
Abstract
Immune checkpoint signaling, such as programmed cell death protein-1 (PD-1), is a key target for immunotherapy due to its role in dampening immune responses. PD-1 signaling in T cells is regulated by complex physicochemical and mechanical cues. However, how these mechanical forces are integrated with biochemical responses remains poorly understood. Our previous work demonstrated that the use of an immobilizing polyethylene glycol (PEG) linker on synthetic microgels for the presentation of a chimeric form of PD-L1, SA-PD-L1, lead to local regulatory responses capable of abrogating allograft rejection in a model of cell-based transplantation. We herein provide evidence that enhanced immune regulating function can be obtained when presentation of SA-PD-L1 is achieved through a longer more flexible PEG chain. Presentation of SA-PD-L1 through a linker of high molecular weight, and thus longer length (10 kDa, 60 nm in length), led to enhance conversion of naive T cells into T regulatory cells (Tregs) in vitro. In addition, using a subcutaneous implant model and protein tethered through three different linker sizes (6, 30, and 60 nm) to the surface of PEG hydrogels, we demonstrated that longer linkers promoted PD-1 immunomodulatory role in vivo through three main functions: (1) augmenting immune cell recruitment at the transplant site; (2) promoting the accumulation of naive Tregs expressing migratory markers; and (3) dampening CD8+ cytolytic molecule production while augmenting expression of exhaustion phenotypes locally. Notably, accumulation of Treg cells at the implant site persisted for over 30 days postimplantation, an effect not observed when protein was presented with the shorter version of the linkers (6 and 30 nm). Collectively, these studies reveal a facile approach by which PD-L1 function can be modulated through external tuning of synthetic presenting linkers.
Impact statement
Recently, there has been a growing interest in immune checkpoint molecules as potential targets for tolerance induction, including programmed cell death protein-1 (PD-1). However, how the mechanics of ligand binding to PD-1 receptor affect downstream activation signaling pathways remains unresolved. By taking advantage of the effect of polyethylene glycol chain length on molecule kinetics in an aqueous solution, we herein show that PD-L1 function can be amplified by adjusting the length of the grafting linker. Our results uncover a potential facile mechanism that can be exploited to advance the role of immune checkpoint ligands, in particular PD-L1, in tolerance induction for immunosuppression-free cell-based therapies.
Introduction
The advent of cancer immunotherapeutics, both checkpoint inhibitors and chimeric antigen receptors, and their unprecedented success in the clinic have resulted in increased interest in targeting T lymphocytes (T cells) for modulation to treat various immune-based disorders.1–4 T cells mature in the thymus and patrol lymphoid organs for the presence of specific agonistic peptides presented by major histocompatibility complexes (pMHCs). 5 The binding of specialized receptors on the surface of these cells, known as T cell receptors (TCRs), to pMHCs presented by antigen-presenting cells (APCs) triggers a signaling cascade that results in T cell activation, expansion, and acquisition of effector function for a productive immune response. 6 These responses are typically driven by the cytokine environment post-TCR engagement, dictating differentiation and T cell responses at many stages of the immune response.
Indeed, T cells are constantly being stimulated by the biochemical environment of the tissues they reside in, and the role of these biochemical cues in regulating downstream T cell triggering events has been extensively investigated.7–10 For example, interleukin-2 (IL-2) plays a vital role in T cell clonal expansion after TCR ligation, while interleukin-15 (IL-15) and interleukin-17 (IL-17) lead to memory T cell generation. 11 But biochemical cues are not the only mechanism involved in T cell activation; T cells continuously receive and respond to physical stimuli found in their microenvironment through their surface receptors, cytoskeleton, and subcellular structures. 12 These receptor–ligand binding events are dynamically regulated through shear stress, contact tension, and substrate rigidity which can influence T cell ligand recognition, as well as regulate the mobility, migration, differentiation, and infiltration to target tissues. Yet, how these mechanical forces can encode information to achieve the exquisite speed and sensitivity of T cell triggering remains unclear.
T cells have the ability to detect a small, low affinity number of antigenic pMHCs in the presence of considerable “noise” (abundant self pMHCs) in the microenvironment. 12 T cells are capable of discriminating small differences in TCR-ligand binding through multiple mechanisms, such as force, receptor abundance, and co-receptor binding. Mechanical force, in particular, can critically regulate the process of T cell sensitivity and recognition of antigens by modulating the lifetime of ligand–receptor complex interactions, a process known as kinetic thresholding.12,13
The lifetime of the ligand–receptor complex interaction has been estimated to be in the transient range of 1–100 s.13,14 However, they are thought to be compensated by signal persistence through ligand rebinding (known as the “sum of binding” model) allowing T cells to integrate information from multiple rebinding events to make rapid and reliable fate decisions. 14 Many factors can modulate the lifetime of these ligand–receptor bonds, including ligand affinity, molecular concentration of ligand and/or receptor, dissociation rate constant, receptor/ligand mobility, and co-receptor expression. Thus, T cell triggering, which can attenuate or boost the differentiation and expansion of T cell responses, is a dynamic process encompassing many variables.
The checkpoint inhibitor pathway involving the programmed cell death protein-1 (PD-1), a negative co-receptor, has taken a prominent role in immunotherapies aiming to balance reactive T cell targeting and tolerogenic regulatory T cell (Tregs) responses. PD-1 is expressed on activated T cells and elicits inhibitory signals once ligated by either of its known ligands PD-L1 or PD-L2.
Evaluation of the molecular mechanisms of interaction between PD-1 and its ligands suggests that its potency in regulating its inhibitory signal on T cells might be dependent on ligand–receptor binding kinetics and not ligand affinity. 9 Indeed, both ligands have been shown to bind to the receptor with comparable affinities, but with striking differences in the level of association and disassociation kinetics. 15 In contrast to PD-L2, PD-L1 has a rapid association with a complex and rapid disassociation phase.15,16 Investigation of the molecular mechanisms of receptor binding has shown that this higher disassociation kinetics allows the ligand to rapidly detach and rebind to the same receptor or competitively bind to additional PD-1 sites, thus differentially affecting the signaling strength perceived by engaging T cells. 17 Yet, it remains unclear if further modulation of PD-L1 binding events through synthetic means can be used to augment PD-1 regulatory function on T cells.
Attachment of polyethylene glycol (PEG) linkers to proteins and peptides, a functionalization known as PEGylation, is a widely used approach in regenerative medicine applications. 18 This process has been shown to increase protein half-life, reduced immunogenicity, and improved solubility, antifouling effect, and stability of the proteins both in vitro and in vivo.19,20 Nonetheless, its effect on association and disassociation constants of protein therapeutics and its downstream effects has been less explored.
