
Abstract
The nasal mucosa functions as a frontline biological defense against various foreign substances and pathogens. Maintaining homeostasis of the nasal epithelium is necessary to promote good health. Nasal epithelia are constantly replaced under normal conditions. However, hereditary diseases, including primary ciliary dyskinesia and cystic fibrosis, can result in intractable dysfunction of the nasal mucosa. Since there is no treatment for this underlying condition, extrinsic manipulation is necessary to recover and maintain nasal epithelia in cases of hereditary diseases. In this study, we explored the use of airway epithelial cells (AECs), including multiciliated airway cells, derived from human induced pluripotent stem cells (iPSCs) on porcine atelocollagen vitrigel membranes, as a candidate of a therapeutic method for irreversible nasal epithelial disorders. To confirm the regenerative capacity of iPSC-derived AECs, we transplanted them into nasal cavities of nude rats. Although the transplanted cells were found within cysts isolated from the recipient nasal respiratory epithelia, they survived in some rats. Furthermore, the surviving cells were composed of multiple cell types similar to the human airway epithelia. The results could contribute to the development of novel transplantation-related technologies for the treatment of severe irreversible nasal epithelial disorders.
Impact Statement
Nasal respiratory epithelia are important for the functions of nasal cavity, including humidifying the air and filtering various toxic substances. However, hereditary diseases, including primary ciliary dyskinesia and cystic fibrosis, can result in intractable dysfunction of the nasal mucosa. Our novel method to transplant airway epithelial cells derived from human induced pluripotent stem cells will be a candidate method to replace malfunctioned nasal respiratory epithelia in such a situation. To secure our method's safety, we used porcine atelocollagen vitrigel membranes, which prevent the immune response and bovine spongiform encephalopathy, as a scaffold.
Introduction
The fundamental role of the airway is to maintain a conduit for air to enter and exit the lungs. In addition, the nasal cavity has important functions in humidifying the air and filtering various toxic substances, including pathogens, allergens, and air pollutants.1,2 The nasal cavity adsorbs 80–90% of microparticles in the air through the nasal mucosa.1,3 Captured toxic substances are cleared out of the nasal cavity by mucociliary clearance. At the same time, the nasal mucosa secretes various cytokines and chemokines to control the innate and acquired immune responses against foreign substances.
As a result, the nasal mucosa has a more critical role in frontline biological defense against various inflammatory and toxic substances and pathogenic microorganisms in the air than the tracheal mucosa. Therefore, maintenance of nasal epithelia is key to ensuring good health. Nasal airway epithelial cells (AECs) are composed of various characteristic cells, 1 including multiciliated airway cells (MCACs), mucus-producing goblet cells, club cells, and basal cells. MCACs have many cilia that continually provide mucociliary clearance. Goblet cells also contribute to mucociliary clearance by secreting mucins.
The pathology of nasal epithelial disorders includes chemical damage, mechanical injury, inflammation due to allergic reactions, and infection. Although the severity varies, these types of nasal epithelial damage are usually reversible. In contrast, pathological conditions caused by hereditary diseases, including primary ciliary dyskinesia (PCD) 4 and cystic fibrosis (CF),5,6 are irreversible because of the lack of effective treatment.
PCD is a heritable autosomal recessive or X-linked disease that occurs in 1 in 10,000 to 20,000 individuals. PCD causes dysfunction of the motile cilia in MCACs. Patients with PCD suffer from chronic sinusitis and otitis media with effusion from childhood due to impaired mucociliary clearance and hypoplasia in the frontal and sphenoid sinuses has been reported. 4
CF has a higher incidence of 1 in 2000 to 3000 individuals. 7 It is an autosomal recessive inheritance caused by mutations in the CF transmembrane conductance regulator gene, 5 which encodes Cl channels in various epithelial cells, including the airway epithelium. Mutations in the gene cause chronic sinusitis and nasal polyps at a high frequency due to the loss of mucus production 7 and ciliary dysfunction in the nasal sinuses. 5 No effective treatment exists for the irreversible dysfunction in the nasal mucosal epithelium of nasal cavity caused by these hereditary diseases.4,5
One of the most promising candidate treatments for the genetic dysfunction of airway epithelia is the transplantation of AECs derived from induced pluripotent stem cells (iPSCs). The transplantation of iPSC-derived cells has been widely studied in regenerative medicine for intractable diseases, including hereditary diseases. 8 Even if the mutation causing the disease remains within patients' iPSCs, recent gene-editing technology can correct the defective phenotypes of iPSC-derived transplanted cells.9,10 Another strategy for the iPSC-derived cell therapy to overcome the hereditary disease is to use allogeneic iPSCs without mutation. If a major histocompatibility complex matches, the transplantation of allogeneic iPSC-derived cells causes only mild immunological rejection against transplanted cells. 11
AECs derived from mouse iPSCs 12 or human induced pluripotent stem cells (hiPSCs) 13 were transplanted with artificial trachea into tracheal defects in nude rats to promote epithelialization of the defects. Okuyama et al. utilized a biodegradable and biocompatible bovine collagen vitrigel membrane as a scaffold for hiPSC-derived MCACs. 13 The authors observed hiPSC-derived cells at the epithelial layer, facing the luminal surface in the transplantation site in the trachea, 13 which share similar airway epithelia with a nasal cavity.
Based on these, we decided to transplant hiPSC-derived AEC sheets into the nasal epithelia as a first step to establish a new therapeutic method for severe irreversible nasal epithelial disorders. Considering potential future clinical applications, we used porcine atelocollagen vitrigel membrane instead of bovine collagen vitrigel membrane to avoid the risk of bovine spongiform encephalopathy.
