
Abstract
Bone marrow–derived mesenchymal stem cells (BMSCs) have been recognized as new candidates for the treatment of serious endometrial injuries. However, owing to the local microenvironment of damaged endometrium, transplantation of BMSCs yielded disappointing results. In this study, Pectin-Pluronic® F-127 hydrogel as scaffolds were fabricated to provide three-dimensional architecture for the attachment, growth, and migration of BMSCs. E2 was encapsulated into the W/O/W microspheres to construct pectin-based E2-loaded microcapsules (E2 MPs), which has the potential to serve as a long-term reliable source of E2 for endometrial regeneration. Then, the BMSCs/E2 MPs/scaffolds system was injected into the uterine cavity of mouse endometrial injury model for treatment. At 4 weeks after transplantation, the system increased proliferative abilities of uterine endometrial cells, facilitated microvasculature regeneration, and restored the ability of endometrium to receive an embryo, suggesting that the BMSCs/E2 MPs/scaffolds system is a promising treatment option for endometrial regeneration. Furthermore, the mechanism of E2 in promoting the repair of endometrial injury was also investigated. Exosomes are critical paracrine mediators that act as biochemical cues to direct stem cell differentiation. In this study, it was found that the expression of endometrial epithelial cell (EEC) markers was upregulated in BMSCs treated by exosomes secreted from endometrial stromal cells (ESCs-Exos). Exosomes derived from E2-stimulated ESCs further promoted the expression level of EECs markers in BMSCs, suggesting exosomes released from ESCs by E2 stimulation could enhance the differentiation efficiency of BMSCs. Therefore, exosomes derived from ESCs play paracrine roles in endometrial regeneration stimulated by E2 and provide optimal estrogenic response.
Impact statement
Endometrial injuries lead to infertility. Bone marrow–derived mesenchymal stem cells (BMSCs) have been recognized as candidates for treatment. However, BMSC transplantation alone always yielded disappointing results. In this study, Pectin-Pluronic® F-127 scaffolds were fabricated to provide three-dimensional architecture for BMSCs. E2 was encapsulated into the W/O/W microspheres (E2 MPs), as a long-term source of E2 for endometrial regeneration. The BMSCs/E2 MPs/scaffolds was proved as a promising therapy for endometrial regeneration. Moreover, it was found that exosomes secreted from endometrial stromal cells play paracrine roles in endometrial regeneration stimulated by E2, which induces BMSCs differentiation into endometrial epithelial cells.
Introduction
Uterine endometrium is a highly dynamic tissue composed of a basal layer and a functional layer.1,2 Endometrial damage to the basal layer caused by curettage, infections, caesarean section, and myomectomy has an obvious impact on endometrium scar formation, fibrosis, or intrauterine adhesion (IUA), which is frequently related to amenorrhea, hypomenorrhea, recurrent pregnancy loss, or infertility.3–5 Several therapies, such as hysteroscopic surgery removal of adhesions and hormonal therapy have been adopted for the treatment of endometrial fibrosis.6,7 However, treatment for these severe cases is difficult, and prognosis is usually unsatisfactory. 8 Therefore, by improving the proliferation and regeneration ability of endometrial epithelium cells (EECs) and endometrial stromal cells (ESCs) to reconstruct the endometrial structure and restore endometrial functioning can fundamentally treat endometrial damage and improve pregnancy rates. 9
Several studies have shown that bone marrow–derived mesenchymal stem cells (BMSCs), which have beneficial effects for the treatment of IUA, could facilitate the proliferation of ESCs and EECs and accretion of endometrium or directly differentiating into EECs.8–13 However, the local microenvironment of the damaged endometrium cannot provide stable three-dimensional (3D) structure and necessary bioactive molecules for BMSCs. It has yielded disappointing results of survival, attachment, differentiation, and proliferation of exogenous BMSCs.14–16 Thus, the therapeutic effect of direct injection of BMSCs into uterine cavity to promote the regeneration of damaged endometrium is currently inadequate.9,11
Given the high-water content and excellent biocompatibility and biodegradability of hydrogels, combining biomaterials with drugs and MSCs has been speculated to have promising therapeutic applications. 17 Pectin is a natural polymer extracted from apple and orange peel with good biocompatibility. 18 Pluronic® F-127, which has been approved by Food and Drug Administration (FDA) for use as food additives and pharmaceutical ingredients, is a copolymer of polyethylene oxide and polypropylene oxide. 19 Pluronic F-127 solution could be transformed into solid through the thermal sol–gel transition to generate and maintain the desired shape. 19 Thus, the Pectin-Pluronic F-127 hydrogel can be injected into the uterine cavity to facilitate endometrial regeneration. In addition, Banks et al. found that Pectin-Pluronic F-127 hydrogels with 3D loose network structures are more conducive to cell adhesion and survival; Pectin-Pluronic F-127 hydrogel as a carrier wrap growth factor can significantly prolong the release time of growth factors.20–22
Estrogen, specifically 17 β-estradiol (E2), is the key to maintain the microenvironment of endometrium and stimulate regeneration of the endometrium.9,12,23 In clinical practice, E2 is commonly used as an adjuvant treatment for patients with IUA. 24 However, the concentration of E2 in the injured site of the uterus by systemic administration is low, which significantly reduces its therapeutic effect. 25 The high levels of E2 in vivo may increase the risk for thrombosis and malignancy tumor, 26 and reduce the receptivity of endometrium.9,12,23,24 In situ administration of E2 has many disadvantages, including limited half-life period and poor solubility in aqueous solutions. 6 In this study, E2 was encapsulated in W/O/W microspheres to construct pectin-based E2-loaded microcapsules (E2 MPs) to address the limitations of E2 usage. E2 MPs could provide sustained E2 release serving as a long-term reliable source of E2 for endometrial regeneration.
Although it has been recognized that E2 plays an indispensable role in promoting the repair of endometrial injury, its mechanism is still unclear. Several studies have demonstrated that stem/progenitor cells in the endometrium do not directly respond to E2, but may indirectly receive E2 signals from surrounding ESCs through paracrine signals to initiate the proliferation and differentiation of cell.12,27,28 Exosomes (exos) are considered the biological medium of intercellular communication and play an important role in regulating cell proliferation and differentiation. 29 Exos are extracellular vesicles with a diameter of ∼30–150 nm, which act as carriers of bioactive proteins, lipid bilayer, and genetic material and transfer them to surrounding cells in the form of paracrine. 30
Zhang et al. found that when MSCs were cocultured with ESCs and E2, it could differentiate into EECs.12,28,31 It has been reported that ESCs also have the capacity to release exos and modify endometrial microenvironments in a paracrine manner. 32 Therefore, we consider that exos derived from ESCs play a paracrine role in E2-stimulated endometrial regeneration and provide optimal estrogenic response.
The objectives of this study were to investigate the beneficial effects of BMSCs/E2 MPs/scaffolding grafts on promoting the recovery of injured endometrium, and to investigate the mechanism of BMSCs and E2 in mouse endometrial regeneration.
Materials and Methods
BMSCs isolation and culture
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Xinhua Hospital Research Ethics Committee, Shanghai Jiao Tong University School of Medicine (reference number: XHEC-F-NSFC-2018-122).
BMSCs were isolated and cultured as previously described. 9 In brief, 5-week-old female C57BL/6 mice were killed. The femora were isolated and rinsed with phosphate-buffered saline (PBS; Gibco, Grand Island, NY). The marrow cells in the bone cavities were flushed out with Dulbecco's modified Eagle medium/Nutrient Mixture F-12 (DMEM/F-12; Gibco). The resultant lavage was passed through 100 μm cell strainer (BD Bioscience, San Jose, CA) and centrifuged at 1200 rpm for 4 min, which were then resuspended in DMEM/F-12 supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin–streptomycin (P/S; Gibco). The isolated cells were seeded in 100 mm Petri dish and maintained at 37°C in a humidified incubator supplied with 5% CO2. The nonadherent cells were removed after 72 h of culture. The medium was changed every other day and passage was conducted when cells reached confluence (80–100%). The BMSCs of passage 3–5 were used for the following experiments.
Flow cytometric analysis
The expressions of cell surface markers on BMSCs were evaluated by flow cytometry. BMSCs were detached from the culture dish using 0.05% trypsin-EDTA (Gibco), centrifuged, rinsed, and resuspended in PBS at a concentration of 105 cells/mL. Resuspended cells were incubated with 5 mL of CD44-FITC antibody (eBioscience, San Diego, CA), CD45-BV510 antibody (eBioscience), CD29-APC antibody (eBioscience), and CD105-PE antibody (eBioscience) in the dark at 4°C for 30 min. After being washed twice with PBS, cytometric analysis was performed with FACSDiva (Canto; BD Bioscience) and data were analyzed with FlowJo software (Tree Star, Ashland, OR) (Supplementary Fig. S1).