In an aqueous environment, PEG molecules do not aggregate but transforms from its gauche configuration to trans conformation due to forces applied by the solvent. Water molecules interact with the repeating ethylene units creating double hydrogen bonds that act as bridges.19,21,22 These bonds transition PEG molecules from its normal entropic spring which is found at low stretching forces to an energetic elastic spring when subjected to higher forces. 22 In addition, a decrease in the packing between the PEG chains, due to the hydrogen bond bridges, can make the PEG linkers more flexible as demonstrated by rheology of elastic PEG gels changing to a fluid-like material when longer chains are used. 23 Thus, by altering the physical properties, such as molecular weight, of PEG chain molecules we can enhance chain flexibility controlling PEG macromolecules bending and deformation in an aqueous environment.
Leveraging the knowledge of the coiling nature of PEG molecules, and its effect on mobility, we examined its ability to affect presentation of PD-L1 both in vitro and in vivo. By varying the molecular weight of a grafting PEG linker, we aimed to promote interactions between the PD-L1 molecule and PD-1 receptor on T cells to increase efficiency of PD-1 immunomodulatory function. In this study, we demonstrated that by increasing the length of a Biotin-PEG-SH linker presenting a chimeric form of PD-1 ligand, SA-PD-L1, on the surface of microgels we can modulate regulatory immune responses in the local cutaneous microenvironment.
Materials and Methods
Microfluidic device fabrication
Polydimethylsiloxane (PDMS) microfluidic devices were made as previously reported. 24 Briefly, PDMS was prepared at a 1:10 curing agent to silicone ratio (Sylgard 184 Silicone Elastomer Kit, Dow) and cast onto epoxy resin molds containing the microfluidic device patterns. The molds were then placed into an oven at 110°C for 1 h. The resulting PDMS Microfluidic devices were then cut and bonded on to glass slides through plasma oxygen bonding. 25
Microgel generation
Crosslinked hydrogel networks were formed through an aqueous phase dispersed in oil using a flow focusing nozzle of 200 μm as previously reported. 26 The aqueous phase consisted of a 5% w/v four armed PEG macromer with maleimide groups at each terminus (four-arm PEG-Mal, 20 kDa, Laysan Bio) that was reacted with Biotin-PEG-Thiol (Biotin-PEG-SH) of different molecular weights (1, 5, and 10 kDa; Nanocs; Catalog No. PG2-BNTH-1k, PG2-BNTH-5k, PG2-BNTH-10k, respectively) providing a biotin functional group. The flow focusing oil phase consisted of mineral oil (Sigma Aldrich) containing 2% SPAN80 (Catalog No. S6760-250ML; Sigma Aldrich). For crosslinking a 15 mM concentration emulsion was generated consisting of dithiothreitol (DTT; Catalog No. 15508-013; Invitrogen). For particle with 0 kDa biotin functional arms, 5% w/v PEG-4 Mal (20 kDa, Laysan Bio) was dispersed through the microfluidics device without previously reacting with Biotin-PEG-Thiol.
After fabrication, microgels were extracted from the oil phase through centrifugation at 1200 rcg and washed five times using a 0.2% bovine serum albumin (BSA; Catalog No. A9418; Sigma Aldrich) in phosphate-buffered saline (PBS; Catalog No. 21-040-CV; Corning) solution. All microgels included in cell culture or animal experiments were washed using 0.22 μm sterile filtered 0.2% BSA solution, subsequently washed in sterile PBS, and kept sterile for the duration of the experiment.
Sizing of microgels
Microgels were sized based on diameter (micrometer) for all conditions both on-chip while the four-arm PEG-Mal was being dispersed through the chip and off chip following the washing protocol. To complete on-chip microgel sizing, images were taken in the crosslinking bath (10 × , EVOS) at 10-min time intervals. For off-chip crosslinking, four 10 μL samples of the microgels in a 10 mL suspension were plated on 2 mm gridded petri dishes (Catalog No. 174926; Fisher) where four representative images of each sample were acquired (4 × , EVOS) for sizing. The images were then imported into ImageJ, and the diameters were measured using the measure analysis function.
Staining and imaging of microgels
For imaging and staining, 1000 microgels were transferred into 1.5 mL Microcentrifuge tubes in 100μL of PBS. When staining for biotin functional arms the samples were dyed using a Streptavidin Alexa Flour 488 Conjugate (Catalog No.S32354; Invitrogen) for 1 h on a tube rotator. For imaging PD-L1 conjugated microgels 1 μg of a streptavidin chimera PD-L1 protein (SA-PD-L1, custom made as previously reported 27 ) was added to the particles for conjugation and incubated for an hour on a tube rotator. Following the incubation, the microgels were spun down at 2000 rcf, and the supernatant was collected for later protein quantification. The microgels were washed once with PBS before staining commenced to remove any residual protein. Microgels grafted with surface tethered SA-PD-L1 were then stained with Anti-Streptavidin DyLight 488 (Catalog No. SP488; Vector Labs) and set to incubate for 1 h. Following staining, microgels were imaged using fluorescent microscopy (25 × , Thunder Leica) to validate presence of functional arm and protein conjugation.
Western blotting
To validate and quantify protein conjugation, free protein and supernatants gathered from protein conjugation experiments were run in a Western blot in a mini blot module following manufacturer's instructions (Catalog No. NW04120BOX). Protein was denatured by incubating at 70°C before running the blot. Immunoblotting was performed using a rabbit anti-PD-L1 Polyclonal antibody (Catalog No. PA5-20343) at 0.5 μg/mL and goat anti-rabbit IgG (H + L) horseradish peroxidase (1:2000 dilution; Catalog No. 65-6120). For imaging a Chemiluminescence Kit was used (Catalog No. 34577; ThermoFisher Scientific), and images were acquired using a G-Box (Syngene). Efficiency of conjugation was calculated using ImageJ, measuring band intensity after background subtraction and calculating ratio from a control protein sample.
Microgel rheology
Formulated microgels were tested using a Microsquisher (CellScale) with a parallel 0.5 mm platen (CellScale) attached to a 0.5588 mm diameter beam with a modulus of 411,000 MPa (Catalog No. W22X2.5; CellScale). The microgels were tested in a bath of 1 × PBS (Catalog No. 21-040-CV; Corning) and were placed under ramp compression testing to 55% strain for 40 s. The data acquired from the testing were analyzed according to CellScale's Compression Testing of soft materials at 10% nominal compression. 28
Maleimide plate functionalization
Pierce Maleimide Activated 96-well plates (Catalog No. 15152; ThermoFisher Scientific) were functionalized with Biotin-PEG-SH arms for cell culture-based assays to evaluate the effects of PD-L1 on isolated naive T cells in vitro. The plates were washed with a wash buffer consisting of 0.1 M sodium phosphate (Catalog No. S5011-100G; Sigma Aldrich), 0.15 M Sodium Chloride (Catalog No. S7653-5KG; Sigma Aldrich), and Tween 20 (Catalog No. P1379; Millipore Sigma) and prepared according to the manufacturer's instructions with some modifications (ThermoFisher Scientific, 2012). Briefly, a 4 mM Biotin-PEG-SH solution was made for the three various molecular weight functional arms (1, 5, and 10 kDa) using a 0.1 M Sodium Phosphate (Catalog No. S5011-100G; Sigma Aldrich), 0.15 M Sodium Chloride (Catalog No. S7653-5KG; Sigma Aldrich), and 10 mM ethylenediaminetetraacetic acid (EDTA; Catalog No. J15694-AP; ThermoFisher Scientific). Solutions were added to the well plate and left to incubate at 4°C overnight.