Materials and Methods
Multiciliated cell differentiation from hiPSCs
The hiPSC line 253G114 was obtained from the RIKEN BioResource Center. The maintenance and differentiation of hiPSCs have been previously described.15,16 On day 1, 1.2 × 106/well hiPSCs were cultured on Geltrex (Thermo Fisher Scientific, Waltham, MA)-coated six-well culture plates using Essential 8™ Medium (Thermo Fisher Scientific). Seeded hiPSCs were cultured in Medium1 consisting of RPMI1640 medium (Nacalai Tesque, Kyoto, Japan), 1 × B27 supplement (Thermo Fisher Scientific), and 50 U/mL penicillin/50 μg/mL streptomycin (Nacalai Tesque) supplemented with 100 ng/mL human activin A (R&D Systems, Minneapolis, MN), 1 μM CHIR99021 (Axon Medchem, Reston, VA), and 10 μM Y-27632 (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan).
On day 2, 0.25 mM sodium butyrate (FUJIFILM Wako Pure Chemical Corporation) was added. On days 3–5, cells were cultured in Medium1 containing 100 ng/mL human activin A, 1 μM CHIR99021, and 0.125 mM sodium butyrate to induce SOX17-positive endoderm cells.
On days 6–9, cells were cultured to induce anterior foregut endoderm cells in Medium2: DMEM/F-12, GlutaMAX (Thermo Fisher Scientific), 1 × B27 supplement, 50 U/mL penicillin/50 μg/mL streptomycin, 0.05 mg/mL L-ascorbic acid (FUJIFILM Wako Pure Chemical Corporation), and 0.4 mM monothioglycerol (FUJIFILM Wako Pure Chemical Corporation) supplemented with 100 ng/mL human recombinant noggin (Human Zyme, Chicago, IL) and 10 μM SB-431542 (Selleck Chemicals, Houston, TX).
On days 10–14, cells were cultured in Medium2 containing 20 ng/mL human recombinant BMP4 (Human Zyme), 10 nM CHIR99201, and 0.2 μM all-trans retinoic acid (Sigma-Aldrich, St. Louis, MO). On day 14, carboxypeptidase M (CPM)-positive ventralized anterior foregut endoderm cells were purified by magnetic activated cell sorting using anti-human CPM antibody (FUJIFILM Wako Pure Chemical Corporation). 15
Then, CPM-positive cells were embedded in Matrigel (Corning, Corning, NY) in a 12-well culture insert with PET membrane (#353292; Corning). CPM-positive cells were cultured in Medium2, including 3.0 μM CHIR99021, 100 ng/mL FGF10 (FUJIFILM Wako Pure Chemical Corporation), and 10 μM Y-27632 to induce AEC spheroids for 2 weeks. Next, cells were cultured in PneumaCult-ALI Maintenance medium (STEMCELL Technologies, Vancouver, Canada) containing 10 μM Y-27632, 10 μM Difluorophenacetyl-L-alanyl-2-phenylglycine t-butyl ester (DAPT, FUJIFILM Wako Pure Chemical Corporation), 4 μg/mL heparin (Nacalai Tesque), and 1 μM hydrocortisone (Sigma-Aldrich) for 14 days. 16
On day 42, spheroids were dissociated by enzymatic treatment and seeded on cell culture insert with porcine atelocollagen vitrigel membrane as our previous report 13 to form a cell sheet by culture under the air-liquid interface (ALI) condition for 14 days.
Preparation of cell culture chamber with porcine atelocollagen vitrigel membrane
The raw material (0.5% atelocollagen sol) was prepared by uniformly mixing equal volumes of 1.0% acidic solution of porcine-derived collagen formulated for regenerative medicine (Nippon Meat Packers, Tokyo, Japan) and serum-free Dulbecco's modified Eagle's medium supplemented with 20 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Rockville, MD). A collagen vitrigel membrane containing 5.5 mg collagen/cm2 unit area was fabricated by gelation, vitrification, and rehydration with phosphate-buffered saline (PBS), as previously described.17,18 Subsequently, the membrane was converted into an atelocollagen xerogel membrane by revitrification on a separable sheet.
The xerogel membrane was pasted onto the bottom-side edge of a plastic cylinder with an inner-outer diameter of 11–15 mm and a length of 15 mm. A couple of hangers were connected to its top edge, resulting in the fabrication of a cell culture chamber with a porcine atelocollagen xerogel membrane, which could be easily converted into an atelocollagen vitrigel membrane by rehydration.
Hematoxylin and eosin staining of induced AECs
Cells were fixed with 4% paraformaldehyde (PFA) overnight at 4°C. Subsequently, the cells were gradually dehydronated with 10%, 20%, and 30% sucrose/PBS overnight for each concentration at 4°C. The cells were then embedded in optimal cutting temperature (O.C.T.) compound (Sakura Finetek Japan, Tokyo, Japan) and frozen at −80°C. The cells were sliced into 10-μm-thick sections by CryoStar NX70 Cryostat (Thermo Fisher Scientific).
For hematoxylin and eosin (HE) staining, the cells were stained by Hematoxylin (FUJIFILM Wako Pure Chemical Corporation) for 60 s and Eosin (FUJIFILM Wako Pure Chemical Corporation) for 30 s, and treated with 70% and 100% ethanol twice and xylene thrice for 3 min at each step. After staining, the cells were mounted in MOUNT-QUICK® (Daido Sangyo, Tokyo, Japan). Images were obtained using a BIOREVO BZ-9000 fluorescence microscope (KEYENCE, Osaka, Japan).
Immunocytochemistry
Cells were fixed with 4% PFA for 15 min at room temperature (RT). Subsequently, the cells were treated with 0.2% Triton/PBS for 5 min and incubated in 1% bovine serum albumin (BSA; FUJIFILM Wako Pure Chemical Corporation)/PBS at RT. The cells were then treated with primary antibodies against acetylated α-tubulin (Mouse, T7451, 1:1000; Sigma-Aldrich), E-cadherin (Rat, 13–1900, 1:1000; Thermo Fisher Scientific), NKX2–1 (Mouse, MS-699-P1, 1:500; Thermo Fisher Scientific), and SOX17 (Goat, AF1924, 1:500; R&D Systems) overnight at 4°C, followed by washing with PBS before treatment with secondary antibodies [Alexa Flour conjugated secondary antibodies (A-11057, A-21121, A-21144, and A-11006), 1:1000; Thermo Fisher Scientific] for 1 h at RT.