Pectin-based E2 MPs fabrication
A double emulsion system (W/O/W) was used to generate the microspheres, as given in previous publications 33 (Fig. 1). A solution of 2.5 mL of 10 mg/L 17β-estradiol (E2; Sigma, St. Louis, MO) in ethanol was added to 7.5 mL of prewarmed 13.33% (w/v) gelatin (Sigma). The first emulsion of gelatin sphere-in-oil (W/O) phase was prepared by adding 1 mL of E2-gelatin solution into 18 mL of prewarmed (45°C) olive oil containing 0.5 mL Tween 20 (Sigma) and 0.5 mL Span80 (Sigma) as stabilizers. The W/O solution was stirred at 450 rpm for 20 min at room temperature to allow for cooling resulting in gelation of the gelatin to produce W/O emulsion. Then, 0.77 mL of the W/O emulsion was added dropwise to 22.23 mL of 3.25% (w/v) low-methoxyl pectin (WillPowder, Miami Beach, FL) solution stirred at 550 rpm to form the double emulsion (W/O/W).

A schematic illustration of the procedure for fabricating Pectin-based E2-loaded microcapsules and Pectin-Pluronic® F-127 scaffolds for endometrial regeneration: (1) preparing microsphere by electrospray and coating with chitosan; (2) incubating cells with E2-loaded microcapsules; (3) mixing with Pectin-Pluronic F-127 solution; (4) evaluating the therapeutic effects of E2 MPs composite scaffolds in vitro; and (5) evaluating the BMSCs/E2 MPs/scaffolds system for in vivo endometrial regeneration in mice. BMSCs, bone marrow–derived mesenchymal stem cells; E2 MPs, E2-loaded microcapsules.
An electrospray set-up (designed by the University of Shanghai for Science and Technology, Shanghai, China) was then used to spray the W/O/W solution into a 0.15 M CaCl2 solution to induce pectin gelation forming hydrogel microspheres. Pectin-based E2 MPs were collected through centrifugation and incubated in 50 kDa chitosan (Zhejiang Golden-Shell Pharmaceutical Co., Zhejiang, China) solutions for 10 min to form the cationic coating. After rinsing in double-distilled water, the E2 MPs were stored in PBS at 4°C.
The encapsulation efficiency was determined, as given by the following equation34,35:
where Ea is the amount of E2 in the W/O/W solution and Eb is the amount of E2 in CaCl2 solution.
Pectin-Pluronic F-127 hydrogel scaffolds fabrication
A mixture composed of 3% pectin (w/v), 20% (w/v) Pluronic F-127 (Sigma) and 0.25 M mannitol solution was used for constructing the hydrogel scaffolds (stored at 4°C). The BMSCs were incubated with E2 MPs in DMEM/F-12 with 10% FBS and 1% P/S for 2 h at 37°C, using six-well ultralow attachment plates. The 2 × 107 cells with or without E2 MPs were then mixed with 2 mL Pectin-Pluronic F-127 solution at 4°C of final concentration 0.5 mg/mL (BMSCs with E2 MPs). The mixtures were transferred into a 10 mL plastic syringe and the needle (0.7 × 30 mm) was attached. The mixtures were dropped slowly onto a petri dish heated at 37°C. When the temperature reached 37°C, the fluid solidified, owing to the thermal sol–gel transition of Pluronic F-127, forming the hydrogel scaffolds.
Pectin-Pluronic F-127 hydrogel scaffolds degradation
The hydrogel scaffold samples with diameter 11 ± 0.5 mm and height 7 ± 0.4 mm were prepared and incubated in DMEM/F12 with 10% FBS and 1% P/S at 37°C with 5% CO2 for 14 days and weighed every day to determine its degradation. The degradation rate was determined as weight remaining (ratio %),
36
as given by the following equation:
where Wa is the dry gel weight after degradation at different time intervals and Wb is the dry gel weight before the start of degradation experiment.
The morphology of the lyophilized gel was examined via scanning electron microscopy (SEM). The scaffolds were immersed in liquid nitrogen and lyophilized at room temperature. The cross-section of the hydrogels was gold-coated and viewed using a microscope (PHENOM, Eindhoven, the Netherlands).
Enzyme-linked immunosorbent assay for E2 detection
E2 MPs/scaffolds/BMSCs were cultured in DMEM/F-12 with 10% FBS and 1% P/S and maintained at 37°C with 5% CO2. Medium was sampled every day during the first 10 days and every other day after that and stored at −80°C for further analysis. The concentration of E2 cumulatively released from MPs was measured using an E2 enzyme-linked immunosorbent assay (ELISA) kit (LDN, Nordhorn, Germany) according to the manufacturer's protocols. In brief, indicated medium was centrifuged to remove cell debris. Twenty-five microliters of supernatant was dispense into each 96-well plate, which then underwent 1 h of incubation at room temperature on a plate shaker. After adding 100 μL of enzyme conjugate, the plate was incubated for 1 h at room temperature on a plate shaker. After complete aspiration of the content, wells were thoroughly washed four times. After adding 200 μL of substrate solution into each well, the reaction underwent 30 min incubation in dark, and 50 μL stop solution was used to terminate the reaction. TECAN Infinite 200 PRO (TECAN) was used to record absorbance at 450 nm with OD 570 nm as the reference.
Acridine orange/ethidium bromide double fluorescence staining for cell viability determination and imaging
E2 MPs/scaffolds/BMSCs were cultured in DMEM/F-12 with 10% FBS and 1% P/S and maintained at 37°C with 5% CO2. Cell viability was evaluated on day 7. Equal amounts of 100 μg/mL acridine orange (AO) (Sigma) and 100 μg/mL ethidium bromide (EB) (Sigma) were used to prepare the AO/EB dye. The AO/EB dye was added into the medium at a ratio of 1:25, and the cells were observed by a fluorescence microscope (Olympus, Tokyo, Japan) after 15 min. Three nonadjacent planes were randomly selected in BMSCs within scaffolds group and three fields were randomly selected in BMSCs without scaffolds group to record cellular viability. Three samples for each group were selected for statistical analysis. The cellular viability was calculated by dividing the number of all live cells in the selected region by the total number of live (green) and dead (red) cells.
Mouse model of endometrial injury and treatment
In total, 72 nine-week-old female C57BL/6 mice with average body weight 25–30 g were randomly and equally divided into six groups: sham-operated group, spontaneous repair group, BMSCs group, E2/BMSCs group, E2 MPs/BMSCs group, and E2 MPs/scaffolds/BMSCs group (12 mice/group). For estrous cycle studies, vaginal smears were obtained daily between 8:00 and 10:00 AM. Only mice with four consecutive 4-day estrus cycles were selected for study. To establish uterine horn damage model, the mice were first anesthetized, and the skin was sterilized with 10% povidone–iodine. The uterine horns were exposed using a low abdominal midline incision. After the distal ends of the bilateral uterine horns near the ovaries were incised, an 18G stainless steel needle with manually grinding rough surface was used to enter the uterine cavity from the incision of the uterine horn and scrape the endometrium repeatedly toward the cervix until the uterine horns were obviously congested.
The opened skin and uterine horn were sutured using absorbable suture. The 0.1–0.2 mL BMSCs with or without E2 MPs and scaffolds solution were injected into and filled uterine cavity. For spontaneous repair, the uterine horns after modeling were allowed healing without further treatments. For sham operation, after exposure by an abdominal midline incision, the uterine horns were left intact in the abdominal cavity without scratch. All animals were intramuscularly administrated with penicillin twice a day for 3 consecutive days after surgery to prevent systematic infection.
In vivo tracing of implanted E2 MPs/scaffolds/BMSCs
To trace implanted E2 MPs/scaffolds/BMSCs, 9 rats (18 uterine horns) were used. At various time intervals (7, 14, and 28 days post-transplantation), 3 mice (6 uterine horns) were killed and uteri were harvested (n = 6). For immunofluorescence tracing of BMSCs labeled with CM-Dil (Invitrogen, Eugene),11,37 the samples were frozen in liquid nitrogen for cryosectioning (Leica, Germany). Slices were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100. They were then incubated with wide spectrum cytokeratin (Pan-CKs; Abcam) and then stained with anti-rabbit IgG-FITC (Abcam). Cell nuclei were stained with 1 μg/mL 4,6-diamidino-2-phenylindole (DAPI; Sigma) and observed using a fluorescence microscope (Olympus).