Following functional arm incubation, the plates were washed again with wash buffer, and streptavidin or SA-PD-L1 was applied to the wells at a concentration of 5 or 10 μg/mL and incubated at 37°C for 1 h. Wells were washed one more time before cells were plated for conversion assay or for imaging. A subset of wells was stained with Anti-Streptavidin DyLight 488 (Catalog No. SP488; Vector Labs) for 1 h, after which they were washed with PBS (21-040-CV; Corning), and 100 μL of PBS was added to each well. Fluorescence staining was validated using a spectrophotometer (Biotek SynergyH1), as well as standard microscopy (10 × , Leica Thunder).
Tregs conversion assay
Spleens were taken from C57BL/6J mice for naive CD4 T cell isolation using the MojoSort Naive CD4 Isolation Kit (Catalog No. 480041; BioLegend). The mice were euthanized according to University of Michigan Institutional Animal Care & Use Committee (IACUC) protocols (IACUC approved protocol PRO00011011), and the spleens were removed through axillary thoracotomy. The spleens were processed, and the tissue and cells were placed through a 0.4 μm cell strainer. Following splenocyte isolation, cells were further enriched, and naive CD4+ T cells were isolated according to manufacturer's instructions. Following isolation, the naive CD4+ T cells were placed in T cell media composed of 95% Roswell Park Memorial Institute (RPMI) (Catalog No. 10-040-CV; Corning), 5% fetal bovine serum (SH30071.03; Cytiva), 100 U/mL Penicillin Streptomycin (Catalog No. 15140-148; Gibco), 10 mM HEPES (25-060-Cl; Corning), 50 mM β-mercaptoethanol (21985-023; Gibco), 1 mM Sodium Pyruvate (11360-070; Gibco), and 1 × minimum essential medium nonessential (Catalog No. 11140-050; Gibco). Cells were seeded into the plate at 150,000 cells/well.
For T cell activation CD3/CD28 Mouse T-Activator beads (Catalog No. 11456D; Gibco) were used according to the product protocol and modified to be added at a concentration of 5 cells:1 bead to each well. 29 Control plates were made without the addition of biotin functional arms and plated accordingly on nontissue culture treated 96-well plates. T cells were harvested after 3 days and washed once with 1 × PBS. The cells were then stained for live/dead using a 1:100 dilution of 1 × PBS and Zombie Violet (Catalog No. B357086; BioLegend) and surface stained for CD4 (APC; Catalog No. 100412; BioLegend), CD25 (PE-Cyanine 7; Catalog No. 102016; BioLegend), and CD3 antibodies (PE Cyanine 5.5 or BV510; Catalog No. 100218 or 100233; BioLegend).
Following surface staining cells were then fixed and permeabilized according to True-Nuclear Transcription Factor Buffer Kit directions (Catalog No. 424401; BioLegend) and further stained for FoxP3 (PE; Catalog No. 320008; BioLegend). Samples were then run for flow cytometry using a Northern Lights Aurora (Cytek) and analyzed using the software Flow Cytometry Standard (FCS) express. Single stain and CD25 and FoxP3 fluorescence minus one (FMO) controls were run with each sample.
Subcutaneous microgel delivery
All animal procedures were performed under approved protocols by the University of Michigan IACUC in accordance with the National Institutes of Health guidelines (IACUC approved protocol PRO00011011). Microgels were injected subcutaneously into 6–8-week-old C57BL/6J mice (n = 6). The 100μL injections consisted of 3000 microgels previously conjugated with 3 μg of SA-PD-L1, and microgels without SA-PD-L1 were used as negative controls. All four microgel conditions were injected into a single animal at separate sites to reduce biological variability across animals.
In vitro PD-L1 protein tracking
In vitro PD-L1 signal tracking was conducted as previously described. 26 Briefly, SA-PD-L1 protein was labeled with an Alexa Flour 750 NHS ester (Catalog No. A20011; Fisher Scientific) for tracking. Labeled SA-PD-L1 (3 μg) was conjugated to the 3000 microgels for an hour before implantation in 100 μL of PBS. The microgels were injected subcutaneously as described above, and signal intensity and distribution were observed longitudinally using an in vivo imaging system (IVIS) SpectrumCT system (Perkin-Elmer). The data were analyzed using Living Image software where regions of interest (ROIs) were drawn in defined areas with microgel implants and quantified using Radiant Efficiency (p/s/sr)/(μW/cm2). These ROIs were always kept the same for each animal, and the relative implant condition was sized to represent the fluorescent signal of the respective area. This allowed for imaging data from each animal to be compared between time points. The intensity measurements were further normalized to the values found at day 0 to evaluate signal decay over time.
Multiparametric flow analysis of tissue responses
Skin tissue samples were obtained from animals with subcutaneous microgel implants on days 3, 7, and 30. Samples were taken using a 12 mm biopsy punch and digested for 1 h at 37°C in a shaking incubator. Tissues were digested in a solution composed of RPMI 1640 (Catalog No. 10-040-CV; Corning), 2.5 U/mL Dispase II (Catalog No. 4942078001; Roche), and 0.2% w/v Collagenase Type II (Catalog No. LS004176; Worthington). Spleen and lymph node tissues were dissected and then mechanically digested. The tissue was passed through a 35 μm strainer, washed with PBS one time, and red blood cells were lysed accordingly using 1 × red blood cell (RBC) lysis buffer (Catalog No. 420302; BioLegend) for 15 min on ice. Cells were washed with fluorescence activated cell sorting buffer composed of HBSS 1 × (Catalog No. 21-022-CM; Corning), 0.5% BSA (A9418-500G; Sigma Aldrich), 2 mM EDTA (Catalog No. J15694-AP; ThermoFisher Scientific) to terminate the RBC lysis and then further washed with 1 × PBS before surface staining.
Cells were stained with the myeloid antibodies described in Table 1 and lymphocyte antibodies described in Table 2. Flow cytometry was performed using a Northern Lights Aurora (Cytek). Single stain and FMO controls (Tables 1 and 2) were run with each timepoint condition. Data were analyzed using the software FCS express.
Myeloid Panel Dyes
Antibodies for Myeloid cell population markers for subcutaneous implant takedown and flow cytometry analysis.
Designates a marker a fluorescence minus one control was made.
APC, Allophycocyanin; BV, brilliant violet; CD, cluster of differentiation; FITC, fluorescein isothiocyanate; PE, phycoerythrin; PerCP, peridinin-chlorophyll-protein.
Lymphoid Panel Dyes
Antibodies for Lymphoid cell population markers for subcutaneous implant takedown and flow cytometry analysis.