Cells were embedded in Fluoromount-G® Anti-Fade (Southern Biotechnology Associates, Inc., Birmingham, AL). Images were obtained using an Eclipse Ti-S fluorescence microscope (Nikon Corporation, Tokyo, Japan) equipped with a model DP73 camera (Olympus, Tokyo, Japan) and BX 50 fluorescence microscope equipped with a DP70 camera (Olympus).
The proportion of α-tubulin-positive MCACs among E-cadherin-positive epithelial cells was calculated by dividing the number of MCACs by the number of epithelial cells. The cell numbers were counted within the microscopic fields with an area of 211.40 × 159.20 μm. The proportion for each cell sheet was determined as the average of three fields from the three cell sheets. Each field contained more than 100 epithelial cells.
Electron microscopy
Cells on the vitrigel membrane were fixed with phosphate buffer containing 4% PFA and 2% glutaraldehyde overnight at 4°C, incubated in 1% osmium tetroxide (Nacalai Tesque) for 2 h, and then dehydrated using ascending concentrations of ethanol.
For scanning electron microscopy (SEM), dehydrated cells were dried using a critical point drying method and coated with platinum palladium. The specimens were observed using a scanning electron microscope (S-4700; Hitachi Co., Tokyo, Japan).
For transmission electron microscopy (TEM), dehydrated cells were embedded in epoxy-resin and DMP-30 (Nacalai Tesque). Thin sections were examined using a transmission electron microscope (H7650; Hitachi).
Rats
Male 7–8-week-old nude rats (F344/NJcl-rnu/rnu; CLEA Japan, Osaka, Japan) weighing 190–220 g were used. The animal experimental protocol was approved by the Animal Research Committee of the Graduate School of Medicine, Kyoto University (MedKyo20131).
Transplantation
A total of 13 male nude rats were used for the transplantation of hiPSC-derived AECs. The surgical procedure (Fig. 1) was performed under anesthesia with an intraperitoneal injection of a mixture of midazolam (2 mg/kg), butorphanol (2.5 mg/kg), and medetomidine (0.15 mg/kg). The center of the facial skin was incised to expose both sides of the nasal bones. The left nasal bone was drilled out and the left side of the roof of the nasal cavity was opened (Fig. 1Ab, Ac and Ba). Mucosal defects measuring 3 × 4 mm (Fig. 1Ba) were created by excising the mucosa, and 2 × 3 mm hiPSC-AEC sheets (yellow arrowheads in Fig. 1Bb) on vitrigel membrane were transplanted into the mucosal defect. Finally, a facial incision was sutured. For histological examinations, heads of recipient mice were collected following decapitation 1 week after transplantation.
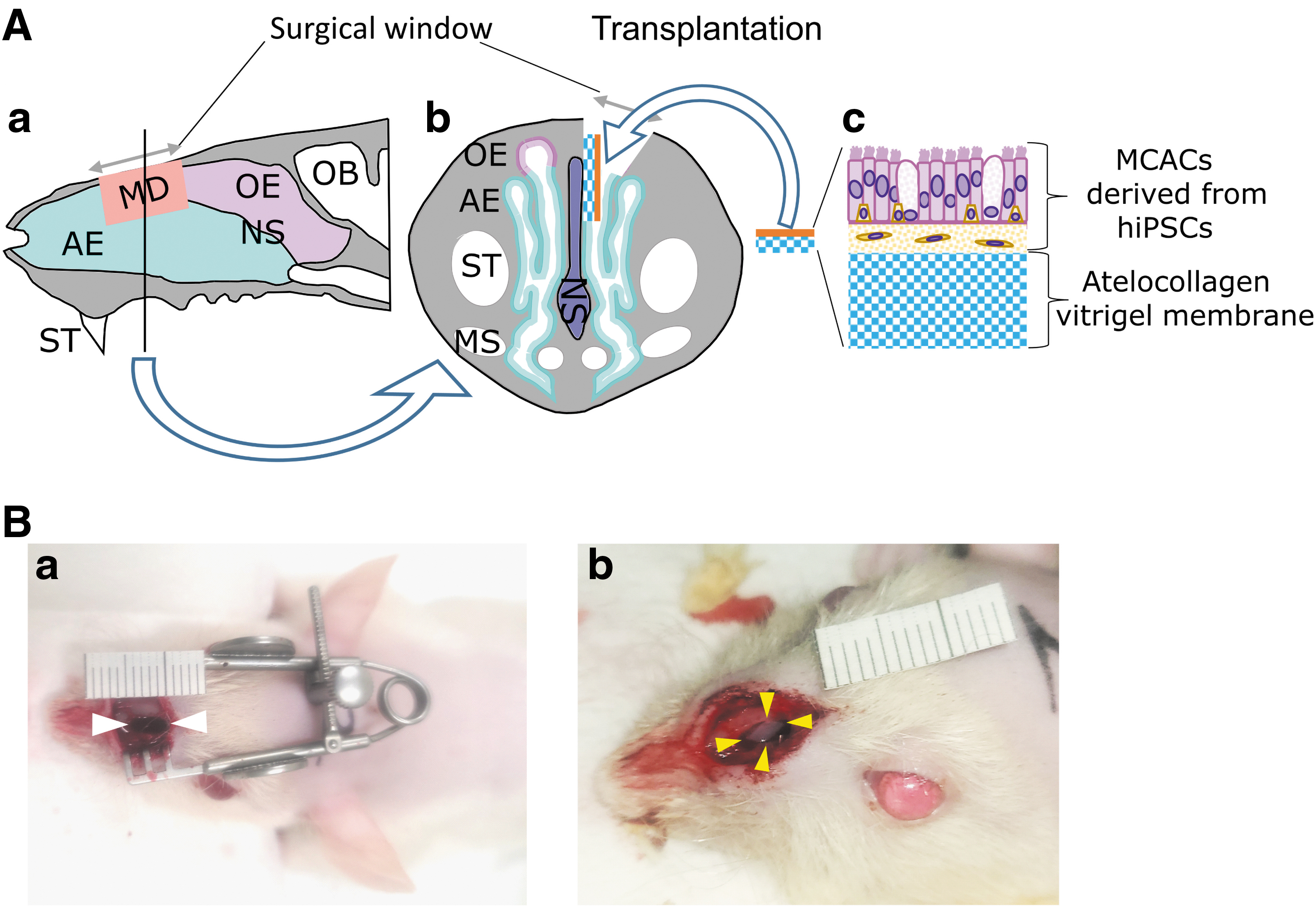
Transplantation of AECs derived from hiPSCs into the nasal cavity of nude rats.