Histological analysis
The uterine horn at 4 weeks postoperatively was fixed with 4% paraformaldehyde overnight, dehydrated in graded alcohols, and embedded in paraffin. Sections of 5 μm were prepared transversally and stained with hematoxylin–eosin and Masson staining. For immunohistochemistry, tissue sections were immunolabeled with anti-von Willebrand factor antibody (vWF; Abcam, Cambridge, MA). The thickness of the cross-sectional area of the endometrium was quantified as the maximum vertical distance from the luminal epithelium to the smooth muscle. Three fields were randomly selected in each sample and the percentage of the endometrium containing fibrosis (the percentage of blue area in relation to total tissue area), overall endometrium of total epitheliums and secretory glands content in the stromal part, and total capillary vessels quantity were all semiquantified using the Image-Pro Plus software. 38 Six samples from each group were selected for statistical analysis.
Fertility test
A total of 36 mice (12 uterine horns/group) were used in the fertility test. Four weeks after the procedure, six female mice per group were mated with two male C57BL/6 mice for 5 days. The day after the first mating day was regarded as gestational day 0.5. At day 14.5–18.5 after gestation, the uteri were exposed under anesthesia to confirm the presence of embryos. Any uterine horn containing at least one embryo >0.5 cm in diameter was counted as pregnancy.
ESCs isolation and culture
Isolation of ESCs was based on the protocol in a previous report. 39 Five-week-old female C57BL/6 mice were injected subcutaneously with 100 μL of the E2 solution (100 ng/100 μL) for 3 consecutive days to stimulate the proliferation of endometrium and killed on the fourth day. The uterine horns were isolated. Uterine horns were transferred to the 0.25% Trypsin-EDTA (Gibco) for 60 min at 4°C, and then incubated for 30 min at 37°C. Then, uteri were transferred into a petri dish containing cold DMEM/F12 medium to inactivate trypsin activity, and then the vortex for 20 s to release the epithelial sheets. The uteri were transferred to 1 mg/mL collagenase I (Sangon Biotech, Shanghai, China) for 30 min at 37°C while shaking (200 rpm). After inhibiting collagenase activity, the stromal cell suspension was centrifuged at 500 g for 7 min. Cells were resuspended in DMEM/F-12 with 10% FBS and 1% P/S and then plated on culture dish maintained at 37°C with 5% CO2. The nonadherent cells were removed after 6 h of culture. The medium was changed every other day and passage was conducted when cells reached confluence (80–100%).
Immunofluorescent staining
Endometrial cells were fixed with 4% paraformaldehyde for 15 min at 4°C and permeabilized with 0.3% Triton X-100 for 1 h at room temperature. They were then incubated at 4°C overnight with primary antibodies including vimentin (Vim; Abcam) and Pan-CKs. The next day, cells were stained with anti-rabbit IgG-FITC and anti-rabbit IgG-PE secondary antibody. Cells were then counterstained with 1 μg/mL DAPI for 5 min and observed with a fluorescence microscope (Olympus).
Differentiation of BMSCs into EEC-like cells by coculture system
BMSCs and ESCs were cocultured in Transwell system (12 mm Transwell with a 0.4-μm pore polycarbonate membrane insert; Corning). 28 In brief, BMSCs were seeded in the bottom of six-well plate at a density of 6 × 105 cells/well and ESCs were seeded on the Transwell membrane at a density of 6 × 104 cells/well to separate the cells but allow soluble factors to pass freely between them. Six experimental groups were set up. In group A (control group), BMSCs were cultured in the bottom of the coculture system alone without ESCs in differentiation medium (DMEM/F-12 with 2% Charcoal-Stripped FBS, BI); in groups B, C, D, E, and F, BMSCs were cocultured with ESCs and different concentrations of E2 (0, 1, 10, 100, 1000 nM, respectively) in differentiation medium. The medium was changed every 2 days over a period of 4 weeks.
Western blot analysis
Differentiated BMSCs and exosomes were lysed in RIPA buffer (Beyotime, Shanghai, China). The protein in the supernatant was quantified using the BCA Protein Assay Kit (Thermo Scientific™), electrophoresed on a 12% SDS-PAGE gel, and transferred to polyvinylidene fluoride membrane (Millipore, Billerica, MA). After blocking, the membranes were incubated at 4°C overnight with one of the following antibodies: CD9 (Abcam), Tsg101 (Abcam), CD63 (Abcam), progesterone receptor (PR) (Invitrogen), estrogen receptor α (ER-α) (Invitrogen), CK19 (Novus, Littleton), CK18 (Abclonal, Wuhan, China), CK13 (ABclonal), β-actin (Cell Signaling Technology, Danvers, MA), or GAPDH (Cell Signaling Technology). Anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology), and anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology) were used as the secondary antibodies. The labeled proteins were visualized with the ChemiDoc™ XRS imaging system (Bio-Rad Laboratories, Inc., Hercules, CA) using an enhanced chemiluminescence kit (Pierce). ImageJ software was used for Western blot grayscale analysis.
Exosome isolation
ESCs at 70% of confluence were incubated in DMEM/F12 containing 10% exosome-depleted FBS (System Biosciences, CA) for 48 h. Subsequently, the medium of ESCs was collected and centrifuged at 2000 g for 10 min at 4°C to eliminate cell debris. The resulting supernatants were filtered through a 0.22-μm filter (Millipore) to remove microvesicles, and then concentrated through an Amicon Ultra-15 100 kDa centrifugal filter (Millipore) at 4000 rpm for 30 min. Exosomes were then isolated using the Total Exosome Isolation kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. In brief, the supernatant was mixed with Total Exosome Reagent and incubated for 24 h at 4°C. Samples were centrifuged at 10,000 g for 2 h and the supernatant removed. The exosome pellet was resuspended in 100 μL of PBS and stored at −80°C.
Identification of ESCs-derived exosomes
The size distribution of ESCs-derived exosomes (ESCs-Exos) was measured by nanoparticle tracking analysis (NTA) with a NanoSight NS3000 instrument (Malvern Instruments, Malvern, United Kingdom), and the exosomes morphologies were observed by transmission electron microscope (TEM) (Hitachi, Tokyo, Japan). The characteristic surface marker proteins of exosomes were analyzed by Western blot.
Exosome uptake
Exosomes were labeled with a membrane-labeling dye (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate, Dil) according to the manufacturer's protocol (Invitrogen). In brief, 15 μg exosomes diluted in DMEM/F12 medium were added to 0.5 μL Dil and incubated for 20 min at 37°C with 5% CO2. Excess dye was removed from the labeled exosomes by ultracentrifugation at 100,000 g for 70 min at 4°C, and the exosome pellets were purified on Exosome Spin Columns MW 3000 (Invitrogen) according to the manufacturer's instructions. The final pellets were resuspended in 100 μL PBS. Dil-labeled exosomes were added to the culture medium of BMSCs, which were seeded at a density of 250 cells/mm2 the previous day and incubated at 37°C for 2 h. Subsequently, BMSCs were fixed with 4% paraformaldehyde for 15 min at 4°C, and then counterstained with 1 μg/mL DAPI for 5 min. The fluorescence was detected under a confocal laser scanning microscope (Leica TCS SP8; Leica Microsystems, Buffalo Grove, IL). Finally, colocalization was performed using the ImageJ Fiji software.
Proliferation assay
The effects of ESCs-Exos on cell proliferation were determined by a cell counting kit-8 assay (CCK-8; Beyotime). In brief, BMSCs were seeded at 1500 cells/well into 96-well plates, cultured in DMEM/F-12 containing 10% FBS and 1% P/S, and allowed to adhere overnight at 37°C under 5% CO2. The cells were washed with PBS and then maintained in a medium supplemented with or without Exos (0, 25, 50, 100, 200, and 500 μg/mL). A group without cells served as the blank. On days 0, 1, 2, and 3, 10 μL CCK-8 reagent was added to each well and incubated at 37°C for 2 h. The absorbance was observed at 450 nm using a microplate reader (Bio-Rad 680; Bio-Rad).
Exosome-mediated stem cell differentiation
BMSCs were plated at a density of 150 cells/mm2. After 24 h, BMSCs were rinsed with PBS and then maintained in a differentiation medium, differentiation medium containing ESCs-Exos with 50 μg/mL, or differentiation medium containing exosomes released from ESCs by E2 stimulation (E2-ESCs-Exos) with 50 μg/mL. The medium was changed every 2 days for up to 4 weeks.
Statistical analysis
All data in this study were analyzed with SPSS 23.0. Continuous variables were shown as the mean ± standard deviation and assessed by Shapiro–Wilk test for normal distribution. Significance of difference between two groups was tested by Student's t-test or analysis of variance. Fisher's exact test (Freeman-Halton) was used to assess the outcome of treatment. Chi-squared tests were performed to compare pregnancy rates. The statistical significances were presented as p-values, and p < 0.05 was considered statistically significant.