Designates a marker a fluorescence minus one control was made.
Immunohistochemistry
After euthanasia on day 14, a skin biopsy was taken from each microgel implantation condition in the animals in the area surrounding the injection site and placed in 10% formalin overnight. Samples were paraffin embedded and subsequently processed through 7 μm sectioning using a microtome (Leica). The samples were then dehydrated in graded ethanol solutions, cleared using xylenes, and hematoxylin and eosin (H&E) staining was completed. For immunohistochemistry, sections were costained with a monoclonal rabbit anti-CD3 (790-4341; Roche) and anti-FoxP3 (ab20034; Abcam) followed by Ventana discovery secondaries in Cy5 (CD3) and Opal 620 (FoxP3). Slides were scanned at 20 × using the University of Michigan In-Vivo Animal Core.
Statistical analysis
All experiments were performed with a minimum of three biological replicates. Sample size for each experimental group and the statistical test are reported in the respective figure legends. p-values are reported in the results section and figure legends of respective experiments. Statistical analysis was conducted using GraphPad Prism 10.0.1. For groups with three or more samples a one-way analysis of variance was performed with a Tukey's multiple comparison test or Dunnett's multiple comparison test.
Results
Physical and mechanical properties of microgels are not altered by linker length functionalization
Previously, we had demonstrated that PEG hydrogels rendered into microspheres, termed microgels, could be effectively implemented for surface tethering and sustained presentation of proteins in vivo.30,31 Herein, we implemented a similar platform to investigate the effect of grafting polymer length on protein flexibility and bioactivity. As previously reported, a Biotin-PEG-SH linker bound to a four-armed PEG-Mal macromer (four-arm PEG-Mal) was implemented for surface capturing of a streptavidin chimera PD-L1 protein (SA-PD-L1). 30
To evaluate the effects of linker length on protein mobility and ligand immunoregulatory function, three molecular weights of a linear PEG linker were tested (1, 5, and 10 kDa). Differences in molecular weight of the PEG chains result in polymer “arms” of 6, 30, and 60 nm, respectively. 32 Following biotinylation, four-arm PEG-Mal macromer was rendered into droplets by dispersing aqueous phase through a microfluidic device and crosslinking using a nondegradable small dithiol linker (DTT) as previously described. 26 As a control, four-arm PEG Mal without biotinylated PEG linkers was used to generate microgels (0 kDa).
To determine the effects of linker addition on crosslinking and droplet formation, microgels were sized both on-chip (Fig. 1A, Supplementary Fig. S1A during droplet generation) and off-chip (Fig. 1B, Supplementary Fig. S1B after swelling occurs). On-chip sizing revealed no difference across all groups tested; 0 kDa: 122.3 ± 17.98 μm, 1 kDa: 122.2 ± 6.244 μm, 5 kDa: 127.5 ± 14.05 μm, and 10 kDa: 132.1 ± 24.15 μm. Furthermore, the incorporation of a biotin linker to the backbone of the four-arm PEG-Mal macromer had no significant effect in crosslinking, as sizing postswelling in an aqueous solution demonstrated no differences in microgel diameter compared to unfunctionalized macromer control (0 kDa: 174.0 ± 20.93 μm, 1 kDa: 175.3 ± 5.909 μm, 5 kDa: 175.5 ± 10.79 μm, 10 kDa: 178.3 ± 6.50 μm; Fig. 1B, C).
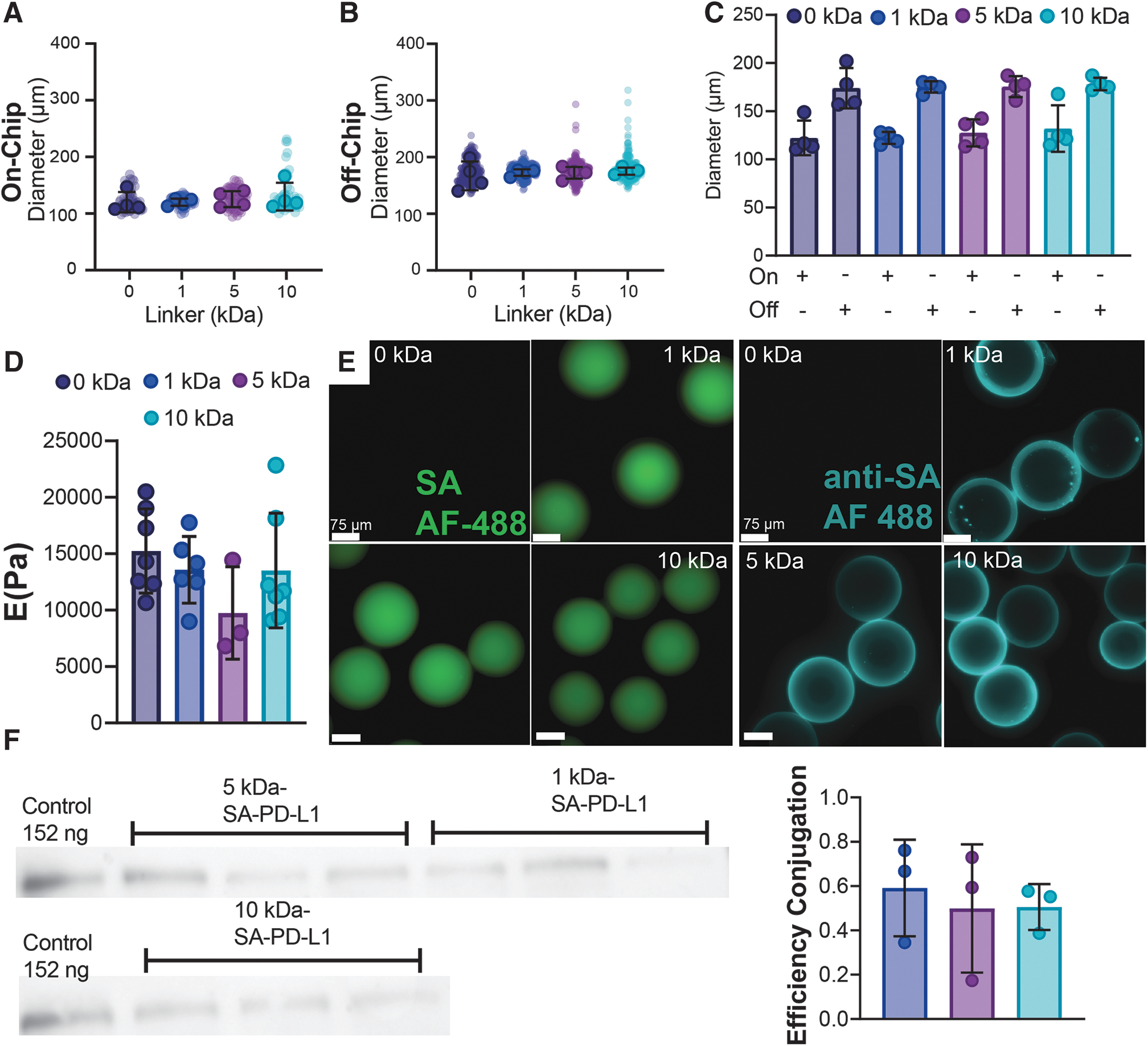
Microfluidic generated microgels with varying length linkers for surface tethering allow controllable capture of SA-modified PD-L1 protein.