Immunohistochemistry
The animals were euthanized using carbon dioxide. The collected heads were immersed in 4% PFA at 4°C for 1 day and decalcified with 10% ethylenediaminetetraacetic acid at RT for 1 week. The nasal tissues were embedded in O.C.T. compound and frozen at −80°C. The specimens were sliced into 10-μm-thick sections at 200 μm intervals and permeabilized with 0.2% Triton X-100. After blocking with 5% BSA/PBS (keratin 5 and SOX2) and 1% BSA/PBS at RT, the sections were incubated with primary antibodies [anti-acetylated α-tubulin (Mouse, T7451, 1:1000; Sigma-Aldrich), human nuclear antigen (HNA) (Mouse, MAB1281, 1:1000; Millipore Darmstadt, Germany), keratin 5 (Rabbit, ab53121, 1:500; abcam, Cambridge, UK), SCGB1A1 (Rat, MAB4218, 1:62.5; R&D Systems), and SOX2 (Goat, AF2018, 1:300; R&D Systems) antibodies] overnight at 4°C.
After washing with PBS, the sections were incubated with secondary antibodies [Alexa Flour conjugated secondary antibodies (A-21124, A-21240, A-11006, A-11057, A32790), 1:1000; Thermo Fisher Scientific] at RT for 1 h. 4′, 6-Diamidino-2-phenylindole (DAPI) was used to stain the nuclei.
For HE staining, sections on slides were rinsed with water for 5 min and were stained by Hematoxylin (FUJIFILM Wako Pure Chemical Corporation) for 60 s and Eosin (FUJIFILM Wako Pure Chemical Corporation) for 30 s. Samples were dehydrated by 70% ethanol for 3 min and 100% ethanol for 3 min twice, and the ethanol in the sample was replaced with xylene for 3 min, thrice. After applying the sealing reagent, the samples were covered by the cover glass (Mount-Quick, Cosmobio, Tokyo, Japan). Images were obtained using a BIOREVO BZ-9000 fluorescence microscope (KEYENCE).
Results
Characterization of each induction step during the induction from hiPSCs to AECs
In previous studies,13–16 253G1 iPS cell line, which we used in this study, had not been used to induce AECs. To confirm the marker protein expression in the cells at each induction step in Figure 2A, induced cells were examined by immunocytochemistry using antibodies of differentiation marker protein at each step, and sphere formation was confirmed using a phase-contrast microscope. On day 6 of AEC induction from hiPSCs, cells that were positive for the SOX17 endodermal cell marker were observed (Fig. 2Ba). On day 14, several cells expressed the NKX2–1 ventralized anterior foregut endodermal cell marker (Fig. 2Bb), and spheroids were observed in the three-dimensional culture on day 28 (Fig. 2Bc). The presence of induced MCACs on the vitrigel membrane sheet on day 56 was confirmed by immunocytochemical analyses and electron microscopy.
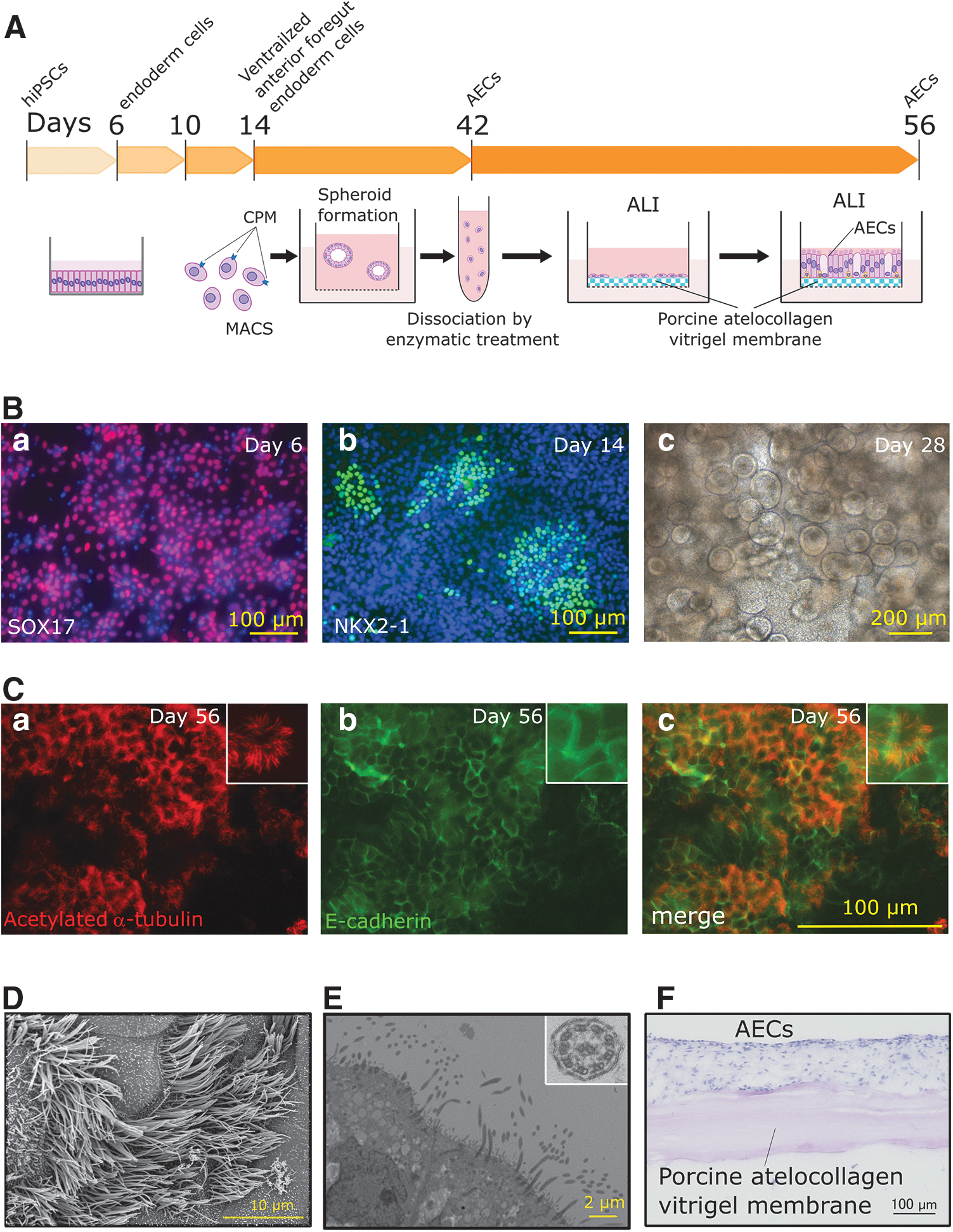
Characteristics of each induction step during the induction from hiPSCs to AECs.