Experiment
Fabrication and characterization of Pectin-Pluronic F-127 hydrogel scaffolds
When the temperature increases from below to above (37°C for this study) its lower critical solution temperature (LCST), Pluronic F-127 with a sol-gel transition behavior in aqueous solution can help generate and maintain the desired shape. Figure 2A shows the images of the Pectin-Pluronic F-127 solution (Left) and hydrogel scaffolds (Right). Then, BMSCs were suspended in Pectin-Pluronic F-127 solution to reach a 1 × 106 cells/mL concentration for forming the hydrogel scaffolds. As revealed in the SEM images, Pectin-Pluronic F-127 hydrogel scaffolds exhibit porous structures, which indicates excellent water absorption capacity and a fair improvement in the 3D network structure of the Pectin-Pluronic F-127 hydrogel scaffolds (Fig. 2B). These continuously interconnected porous structures are also conducive to the nutrient supply for cell growth in tissue engineering. BMSCs could get attached to the scaffold and distribute to the inner space of the scaffold (Fig. 2C).
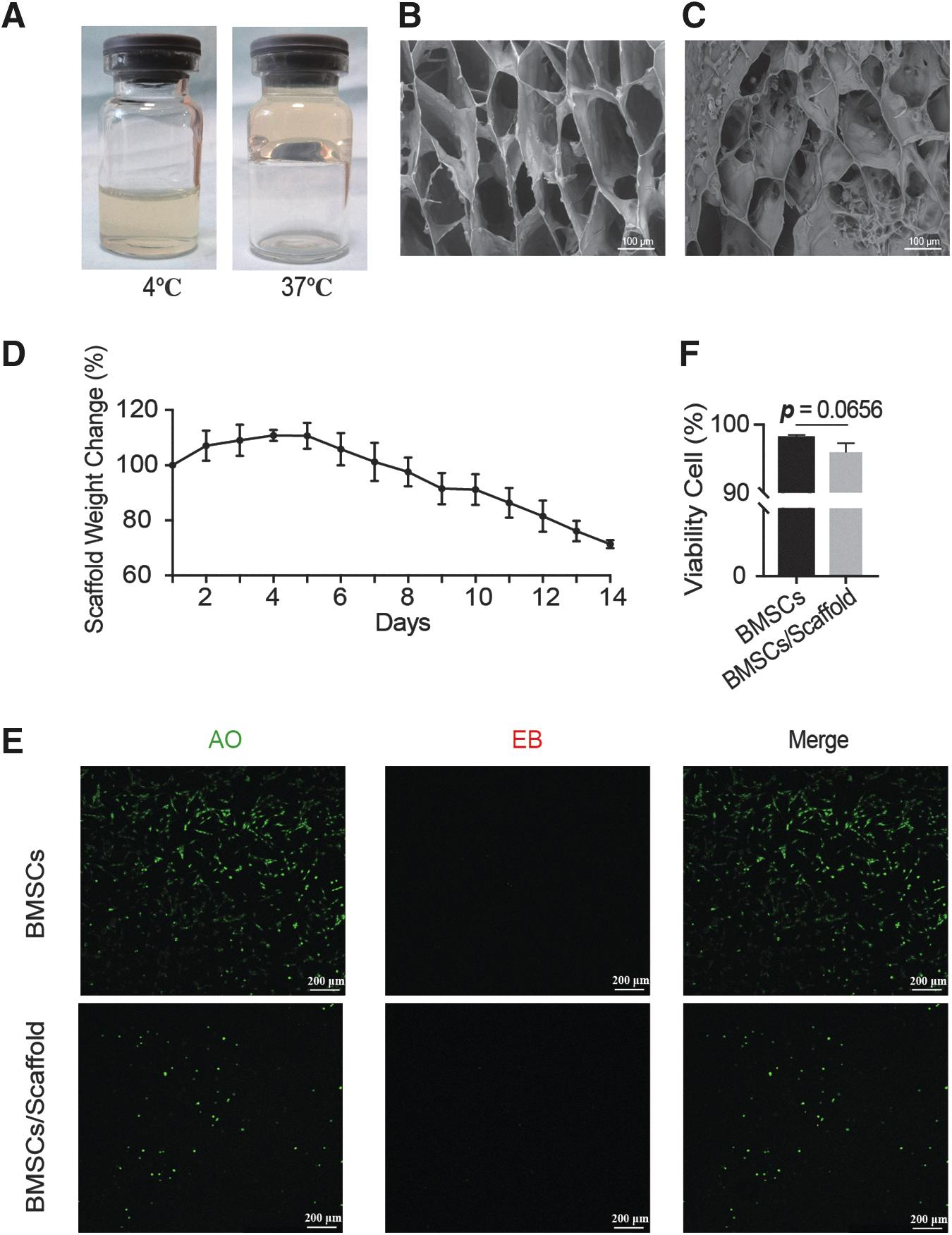
Pectin-Pluronic F-127 scaffolds could provide 3D architecture for the attachment of BMSCs for the culture in vitro.
As given in Figure 2D, the hydrogel scaffold can maintain a relatively stable state within 2 weeks. It began to degrade after day 5, and ∼28% of scaffold degraded at a consistent and sustained pace at day 14, whereas the hydrogel scaffold broke into several pieces at about day 14–18, and then accelerated the disintegration.
The cytotoxicity of scaffolds was further explored. After 7 days of culture, the even distribution of BMSCs within the scaffold can be viewed in focus and out of focus as they are at different planes by AO/EB staining (Fig. 2E). BMSCs had a high cellular viability rate of 96% ± 1.3% within the bioprinted scaffold compared with 98% ± 0.5% for control, indicating that the hydrogel scaffold could provide 3D architecture for the attachment of BMSCs and exhibit negligible toxicity of BMSCs for the culture in vitro (p = 0.0656) (Fig. 2E, F).
Preparation and characterization of Pectin-based E2 MPs
Figure 3A and B presents that the double emulsion (W/O/W) microspheres were successfully created with the diameters averaging ∼100 μm. Encapsulation efficiency of E2 was 90.8% ± 0.8%. Then, BMSCs were coincubated with E2 MPs for 72 h. The CCK-8 assay revealed that BMSCs incubated with E2 MPs proliferated faster than those without E2 MPs (n = 3) (Fig. 3C). Furthermore, Pectin-based E2 MPs (equivalent concentration 10 nM) were incubated in medium, which was harvested for 30 days to assay for the release of E2 using ELISA. As given in Figure 3D, the E2 in medium would degrade rapidly, and the effective concentration of E2 was reduced to 19.7% of the original concentration in the first 24 h and almost completely degraded within 72 h.
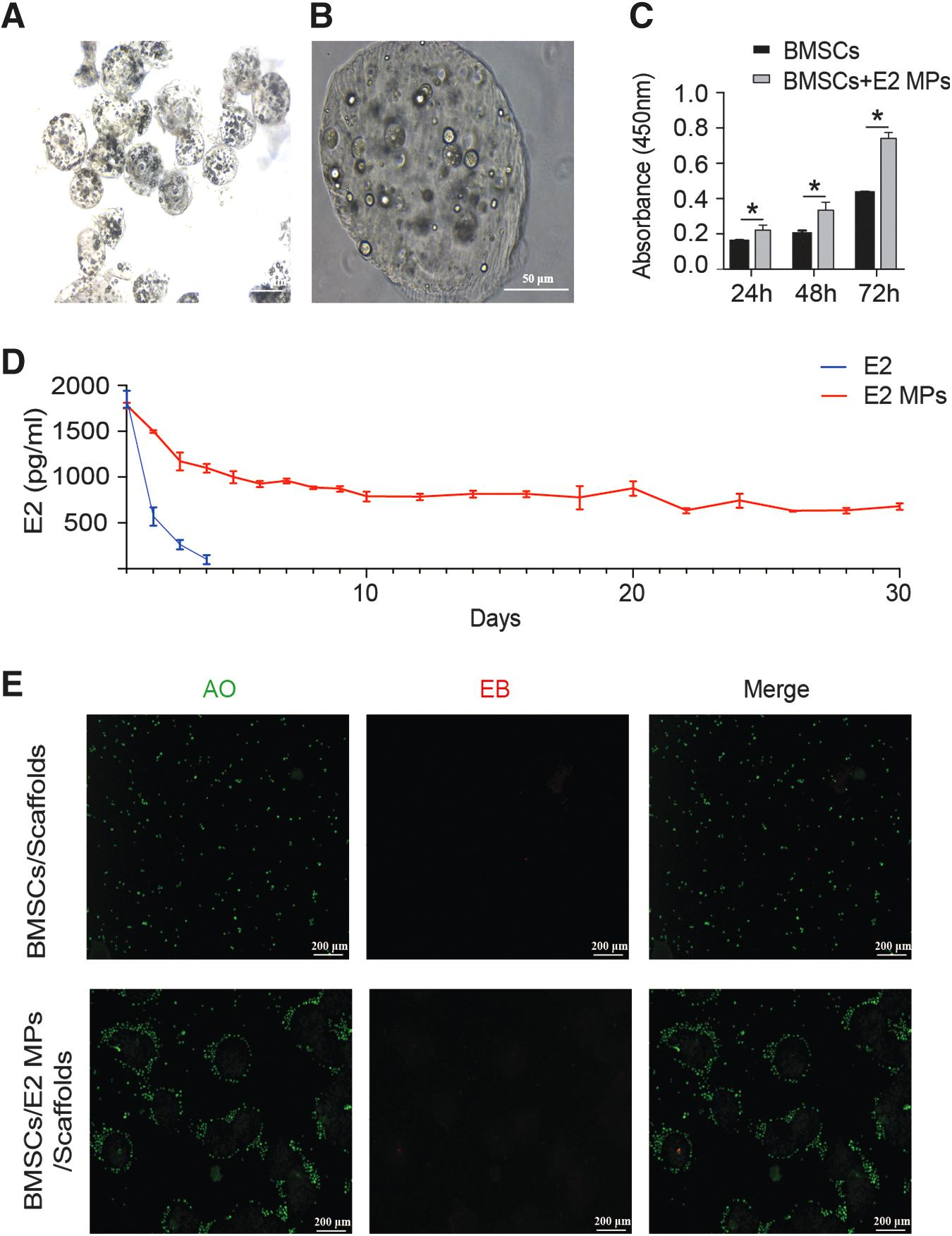
Pectin-based E2 MPs serve as a reliable source of E2 to promote endometrial regeneration.