To further validate the effect of linker addition on microgel mechanical properties, swollen samples underwent a compression test to 55% strain using a CellScale MicroSquisher. Stiffness measurement at 10% nominal compression (Supplementary Fig. S2) 28 revealed no differences compared to controls, with Young's modulus averaging (13018.339 ± 2324.42 Pa) for samples tested (Fig. 1D; 0 kDa: 15240.286 ± 3726.70 Pa, 1 kDa: 13573.833 ± 2957.47 Pa, 5 kDa: 9744.667 ± 4097.65 Pa, and 10 kDa: 13514.571 ± 5088.58 Pa, p = 0.8820, p = 0.2403, p = 0.8573 vs. 0 kDa control, respectively). Overall, no differences were found among the different PEG linkers grafted demonstrating that PEG linker length does not have an effect on the physical or mechanical properties of the microgel carrier platform.
Microgels efficiently capture SA-modified PD-L1 protein despite variance in linker length
To evaluate if PEG linker length had any effect on protein capturing and presentation, microgels were initially stained with a streptavidin based Alexa Fluor 488 dye to visualize biotin presentation. Fluorescent imaging demonstrated that microgels conjugated with Biotin-PEG-SH had homogeneous biotin presentation despite differences in linker length (Fig. 1E, SA-AF488 dye green, scale 75 μm). To evaluate the effects on protein capturing, microgels were conjugated with 1 μg of SA-PD-L1 protein and then stained with an anti-streptavidin DyLight 488 dye. As expected, no conjugation of PD-L1 protein was present in control condition as it lacked any biotin for surface capturing (Fig. 1E, 0 kDa anti-SA aqua, 75 μm). In contrast, all three conditions with biotin functionalization (1, 5, and 10 kDa) show homogenous fluorescence confirming presence of bound SA-modified PD-L1 protein on the surface (Fig. 1E).
To further validate and quantify the presence of bound SA-PD-L1 protein onto the surface of the microgels, supernatants from protein conjugation were run through a Western blot (Fig. 1F). Quantification of the immunoblot revealed a conjugation efficiency averaging (0.531 ± 0.051) for all conditions tested. Importantly, no differences in conjugation efficiency were observed across different linker lengths demonstrating that length of PEG chain polymer does not affect protein capturing efficacy (Fig. 1F; 1 kDa vs. 5 kDa p = 0.8645, 1 kDa vs. 10 kDa p = 0.8819, and 5 kDa vs. 10 kDa p = 0.9992).
Length of PEG chain linker boost PD-L1 capacity to promote T regulatory conversion in vitro
PD-L1 is known to be a key contributing signaling molecule to induce Treg development.33–35 Thus, we set to investigate whether PD-L1 presentation through PEG linkers of different mobility could influence the conversion of naive CD4+ T cell population (defined as CD4+CD25-) into Tregs (defined as CD4+CD25+FoxP3+) in vitro. After grafting Biotin-PEG-SH chains of length 6, 30, and 60 nm to a Maleimide conjugated plate, 2 μg of SA-PD-L1 or wild-type streptavidin (SA, as a protein control) was then bound to the functional group on the PEG chain for presentation (Fig. 2A, Step 1). Cells supplemented with transforming growth factor (TGF)-β only were used as a positive control, as TGF-β has been shown to be a necessary cytokine for Treg generation. 33 Freshly isolated naive CD4+ T cells were plated and cultured for 3 days before being harvested, stained, and analyzed using flow cytometry (Fig. 2A, Step 2).
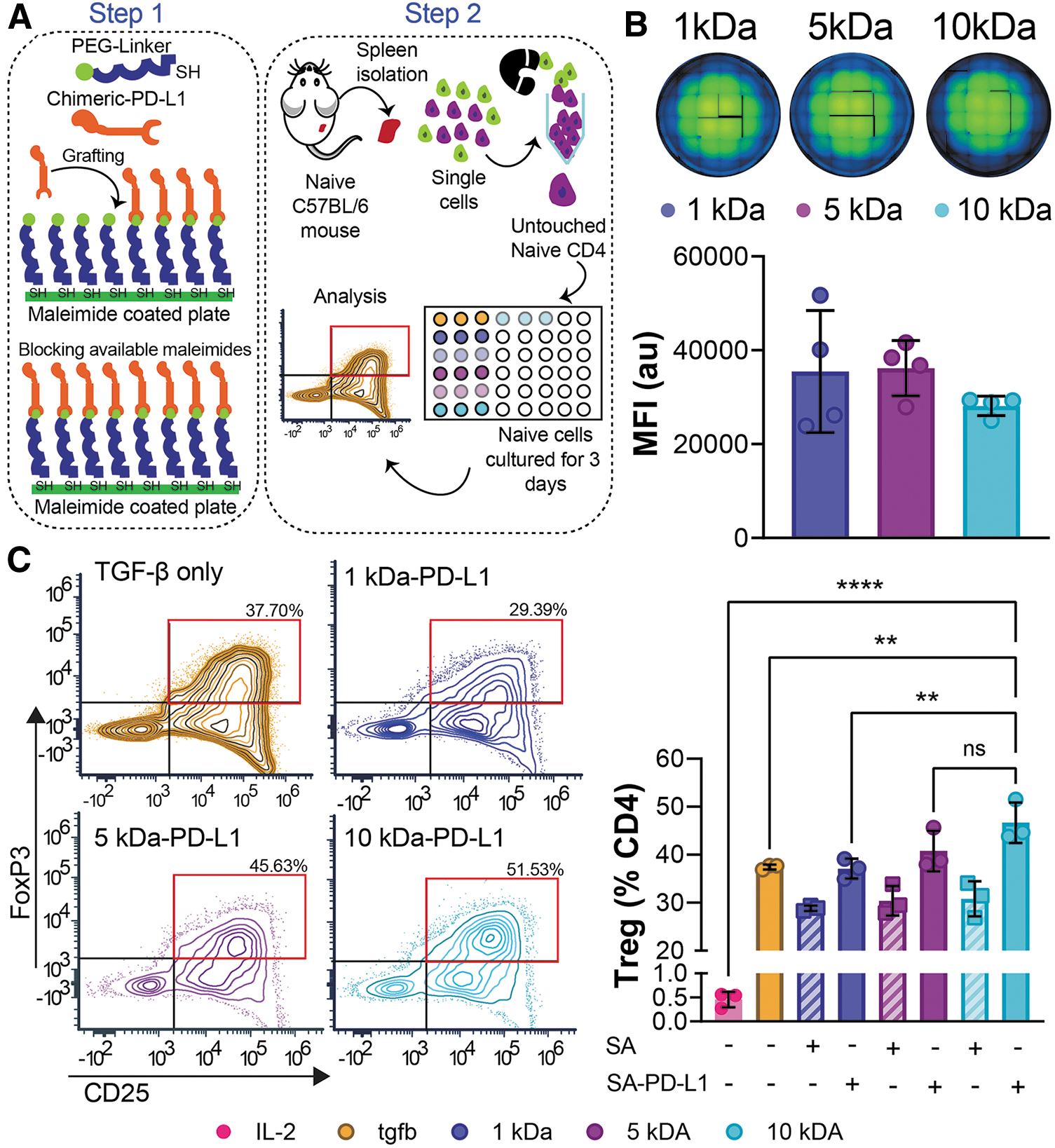
In vitro presentation of SA-PD-L1 on higher molecular weight PEG linkers boost naive CD4+ T cell conversion into Tregs.