Acetylated α-tubulin-positive cells with cilia-like structures (Fig. 2Ca) were observed in a part of the E-cadherin-positive epithelial cell sheet (Fig. 2Cb, Cc). Cilia-like structures were also observed by SEM (Fig. 2D). TEM (Fig. 2E) revealed the existence of “9 + 2” structures (Fig. 2E, inset), consisting of nine doublet microtubules arranged in a circle around one central doublet microtubules with dynein arms. The structures are a specific motif in motile cilia. HE staining image of cryo-cross-section of induced cells on day 56 (Fig. 2F) shows the induced cells are on vitrigel membrane as shown in the illustration of the AEC on day 56 in Figure 2A.
We performed the induction three times to calculate the induction ratio of α-tubulin-positive MCACs in total cells on the vitrigel membrane. The ratios were 50.9%, 32.6%, and 50.5% (44.7% ± 10.4%. The value of each experiment was obtained from three fields of view for each sample). These results indicated that the induced cells contained sufficient MCACs and the induction efficiency was good.
Survival of transplanted cells in nasal cavities of nude rats
We transplanted hiPSC-derived AECs on vitrigel membranes into the left nasal cavities of 13 nude rats through an incision made on the dorsum of the nose (Fig. 1). One week after transplantation, we examined the site of transplantation histologically (Figs. 3 and 4). HE staining (Fig. 3B, D) showed that transplanted cells, identified as HNA-positive cells (Fig. 3C), were surrounded by granulation tissue, existed within cysts, and were isolated from the recipient respiratory epithelia. Transplanted hiPSC-derived MCACs were observed in the nasal cavities of 4 of the 13 (30.7%) rats 1 week after transplantation.
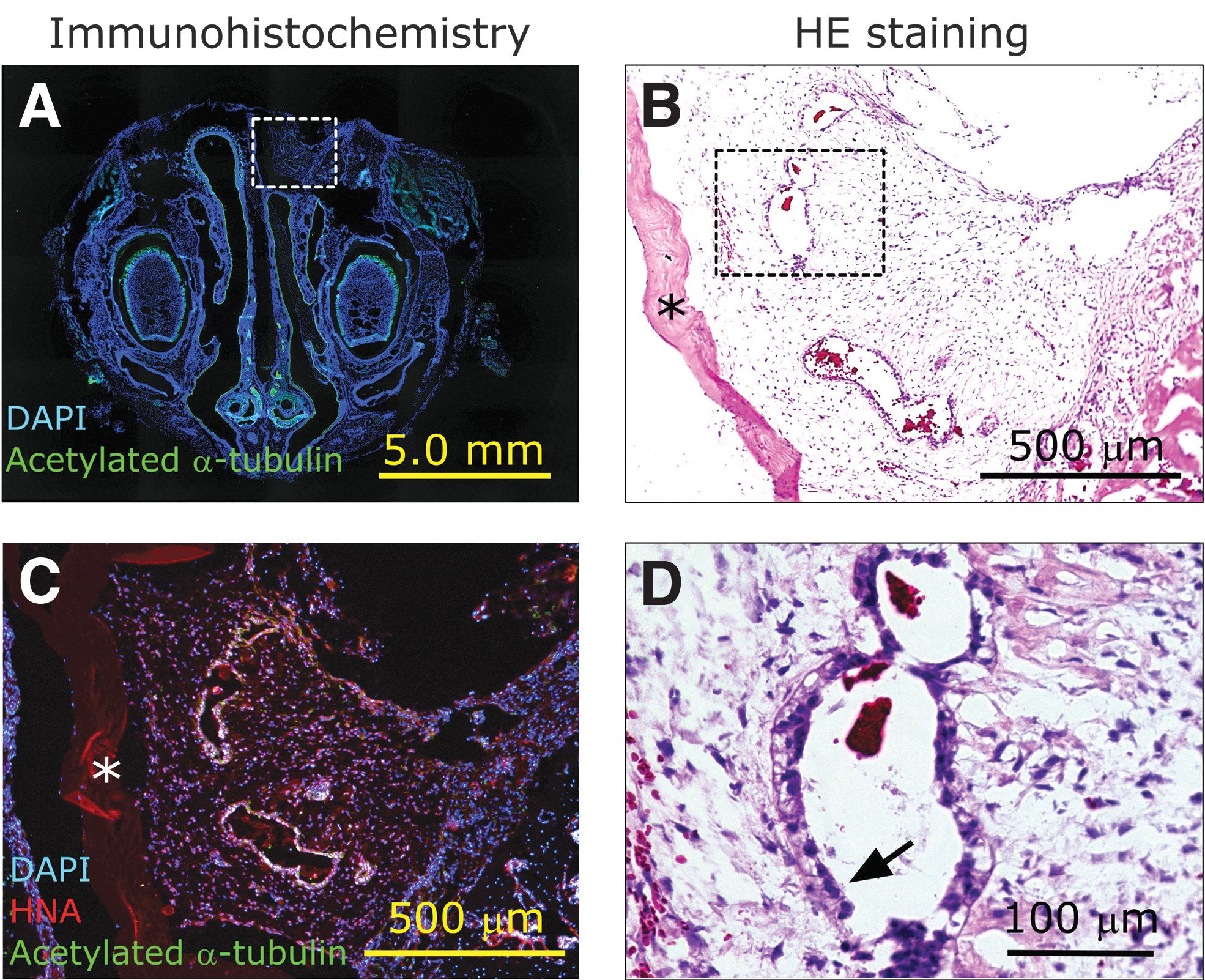
Microscopic morphology of the site of transplantation in the nasal cavity.