However, the microcapsule could maintain the effective concentration of E2 in medium for 30 days by controlling the release of E2. Diffusion is the main driving mechanism of E2 release. These results supported that E2 was shown to be successfully encapsulated into the W/O/W microspheres, which provided a sustained E2 release and served as a reliable source of E2 to promote endometrial regeneration.
The prepared E2 MPs and BMSCs were mixed with Pectin-Pluronic F-127 solution to generate the hydrogel scaffolds for investigating the influence of E2 MPs on microvascularized 3D tissue formation. The presence of the W/O/W microspheres does not impact the hydrogel scaffold formation process. Without microcapsule, the BMSCs are evenly distributed within the scaffolds. In the presence of E2 MPs, the BMSCs self-assembled around the E2 MPs and interacted to form a 3D network (n = 3) (Fig. 3E).
BMSCs/E2 MPs/scaffolds system for in vivo endometrial regeneration in mice
To investigate the potential therapeutic effect of BMSCs/E2 MPs/scaffolds transplantation in endometrial regeneration, BMSCs/E2 MPs/scaffolds were injected into the uterine cavity of mouse endometrial injury model. At 28 days after surgery, E2 MPs and scaffolds were completely degraded. Sections of all groups were lined by simple columnar epitheliums that were similar to the Sham group. Endometrial tissue in BMSCs/E2 MPs/scaffolds group appeared well organized with epitheliums, secretory glands, and apparent neovascularization, whereas endometrial of spontaneous regeneration group formed the scar tissues with collagen deposition (Fig. 4). The thickness of endometrial in BMSCs/E2 MPs/scaffolds (877.95 ± 23.95 μm) was similar to that in sham group (896.41 ± 62.65 μm) and was higher than the spontaneous group (574.87 ± 149.43 μm), BMSC group (612.78 ± 29.05 μm), BMSCs/E2 groups (723.89 ± 108.75 μm), and BMSCs/scaffolds groups (728.78 ± 111.75 μm) (p < 0.05, respectively) (Fig. 4A, B).
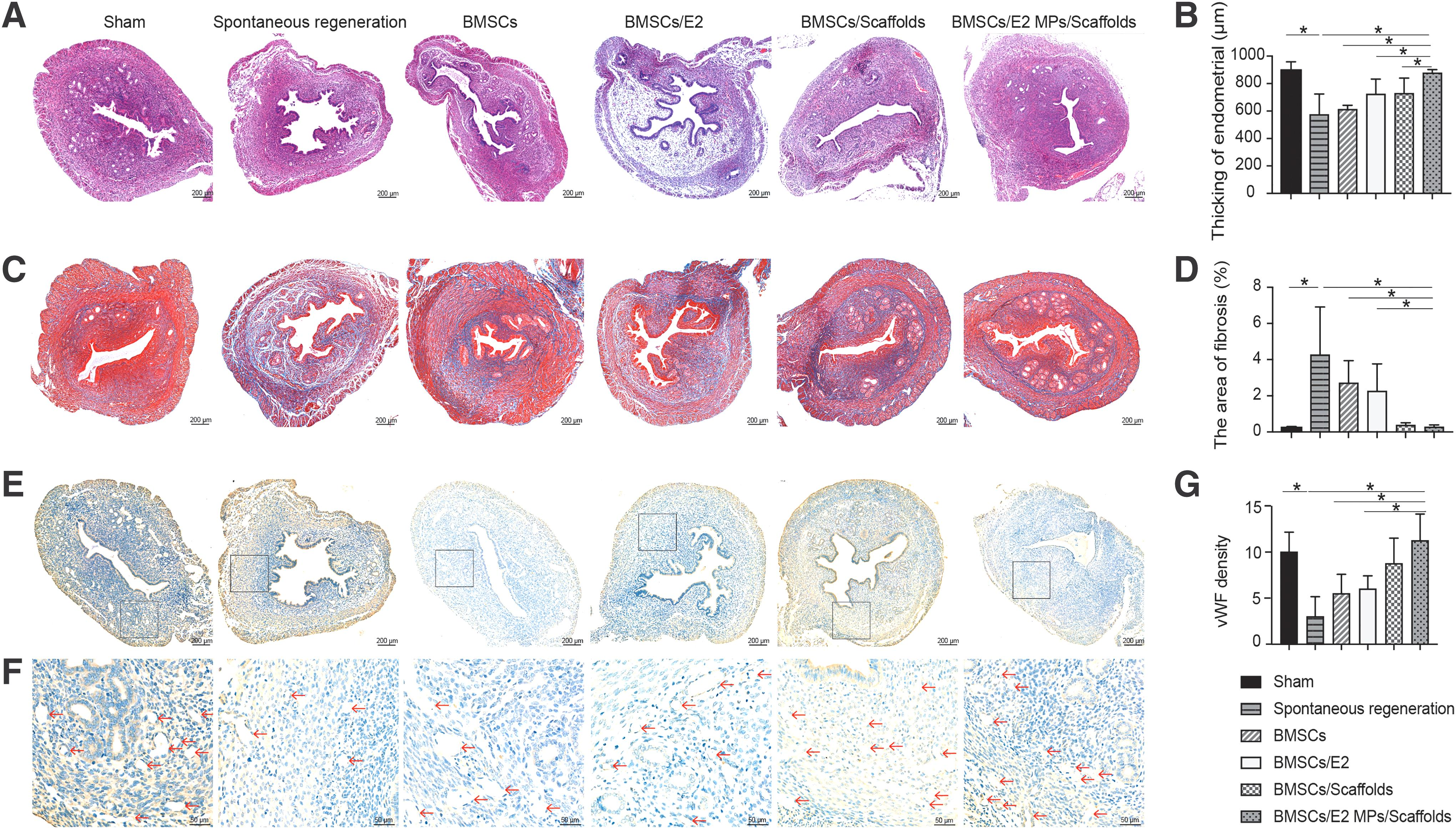
Transplantation of BMSCs and pectin-based E2-loaded microcapsules on Pectin-Pluronic F-127 scaffolds for mouse endometrial regeneration.
In addition, although the rate of endometrial fibrosis in mice receiving BMSCs/E2 MPs/scaffolds (0.27% ± 0.11%) was slightly higher than in the sham-operated group (0.23% ± 0.07%), it was significantly lower than in the spontaneous regeneration group (4.26% ± 2.65%), the group receiving BMSCs alone (2.71% ± 1.24%) and the group receiving BMSCs/E2 (2.26% ± 1.50%) (p < 0.05) (Fig. 4C, D).
Neovascularization in regenerating endometrium is important for the restoration of functional fertility. At 28 days, blood vessel density in the endometrial tissue after implantation of BMSCs/E2 MPs/scaffolds grafts (11.3 ± 2.9) was significantly higher than that in spontaneous group (3 ± 2.2), BMSCs group (5.5 ± 2.1), BMSCs/E2 groups (6 ± 1.4) (p < 0.05, respectively) (p < 0.05), while still comparable to that in BMSCs/scaffolds groups (8.8 ± 2.8) and sham-operated group (10 ± 2.2) (Fig. 4E–G).