To confirm that PEG chains could be grafted to the plates at similar densities, additional wells were stained with a streptavidin Alexa Fluor 488 dye, and fluorescent intensity was quantified through both imaging and spectrophotometer measurements (Fig. 2B). Mean fluorescent intensity ranged from 28,131 to 36,185 across all groups with no statistical difference observed across conditions (1 kDa vs. 5 kDa p = 0.9919, 1 kDa vs. 10 kDa p = 0.4575, and 5 kDa vs. 10 kDa p = 0.3962). This confirms that the presence and distribution of PEG chains in the wells were similar across all conditions.
As expected, coculture of naive CD4+ T cells in the presence of IL-2 alone did not lead to any in vitro conversion (Fig. 2C, 0.5%, pink bar), while the addition of TGF-β resulted in the induction of naive T cells into Foxp3+ Tregs (Fig. 2C, 38.4%, yellow-orange) at similar levels to what has been reported previously. 33 Presentation of immobilized SA in the culture led to a lower percentage of Tregs in comparison to soluble TGF-β condition (1 kDa-SA p = 0.0312). This is consistent with our previous results where SA at an equimolar concentration to PD-L1 led to lower induction of regulatory T cells in vitro, which we attribute to nonspecific inhibition by the SA protein. 27 In contrast, when naive cells were cocultured with surface bound PD-L1 through PEG linkers, and at the same range of soluble TGF-β concentration, similar levels of Treg conversion were observed in the group displaying PD-L1 protein at the shorter linker lengths (1 kDa PD-L1 vs. TGF-β, 37.107 ± 2.099 and 37.437 ± 0.508, respectively, p > 0.99).
Notably, the presentation of PD-L1 through a longer more flexible linker (10 kDa) led to a greater number of FoxP3+ Tregs in culture compared to soluble TGF-β alone and a similar amount of protein presented through a shorter linker (p = 0.0075 vs. TGF- β, and p = 0.0057 vs. 1 kDa, respectively). This suggests a role for polymer length in PD-L1 presentation and regulation of Treg induction in vitro.
In vivo protein clearance is not affected by length of tethering linker
To evaluate if linker length affects the kinetics of protein clearance in vivo, a near-infrared fluorescent dye (Alexa Fluor 750 NHS ester) was conjugated to SA-PD-L1 protein for in vivo tracking as previously reported. 30 Microgels containing labeled protein were subcutaneously injected into the back of C57BL/6J mice (3000 microgels per condition). Each animal received all conditions: 1, 5, and 10 kDa into three separate subcutaneous locations to remove biological variance between animals (n = 6, Fig. 3).

SA-PD-L1 is cleared from the site of implantation at a similar rate despite linker length.
Tracking of signal intensity over time demonstrated a decrease in signal of 55.9 ± 9.3%, 58.2 ± 7.7%, and 71.2 ± 7.7%, respectively, by 24 h postimplantation (Fig. 3C). By day 3, signal in the 10 kDa group was reduced by 85%, a significant reduction compared to the other two linkers evaluated (Fig. 3D, 1 kDa vs. 10 kDa p = 0.0006, and 5 kDa vs. 10 kDa p = 0.0028). Yet by day 7, signal had decayed to similar background levels in all groups tested (Fig. 3E). Protein half-life, as estimated through an exponential decay curve fit, was 1 kDa- 0.945 ± 0.372, 5 kDa- 1.082 ± 0.923, and 10 kDa- 0.530 ± 0.156 for all conditions tested. No statistical difference between linker lengths was observed (1 kDa vs. 5 kDa- p = 0.9800, 1 kDa vs. 10 kDa- p = 0.1054, 5 kDa vs. 10 kDa- p = 0.4567). Thus, presentation of proteins through differential PEG chain lengths does not lead to exacerbated protein clearance rates in the subcutaneous space.
PD-L1 presentation through a longer linker boost regulatory T cell response in vivo
To assess whether the effects seen in our in vitro assay for Treg generation could be replicated in an in vivo setting, we injected naive C57BL/6J mice (n = 4) with microgels used to immobilize SA-PD-L1 protein through different PEG linkers (1, 5, and 10 kDa). Microgels not presenting any protein (0 kDa) were used as controls to determine immunological response to the material alone. Skin biopsies were taken for each condition on predetermined time points (3, 7, and 30 days) to assess acute and long-term effects of protein delivery and material residence time (Supplementary Fig. S3–S6).
Presentation of a set amount of SA-PD-L1 protein through a flexible/longer linker (10 kDa) led to increase in presence of Treg cells at the implant site, to extents greater than protein presented through an intermediate linker (Fig. 4A, 5 kDa, five-fold greater, p = 0.0152) or through a short linker (Fig. 4A, 1 kDa, 15-fold greater, p = 0.0098) after 7 days postimplantation. Notably, this trend was sustained for over 30 days (Fig. 4A, inset right; 10 vs. 5 kDa 6-fold difference, p = 0.0167; 1 vs. 10 kDa 7-fold difference, p = 0.0130), weeks after protein had been cleared from the injection site as determined by the IVIS imaging.