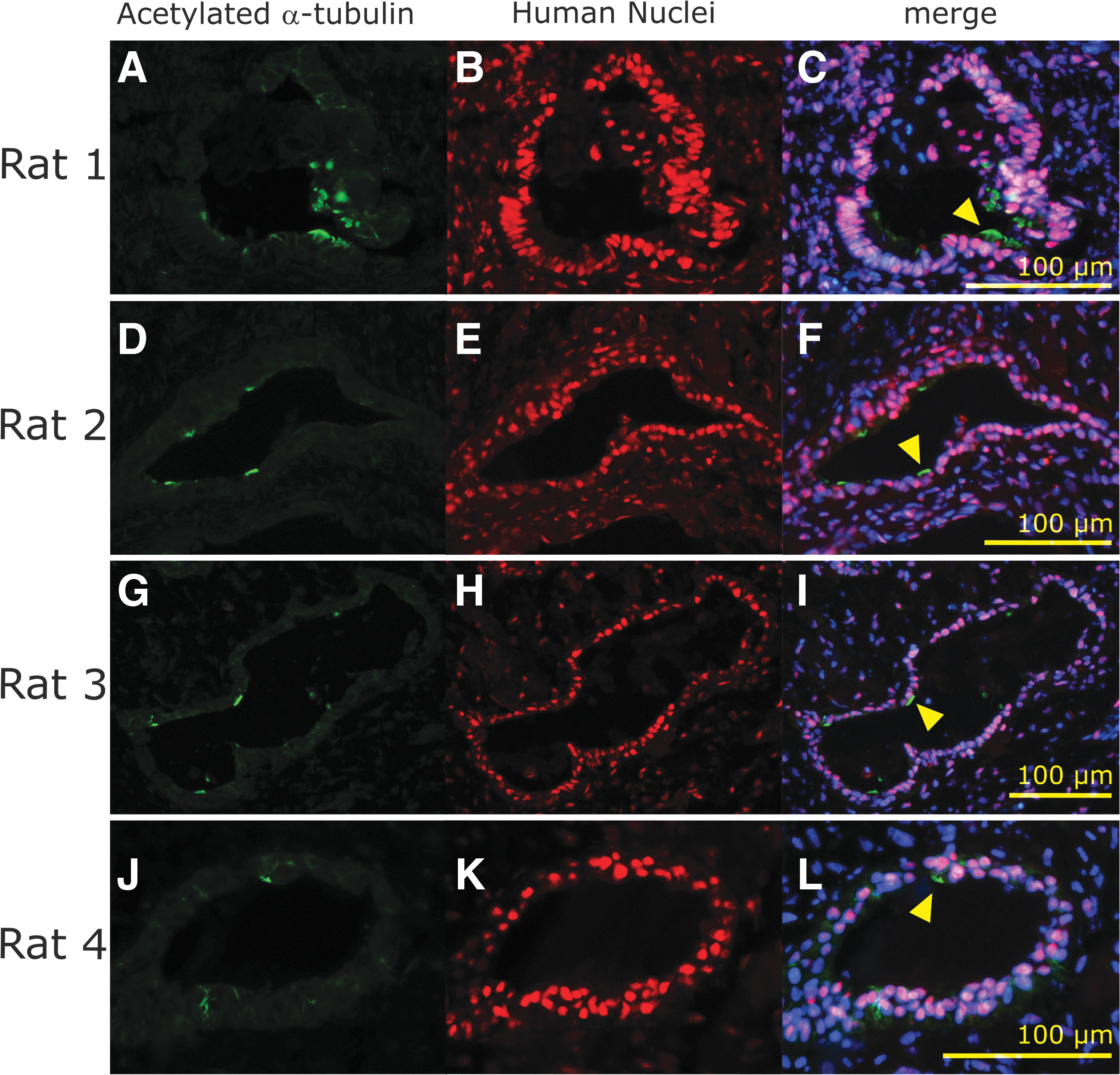
Immunohistochemistry of surviving transplanted cells in granulation tissue of the nude rat nasal cavity. Images of acetylated α-tubulin
Transplanted cell-derived cysts were observed in the nasal cavities of the four transplanted animals. The other nine rats did not display viable hiPSC-derived MCACs, although the vitrigel membrane remained inside the granulation tissue of all nine animals. Some HNA-positive epithelial cells had acetylated α-tubulin-positive cilia-like structures (Figs. 3A, C, and 4). These results indicated the presence of AECs, including MCACs, in the cysts, as observed in the cell sheet before transplantation.