To track the transplanted BMSCs in the regenerated endometrium, immunofluorescence of uteri at each time interval was analyzed (Fig. 5A). At 7 days post-transplantation, some CM-Dil–labeled BMSCs migrated to the wounded endometrium edges. With longer time post-transplantation, the number of CM-Dil–labeled BMSCs decreased and a few BM-MSCs directly differentiated to endometrial epithelium cells, which were positive for Pan-CKs staining.

Pregnancy within the regenerated uterine horns
Four weeks after surgery, pregnancy was observed in some of the regenerated uterine horns. Embryos were present in some of the regenerated uteri after grafting with BMSCs, BMSCs with E2 or scaffolds, but were found in all regenerated uterine horns in BMSCs/E2 MPs/scaffolds groups. The pregnancy rate of BMSCs/E2 MPs/scaffolds groups (100%) was much higher than those of spontaneous regeneration group (25%), BMSCs group (41.7%), BMSCs/E2 group (50%), BMSCs/scaffolds group (66.7%), and comparable with that of sham-operated group (100%) (Fig. 5B). This suggested a nearly full uterine recovery from injury in pregnancy rate. These results supported the beneficial effect of BMSCs/E2 MPs/scaffolds in promoting the recovery of injured endometrium to functional endometrium.
Culture and identification of endometrial cells
Endometrial cells were isolated from mice. ESCs were spindle-shaped fibroblast-like cells, whereas EECs were polygonal cells rounder than stromal cells (Fig. 6A). A vimentin/cytokeratin double staining was performed and indicated that stromal cells were positive for Vim, and negative for Pan-CKs, whereas epithelial cells do express Pan-CKs but not Vim. These immunofluorescence staining showed that ESC culture has a purity of up to 90% (Fig. 6B).
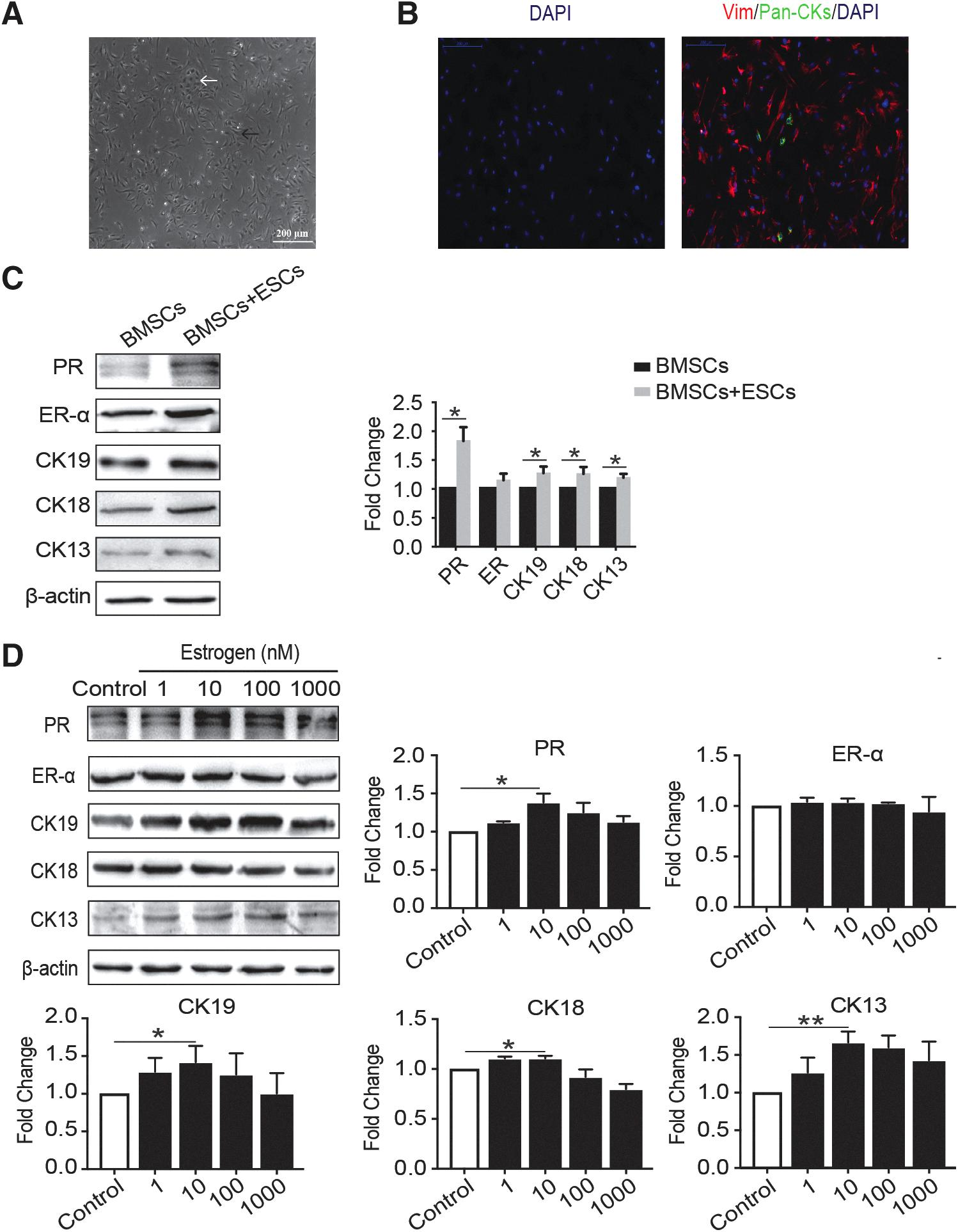
BMSCs differentiate into EEC-like cells in the coculture system.
Evaluation of the effects of E2 on epithelial differentiation of BMSCs
BMSCs were cocultured with ESCs in Transwell system, the system is characterized by separating the cells but allowing soluble factors to pass freely between them. After 6 weeks of culture, the expression of EEC markers, CK19, CK18, CK13, ER-α, and PR on BMSCs were tested by Western blot. Of note, these marker expressions were upregulated in BMSCs cocultured with ESCs compared with that in BMSCs cultured alone (Fig. 6C). This suggests that endogenous factors secreted by ESCs provide an environment for BMSCs to differentiate in the direction of epithelial cells.
Then, BMSCs were treated with gradient concentrations of E2 (0, 1, 10, 100, 1000 nM, respectively) in the coculture system. A Western blot showed that E2 dose-dependently promoted BMSC differentiation. After 6 weeks in culture, the expression levels of the EEC markers except ER-α were highest in cells cultured with 1 × 10−8 mol/L E2 (p < 0.05). These data suggested that 10 nM E2 provided the optimal estrogenic response on the cultured cells in the microenvironment (Fig. 6D).
Characterization of ESCs-Exos
ESCs-Exos were characterized in terms of size, morphology, and surface markers. NTA revealed that the diameters of these particles predominantly ranged from 30 to 150 nm (Fig. 7A). ESCs-Exos exhibited a cup- or sphere-shaped morphology (Fig. 7B), as shown by TEM. The identity of these particles was further confirmed as exosomes by Western blot, which showed the presence of exosomal surface markers including CD63, CD9, and TSG101 (Fig. 7C). All these data suggested that these nanoparticles were indeed exosomes. In addition, the numbers of the exosomes released from ESCs with E2 stimulation was similar to those of exosomes isolated from ESCs without E2 stimulation (47.2 ± 2.8 μg vs. 42.1 ± 5.0 μg crude exosomes/mL/106, p = 0.073).