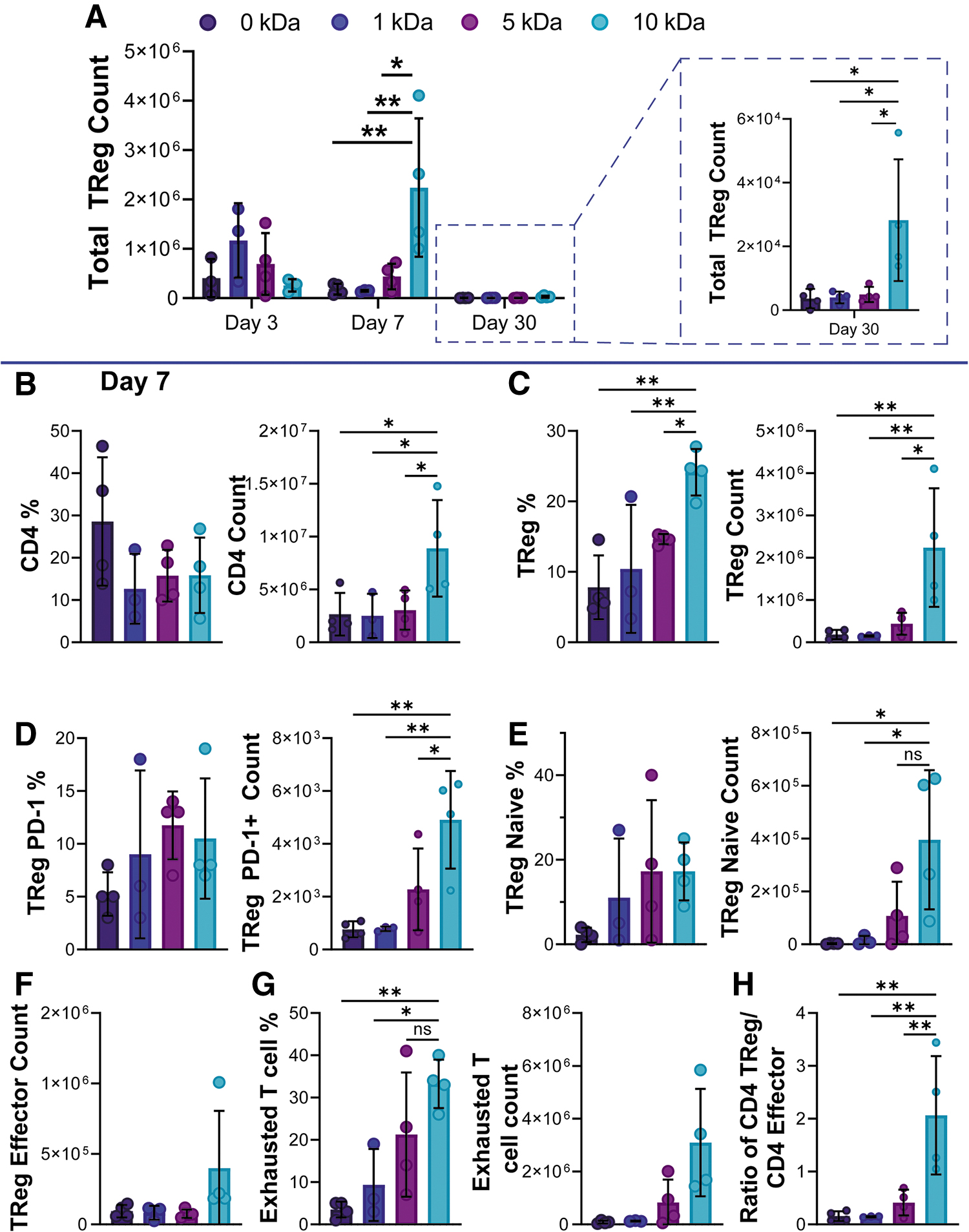
Longer PEG linker enhances T regulatory presence at subcutaneous implant site.
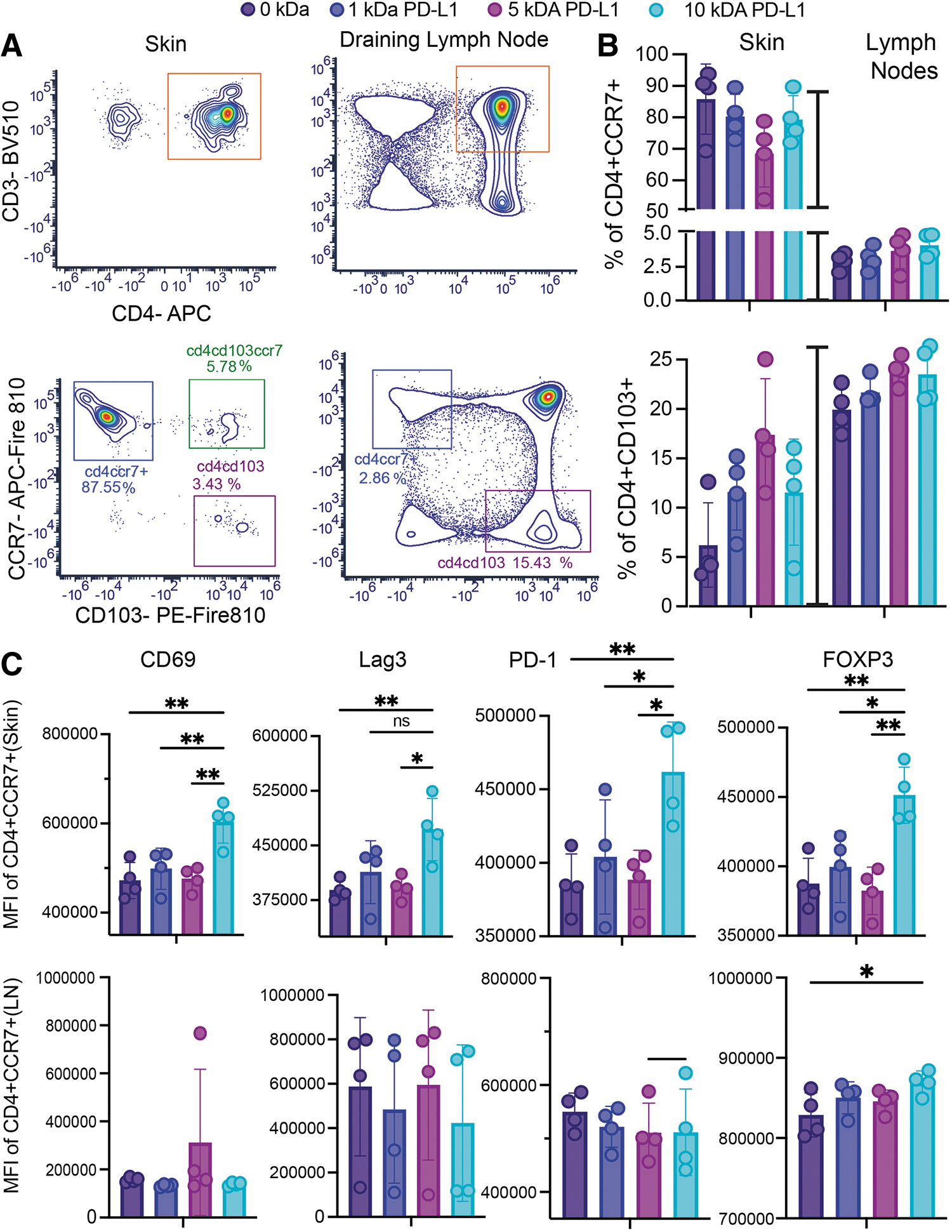
Infiltrating cells migrate to the implant site in a CCR7-dependent manner.