Characterization of surviving transplanted AECs in nude rat nasal cavities
Next, we determined whether cysts consisting of HNA-positive cells included other types of AECs than MCACs by detecting the specific markers of each cell type 16 (Fig. 5). Several HNA-positive cells were also positive for the SCGB1A markers of club cells (Fig. 5A) and the SOX2 and keratin 5 markers of basal cells (Fig. 5E, F, I, J). These results indicated the similar cellular composition of HNA-positive cell cysts to that of human airway epithelia.
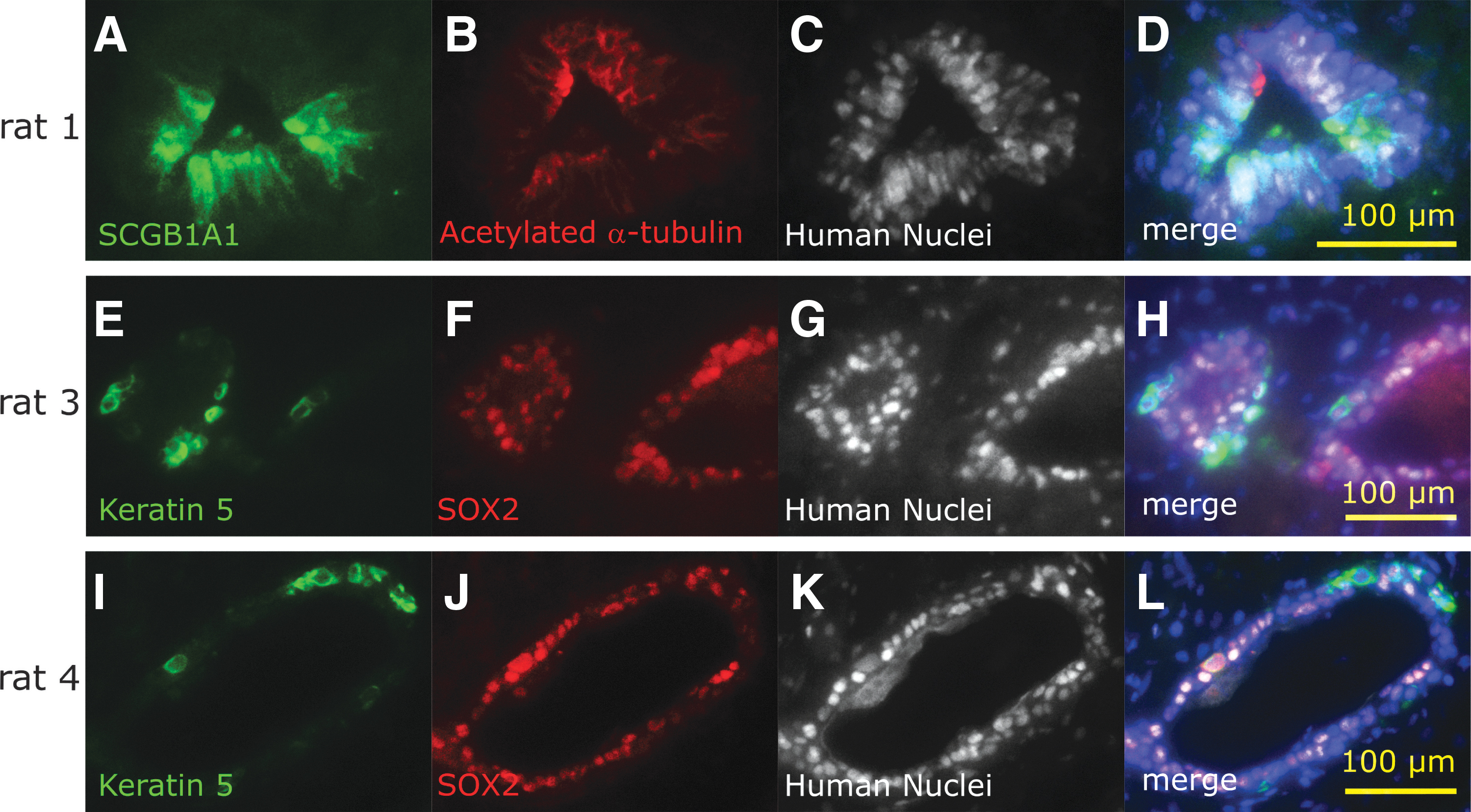
Characterization of the surviving transplanted cells in the nasal cavity of each nude rat. Club cell marker
Discussion
We report two novel results. First, we successfully induced MCACs from hiPSCs on porcine atelocollagen vitrigel membrane. The vitrigel membrane is a scaffolding material first developed by Takezawa et al. 19 Many researchers have used it in regenerative medicine research to induce organoids from pluripotent stem cells. 20 Although a previous study used bovine vitrigel membrane, 13 we successfully cultured hiPSC-derived MCACs on the porcine atelocollagen vitrigel membrane. This is important, as the membrane is free from bovine-derived infectious disease and can be safely applied in clinical treatment.
The second novel result is the survival of transplanted hiPSC-derived AECs in the nasal cavities of nude rats. The transplanted cells were viable, although they were observed within cysts, and we confirmed the cellular characteristics of three major cell types of AECs1,21,22 in surviving cells using distinctive immunostaining markers. These markers included α-tubulin in MCACs, SCGB1A1 in club cells, and keratin 5 in basal cells.21,23 SCGB1A1 is a multifunctional secreted protein with anti-inflammatory effects. 24 SCGB1A1 expression in the nasal mucosa has been reported to be reduced in chronic sinusitis. 25 In this study, the expression of the keratin 5 basal cell marker of AECs was also observed within the transplanted AECs, as in a previous study. 16
This indicates that the implanted AECs were composed of multiple cell types similar to the human airway epithelia. However, the ratio of components within the induced cells is an issue. The percentage distribution of each type of AECs differs between the nasal cavity and trachea. The ratio of MCACs in the mouse airway epithelium is reportedly over twofold higher in the nasal septum (90.1%) than in the trachea (38.7%). 26 To apply these results clinically, a method is needed to control the percentage of each cell type in the population of induced AECs.