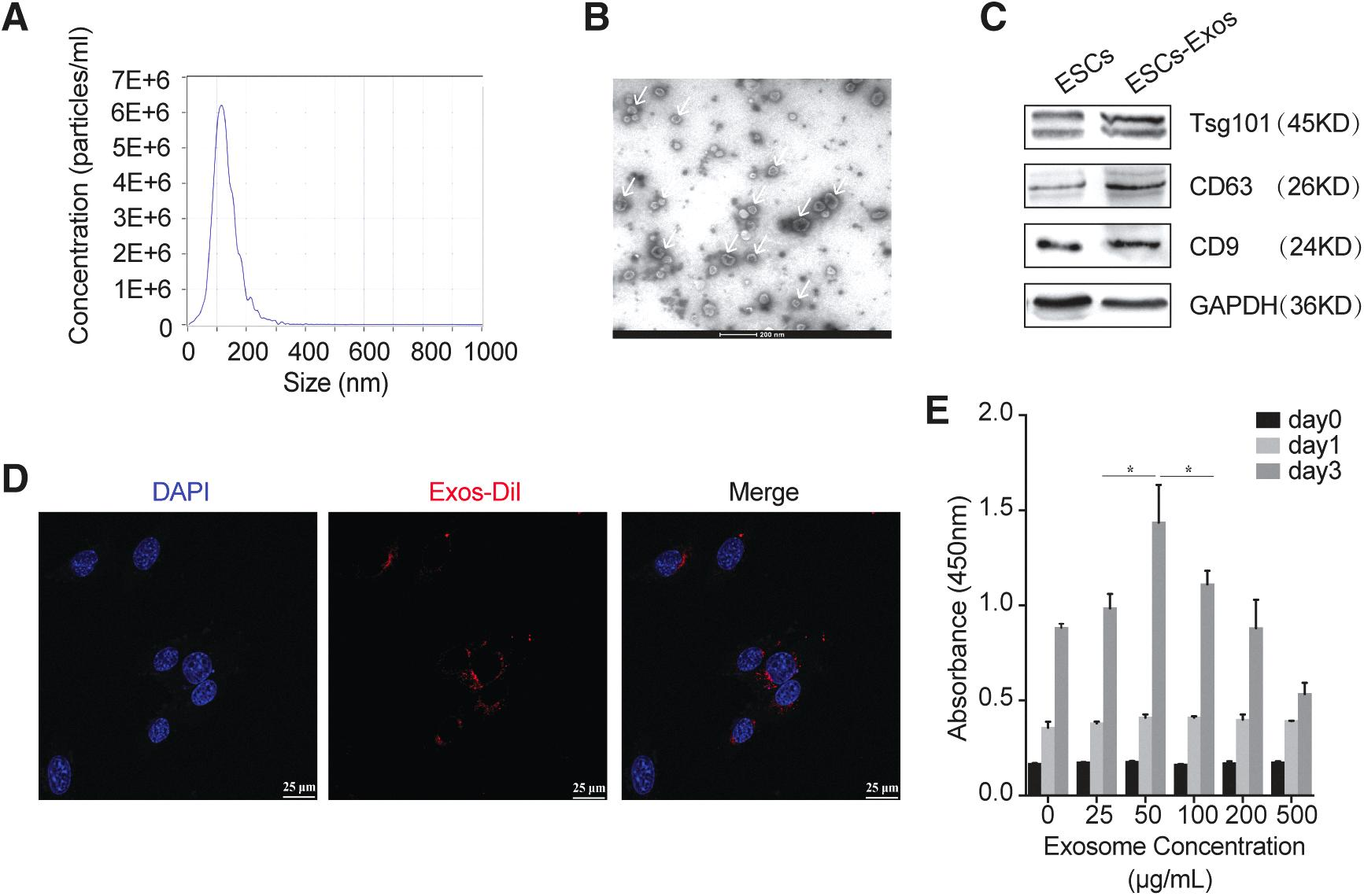
Characterization of exosomes secreted by ESCs:
Responses of BMSCs to ESCs-Exos in vitro
The confocal microscope observed that exosomes were internalized into the cytoplasm of BMSCs and were partly colocalized within the nuclei (Fig. 7D), which implied that ESCs-Exos can be transferred into stem cells with high cellular uptake efficiency. Then, the proliferative effect of ESCs-Exos on BMSCs showed a dose-dependent response by the CCK-8 assay (for 3 days) (Fig. 7E). In particular, an exosome concentration of 50 μg/mL was the most beneficial for the BMSCs. However, high concentrations (500 μg/mL) of exosomes resulted in high cell death.
Furthermore, BMSCs were coincubated with exosomes released from ESCs with or without E2 stimulation. After 21 days of culture, the expression of the EEC markers, CK19, CK18, CK13, ER-α, and PR significantly augmented in BMSCs treated with 50 μg/mL of ESCs-Exos. However, exosomes derived from E2-stimulated ESCs (E2-ESCs-Exos) could further promote the expression level of these markers in BMSCs (Fig. 8A). Higher proportion of Pan-CKs staining–positive cells were also observed in this group according to immunofluorescence analysis (Fig. 8B). These results indicated that ESCs-Exos provided biochemical cues to stimulate differentiation of BMSCs toward EECs, and involvement of E2 could enhance its differentiation-promoting capacity.
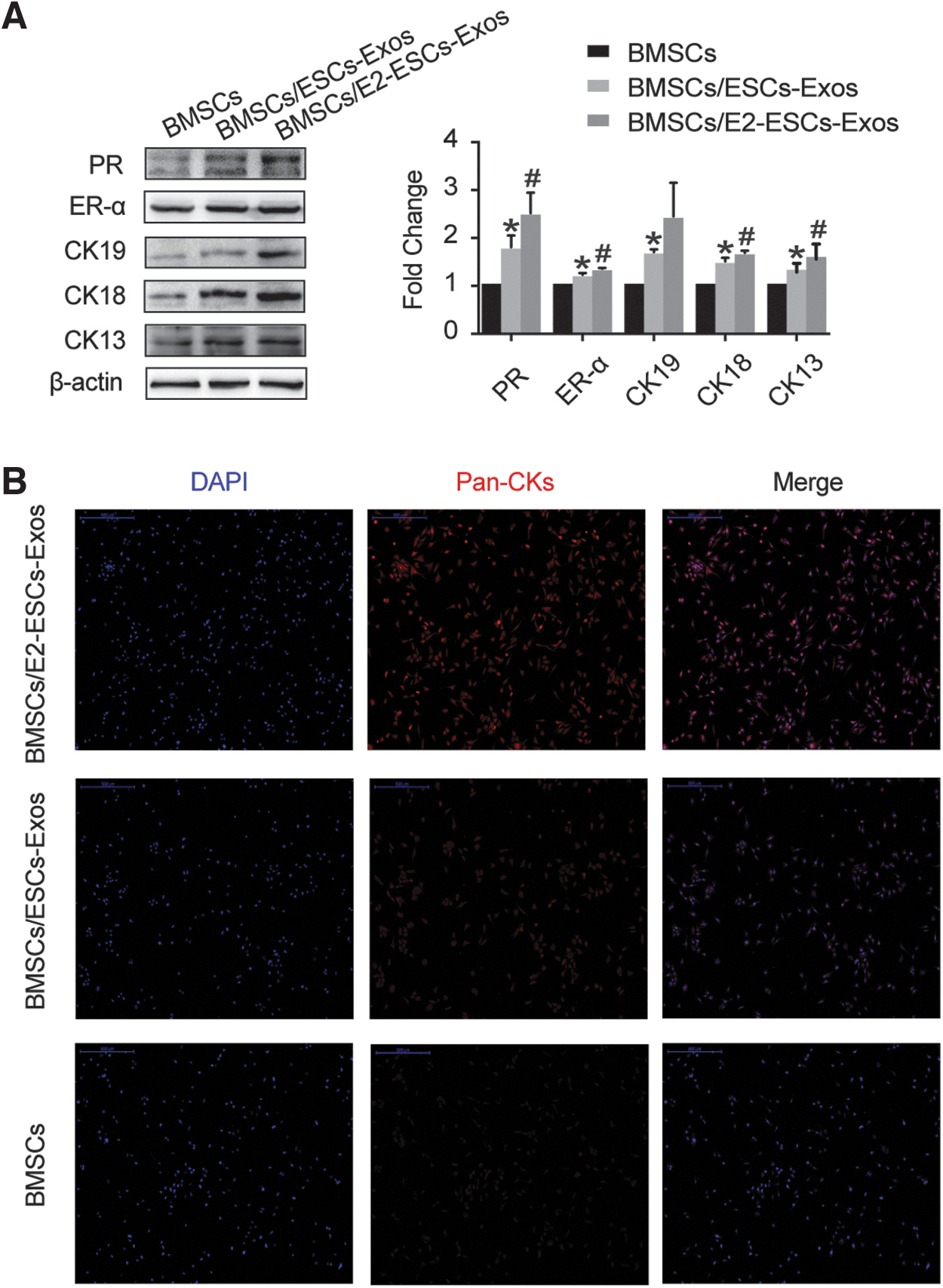
E2-Exos promoted BMSCs differentiation.
Discussion
IUA, which may lead to infertility and/or pregnancy complications, are frequently caused by infectious or mechanical injury to the endometrium.3–5 Several therapies, such as hysteroscopic surgery removal of adhesions and hormonal therapy have been adopted for the treatment of endometrial fibrosis.6,7 However, severe damages related to the basal layer endometrium results in the loss of resident stem cells in it and injures the microenvironment of endometrium, which is followed by the failure of endometrial functional layer regeneration.3,4 Treatment for these severe cases is difficult, and prognosis is usually unsatisfactory. 8 BMSCs have been recognized as a strong candidate tool for treating severe endometrial injuries.13,40,41 Studies have proved that BMSC transplantation based on its autocrine or paracrine effects at the lesion site can inhibit the excessive fibrosis and inflammatory response of endometrium, promote the proliferation of endometrial cells and revascularization, to repair endometrial damage, and restore fertility to a certain extent.8,11,37,38,42,43
Besides, BMSCs could directly differentiate to endometrial epithelium cells to regenerate endometrium.11,43 Donor-derived bone marrow cells have been shown to differentiate into EECs and ESCs.13,14 Donor-derived endometrium was detected accounting for 0.2–48% and 0.3–52% of the endometrial epithelium and stroma, respectively. 14 Ding et al. used collagen scaffolds laden with BMSCs for the rat damaged uterus tissues and traced implanted BMSCs. They found that implanted BMSCs migrated the vicinity of wounded region and distributed in the submucosa and basal layer of the regenerated endometrium.11,43 Similarly, in this study, some CM-Dil–labeled BMSCs migrated to the edge of the injured endometrium at 7 days after transplantation, and over time, a few BMSCs could directly differentiate into Pan-CK staining–positive endometrial epithelium cells.
