.: Collection and/or assembly of data. J.W., P.D., and C.H.: Provision of study material. Z.Y., G.D., and R.C.: animal feeding. Z.X.: Administrative support.
Footnotes
Acknowledgments
The authors would like to thank the National Natural Science Foundation of China (No. 31872529) for providing fund, the Experimental Animal Center of Northwest A&F University for providing test animal, and the Wiley Editing Services for helping with English writing.
Disclosure Statement
No competing financial interests exist.
Funding Information
The authors would like to thank National Natural Science Foundation of China (No. 31872529) for providing fund.