The transplanted cells were observed in the nasal cavities at 1 week after transplantation in 4 out of 13 animals (30.7%), although observed AECs, including MCACs, were found within cysts, did not face the luminal surface of the nasal cavity in all animals (0%), and may not contribute to the functions of the nasal cavity. In contrast, the transplantation of hiPSC-derived MCACs into the trachea caused their engraftment with the apical side of the epithelia facing toward the tracheal lumen in two out of seven animals (28.6%). 13
The difference in the rate of luminal facing of transplanted MCACs may be due to anatomical factors. The nasal cavity is the frontline of biological defense against various inflammatory and toxic substances and pathogenic microorganisms. As a result, the nasal cavity has a more challenging environment than the trachea, causing an inflammatory response. Moreover, the space of the lumen is narrower in the nasal cavity. These factors may contribute to cyst formation.
In vivo studies of AEC regeneration have reported that allogeneic mucosal transplantation in the rabbit maxillary sinus improves wound epithelialization. 27 However, allogeneic transplantation induces an immune response that results in the invasion of donor-derived cells into the recipient. Since the transplantation of hiPSC-derived cells is xenogeneic, the transplanted epithelia in our study may also induce a severe inflammatory reaction, resulting in cyst formation. hiPSC-AECs disappeared from the nasal cavity of nude rats after transplantation due to several possible mechanisms. One is the dropout of transplanted cells by the nasal airflow after transplantation. The other is phagocytosis by an immune response.
Therefore, it will be necessary to evaluate phagocytosis histologically before hiPSC-AECs disappear. Considering that the transplantation of AECs induced from hiPSCs is an autograft/allograft in a clinical setting, cyst formation may not occur.
The ciliary function of hiPSC-derived MCACs has already been confirmed in the previous study.13,16 Therefore, to achieve more functional regeneration of nasal airway epithelia in our experimental system, it is necessary to inhibit the immune response and granulation tissue formation around the transplanted tissues. For this purpose, several methods have been considered, including improving the surgical technique, optimizing the scaffold materials for transplanted cells, and controlling the immune response.
Regarding the scaffold materials, the high concentration (5.5 mg/cm2) of porcine atelocollagen was the possible reason for the observed lack of transplanted cells on the luminal surface of the nasal cavity. At this concentration, the scaffold was so thick that a large gap was formed between the transplanted cells and the mucosal fracture of the transplanting wound. Although a vitrigel membrane with lower concentrations of porcine atelocollagen is preferable, this will reduce the rigidity of the vitrigel membrane and transplantation becomes difficult. To overcome this issue, the rigidity of the atelocollagen vitrigel membrane can be increased by irradiation with ultraviolet light. 28
To avoid the effects of immune response, it is necessary to collect data earlier or to use immunosuppressing reagents or more severe immune-deficient animals, such as X-SCID rats. 29 Our experimental model was xenotransplantation (human cells in rats). We found only scaffolds, and not any transplanted cells, in 9 out of 13 experiments. This suggests that transplanted cells disappeared before they were engrafted into the rat nasal mucosa or that they were replaced by granulation tissue. Severe immune responses, including phagocytosis by inflammatory cells, may cause the transplanted cells to disappear. The X-SCID rat lacks T and B cells, as well as natural killer cells, which are essential for immune function against transplanted cells. 29 Thus, there may not be an immune response to transplanted human-derived cells.
Tumor formation is another issue to be considered in iPSC-based cell therapy. In this study with 1-week observation, it is difficult to confirm the lack of tumor formation because it was reported after more extended observation, more than 40 days, 30 in neurospheres derived from the iPSC line, 253G1, which we used in this study. Sorting induced cells with differentiation markers and removing undifferentiated cells from hiPSC-derived cells contribute to the prevention of tumor formation. 31 In this study, we used the induction protocol15,16 that includes sorting CPM-positive ventralized anterior foregut endoderm cells on day 14 of induction. This protocol presumably contributes to inhibiting tumor formation.
If the transplantation method is improved, we will be able to apply this method to a humanized animal model in which part of the nasal epithelium is replaced with hiPSC-derived AECs. It can be used as an in vivo model for human-specific infectious diseases that cannot be studied in human bodies, as well as a model for the development of drugs and other therapies. 29 Gene editing of hiPSCs will permit the reproduction of human hereditary diseases in the animal nasal cavity. This will enable animals to be used as models of human diseases.
Conclusion
hiPSCs were differentiated into AECs on porcine atelocollagen vitrigel membranes and hiPSC-derived AECs were transplanted into the nude rat nasal cavity for engraftment. The transplanted cells survived within the cysts isolated from the recipient nasal respiratory epithelia. Signals of nasal respiratory epithelial cell markers were detected in the survival cells after transplantation. The findings provide a basis for the development of technologies for the treatment of severe nasal airway epithelial disorders, including genetic disorders, using transplantation.
Footnotes
Acknowledgments
We thank Ms. Keiko Okamoto-Furuta and Mr. Haruyasu Kohda from the Division of Electron Microscopic Study, Center for Anatomical Studies, Graduate School of Medicine, Kyoto University, for their technical support in electron microscopy and Ms. Mizuki Tayama-Abe from the Vitrigel Project Research Team, Institute of Agrobiological Sciences, National Agriculture and Food Research Organization, for their technical support in the preparation of collagen vitrigel.
Authors' Contributions
N.Y., H.O., and K.O. designed the study. T.T. prepared the cell culture chamber with a porcine atelocollagen vitrigel membrane. M.Y., Y.K., and M.K. provided administrative support and approved the final article. H.O., H.O., and F.K. prepared hiPSC-derived MCACs, and F.K. performed the transplantation. F.K., Y.H., M.Y., and H.O. performed the histological analyses. F.K., N.Y., and H.O. wrote the main article text and all figures and the table. All authors participated in revising the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (20K18279) and alumni otolaryngology fund from the Department of Otolaryngology-Head and Neck Surgery, Graduate School of Medicine, Kyoto University.