However, low differentiation efficiency and poor proliferation capacity are still the major obstacles of BMSC transplantation.8,9,11 Thus, a therapeutic strategy is needed for the restoration of endometrium by modifying the microenvironment of endometrium with promotion of survival, attachment, proliferation, and differentiation of BMSCs.
In this study, Pectin-Pluronic F-127 hydrogel scaffolds were used for providing 3D architecture for BMSCs. Both Pectin and Pluronic F-127 show well-proved biocompatibility and bioactivity. 18 In addition, Pluronic F-127 exhibits a sol–gel transition behavior in aqueous solution when temperature increases from below to above (37°C used in this study) its LCST. 19 Thus, the Pectin-Pluronic F-127 solution can completely fill the uterine cavity, reach the injured area, and form the hydrogel scaffold in vivo. In this study, it was found that a Pectin-Pluronic F-127 hydrogel with slow degradation time and 3D loose network structure is more conducive to the adhesion and survival of BMSCs. Low degradation rate is conducive to its application for long-term delivery. BMSC/scaffold groups show better integration into adjacent tissues and more effective tissue regeneration than BMSC groups, such as marked cell proliferation and massive neovascularization at 4 weeks after operation.
E2 is the key to maintain the microenvironment of endometrium. E2 exerts its effects via ER-α and ER-β, in which ER-α predominates and mediates the majority of E2 signals in mouse endometrium.44,45 However, several studies have demonstrated that epithelial stem/progenitor cells in the endometrium do not directly respond to E2, but may indirectly receive E2 signals from surrounding ER-α stromal cells through paracrine signals to initiate the proliferation and differentiation of cells. 44 Similarly, some research found that the angiogenesis effect of E2 on endometrial vascular endothelial cells is also indirect, where E2 promotes the expression of key regulators of angiogenesis such as VEGF, cystathionine-β synthase, and H2S via ER-α or Erβ of ESCs.46,47 Besides, E2 also plays a part in cell viability of endometrium.24,48
Zhang et al. demonstrated that E2 could elicit its protective effect on endometrium regeneration by blocking cell apoptosis in the basal layer of endometrium.24,48 They suggested that endoplasmic reticulum stress-induced cell apoptosis also participates in the progress of IUA, and E2 could inhibit endoplasmic reticulum stress by activating PI3K/Akt and ERK1/2 signaling pathways. 24 Moreover, E2 could also elevate the expression of kisspeptin on the damaged endometrium by activating ERK1/2 and MAPK p38 signals. 48 In this study, E2 is responsible for uterine endometrial regeneration through stimulating the proliferation and differentiation of stem cells and forming new capillaries after injury.
However, owing to its low concentration at the injured site, the risk of thrombosis and malignancy by systemic administration, short half-life period, and poor solubility in aqueous solutions, there are many restrictions on the usage of E2 in vivo direct. Therefore, it is difficult to maintain the effective E2 concentration in the injured endometrium when administered only by injections.9,12,23–25 In this study, we developed a new sustained-release system intended for in situ administration of E2. E2 was encapsulated into the W/O/W microspheres to construct Pectin-based E2 MPs, which could maintain the effective concentration of E2 in the culture medium within 30 days by the controlled release of E2 to promote the differentiation of BMSCs and endometrial regeneration. The therapeutic effect of BMSCs/E2 MPs/scaffolds group in promoting functional endometrial recovery was also confirmed. Four weeks after the transplantation, BMSCs/E2 MPs/scaffolds system increased proliferative abilities of uterine endometrial and vascular endothelial cells.
Furthermore, Zhang et al. proved that when coincubated with HUVECs, microspheres combined with scaffolds have the potential to generate vascularized tissue, and human umbilical vein endothelial cells (HUVECs) self-assembled around MPs and formed a 3D vascular-like network. 49 In our mouse endometrial injury model, the BMSCs/E2 MPs/scaffolds group also showed an increased expression of the blood vessel marker vWF and more vessels within regenerated endometrium. BMSCs/E2 MPs/scaffolds could promote the restoration of the endometrial construction, to some extent, which may result from increased vascularity, thus improving the access of injured tissues to nutrients, oxygen, and hormones. After the transplantation, the pregnancy rate of BMSCs/E2 MPs/scaffolds group was significantly higher than that of other groups. In addition, endometrial glands and uterine vasculature were well developed, and reproductive hormone response of regenerated tissue was normal, close to normal tissue. Therefore, BMSCs/E2 MPs/scaffolds system is a promising strategy for endometrial tissue regeneration.
Although it has been recognized that E2 plays an indispensable role in promoting the repair of endometrial injury, its mechanism remains poorly understood. Based on previous studies, the proliferative and differentiation response of epithelial cells and endometrial stem cells to E2 is mediated by ESCs, which synthesize paracrine mediators under the action of E2 and transmit them to surrounding cells.27,28,31 In this study, BMSCs and ESCs were cocultured in Transwell system. After 6 weeks of culture, the expression of EEC markers in BMSCs cocultured with ESCs was upregulated, and E2 could further facilitate the differentiation of BMSCs toward epithelial lineages. It has been noted that the exogenous BMSCs implanted in the endometrium are surrounded by endometrial stroma, which provides a local environment for survival of BMSCs, enabling it to reconstruct the injured uterine endometrium through the autocrine and paracrine pathways or self-proliferation and differentiation into epithelial cells. 50
It has been reported that ESCs can secrete a large number of vesicles, that is, exosomes, which affect the growth, function, and development of endometrial tissue.32,51–54 As a carrier of bioactive proteins, lipid bilayer and genetic material, exosomes are considered as the biological mediator of intercellular communication and play an essential role in regulating cell proliferation and differentiation.29,30,55,56 In this study, we found that ESCs could secrete exosomes, which are successfully delivered into BMSCs with high cellular uptake efficiency and significantly affect cell proliferation in a dose-dependent manner. ESC-derived exosomes could also induce BMSCs differentiate toward endometrial epithelium, which was more significant than ESCs itself. In addition, exosomes derived from E2-stimulated ESCs could enhance the differentiation efficiency of BMSCs. These results indicated that the exosomes derived from ESCs play a paracrine role in E2-stimulated endometrial regeneration by inducing BMSCs to differentiate into EECs.
Greening et al. proved that E2 treatment could reprogram the components in the endometrial-derived exosome cargo, accompanied by changes of its regulatory effect on cells.57,58 Based on our and other previous studies, exosomes were released in numbers independent of E2 regulation. 58 Therefore, it is speculated that E2 enhance the ability of BMSCs to differentiate into epithelial cells via changing the specific composition of exosomes released by ESCs.
Conclusion
The BMSCs/E2 MPs/scaffolds therapeutic strategy may be beneficial in the treatment of severely damaged endometrium. The BMSCs/E2 MPs/scaffolds system could provide a 3D architecture for the attachment, growth, and migration of BMSCs and vascularization promotion while serving as a reliable source of E2 to promote endometrial regeneration. Furthermore, endometrial regeneration depends, at least partly, on engraftment of BMSCs and their subsequent differentiation into endometrial epithelium, which might be induced by ESCs in the local microenvironment through exosomes. Exosomes derived from ESCs play paracrine roles in endometrial regeneration stimulated by E2 and provide optimal estrogenic response.
Footnotes
Acknowledgments
A preprint of the article has previously been published in Research Square. 59 The authors thank Gina Mazzone (MSOE) for proofreading the article.
Authors' Contributions
Y.W. and S.G. performed experiments. Y.W. and S.G. performed the data analysis. Y.W., S.G., J.M.C., G.H.D., T.A.M. and C.J.S. contributed to data interpretation. B.L., W.Z. and X.H. designed experiments. Y.W. and S.G. wrote the article. J.M.C., G.H.D., T.A.M., and C.J.S. helped make critical revisions to the article. All authors have read and approved the final version of the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by National Natural Science Foundation of China (81873816; 82071629); Foundation of Shanghai Municipal Health Commission (202040128) and Pudong Commission of Health and Family Planning (PW2019D-13).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.