In-depth analysis of CD4+ T cell compartment at day 7, just as protein was being cleared from the site, revealed significant differences in the total CD4+ T cell population only when protein was presented through a longer linker (Fig. 4B). On day 7, over 20% of the CD4+ T cells present at the injection site were Tregs in the 10 kDa group, a significant increase compared to controls and the shorter linker versions tested (Fig. 4C).
PD-1 expression on Tregs has been associated with metabolic changes such as enhanced phosphorylation of FoxO-1, which has been linked to the migratory history of the parent CD4+ cell.36,37 Evaluating the expression of PD-1 on the Tregs present in the subcutaneous implant site demonstrated an elevated expression of this marker only when the ligand was presented through a longer flexible linker (Fig. 4D, 10 kDa vs. 0 kDa p = 0.0024, 10 kDa vs. 1 kDa p = 0.0090, 10 kDa vs. 5 kDa p = 0.0232). Likewise, a higher proportion of the induced Tregs in the PEG 10 kDa group express the CD62L marker vs CD44 (Fig. 4E, CD62Lhigh CD44low, Fig. 4F CD62Llow CD44high) suggesting a higher capacity to migrate and accumulate in peripheral sites. 38
Evaluation of the frequency and total cell count of CD4+ T cells with surface expression of exhaustion marker PD-1 demonstrated significantly higher frequencies of an exhausted phenotype (defined as CD4+CD25+PD-1+) when protein is presented through the longer PEG 10 kDa linker compared to intermediate and shorter lengths, as well as when no protein was delivered (Fig. 4G, 10 kDa vs. 0 kDa p = 0.0094, 10 kDa vs. 1 kDa p = 0.0163, 10 kDa vs. 5 kDa p = 0.0457). This effect peaked at day 7, as no differences in the CD4+ (aside from Tregs) compartments were observed acutely (Supplementary Fig. S4, day 3; Supplementary Fig. S5 day 7), or after long implantation periods (Supplementary Fig. S6, day 30).
Finally, as Tregs have been shown to control T effector proliferation and inflammatory potential,39,40 we set to calculate the ratio of Tregs present at the injection site compared to effector CD4+ cells (defined as CD4+CD62L lowCD44high). A boost in Tregs versus effector cells was observed only when SA-PD-L1 ligand was presented in the long flexible linker version (Fig. 4H, 10 kDa vs. 0 kDa p = 0.0025, 10 kDa vs. 1 kDa p = 0.0041, 10 kDa vs. 5 kDa p = 0.0067).
To elucidate the source of the immune cells infiltrating the graft we performed standard flow cytometry, focusing on markers involved in cell migration. In skin samples and afferent lymph nodes, CD103 and CCR7 expression delineated three populations of CD4 cells, with dynamic distribution between sites. Implants were prominently infiltrated by CD4 cells expressing CCR7 receptors (Fig. 5A, B), which allow them to migrate between cutaneous and lymphoid tissues. 41 Contrariwise, the number of skin-resident cells (CD4+CD103+CCR7–) at the implant site was lower, representing on average <10% of total CD4+ cells. In addition, CD4+CCR7+ cells in the skin were affected by the ligand linker length with the longer linker promoting the expression of adhesion molecule CD69 (Fig. 5C), whose expression has been shown to scale to TCR signaling strength. 42
Finally, immunomodulatory and regulatory molecules, Lag3, PD-1, and FoxP3, were also sensitive to linker length. Similar examination of marker expression in the afferent lymph nodes revealed no differences among the different linkers tested, with cells maintaining comparable levels of expression of adhesion receptors and regulatory molecules (Fig. 5C). These results suggest that the phenotypic changes induced by SA-PD-L1 presentation are happening locally at the implant site and involve T cells that undergo CCR-7 dependent migration between the skin and draining lymph nodes. Finally, this effect was local and did not impact systemic immunity as no differences in CD4 and CD8 distributions were observed in isolated splenocytes from recipient mice (Supplementary Fig. S7). Overall, these data hint at a role for linker length in mechanically manipulating PD-L1 presentation, helping turn the response from a digital (on or off) to an analog signal (“scaled”), modulating not just frequency of cells but also the strength of response to TCR signaling.
As PD-L1 engagement with the PD-1 receptor plays an important role in inhibiting the function of CD8 cytotoxic T cells,9,43 we hypothesize that PD-L1 presentation could also dampen cytotoxic T cell responses. Consistent with our previous observations no differences occurred in the recruitment of CD8 cells to the graft at any of the time points tested (Day 3, 7, and 30; Supplementary Figs.S4–S6). Yet immune activation status, as evidenced by the expression of cytotoxic granzyme B, was dampened in the presence of SA-PD-L1 only when presented through a longer linker (Supplementary Fig. S8). This effect was localized to the graft as similar analysis in draining lymph nodes near each tissue implant demonstrated no difference in this population (Supplementary Fig. S8B).
In addition, no effects were seen in perforin expression neither at the graft nor in the draining lymph nodes (Supplementary Fig. S8C, D). These results agree with previous work which has suggested no correlation in the effects of PD-L1 immunotherapy on inhibitory molecules (TIM-3, Lag-3, and CTLA-4), as well as in secretion of cytokines IL-2, TNF-α, and IFN-γ, while only granzyme B secretory activity acting as a predictive biomarker for PD-L1 based immunotherapy. Overall, these data suggest that the increase in linker length augmented the effect of SA-PD-L1 presentation in vivo allowing a reduction of effector cells and dampening cytotoxic T cell activation in the infiltrating immune cell population.44,45
The effect of linker length and PD-L1 presentation was also evaluated on the infiltrating myeloid cell population (Supplementary Fig. S9). Despite our previous evidence demonstrating an effect on PD-L1 presentation on the myeloid compartment, under this setting no differences were observed in infiltrating CD45+CD11b+ immune cells or phenotypes.30,46 However, in this model no antigen was presented which might explain the contradictory responses, as APCs such as DCs require antigen to upregulate this pathway. 47 Overall, these data indicate that and increase in linker length did not affect infiltration and phenotypic polarization in the myeloid compartment.
Visible cell infiltration in all linker length conditions was observed following subcutaneous implantation
To confirm cell infiltration at the site of subcutaneous implantation skin biopsies surrounding the implantation site were taken from C57BL/6J mice for 0 kDa, 1, 5, 10 kDa, and skin control. Staining through H&E showcases clear visible cell infiltration at the site of microgel implantation in subcutaneous tissue, with a qualitative increase in cell infiltration in the 10 kDa implant site (Fig. 6). Dual immunofluorescent staining for CD3 and FoxP3 demonstrates the presence of regulatory cell phenotypes in the immediate area surrounding the microgels, with cell density affected by the linker size used to present the immunomodulatory protein. These results agree with our flow cytometry data which demonstrated a significant increase in immune cell infiltration as a function of linker length (Fig. 4). These data together suggest that immune infiltration by Tregs at the implant site can be modulated by the length of the grafting linker implemented for protein presentation.
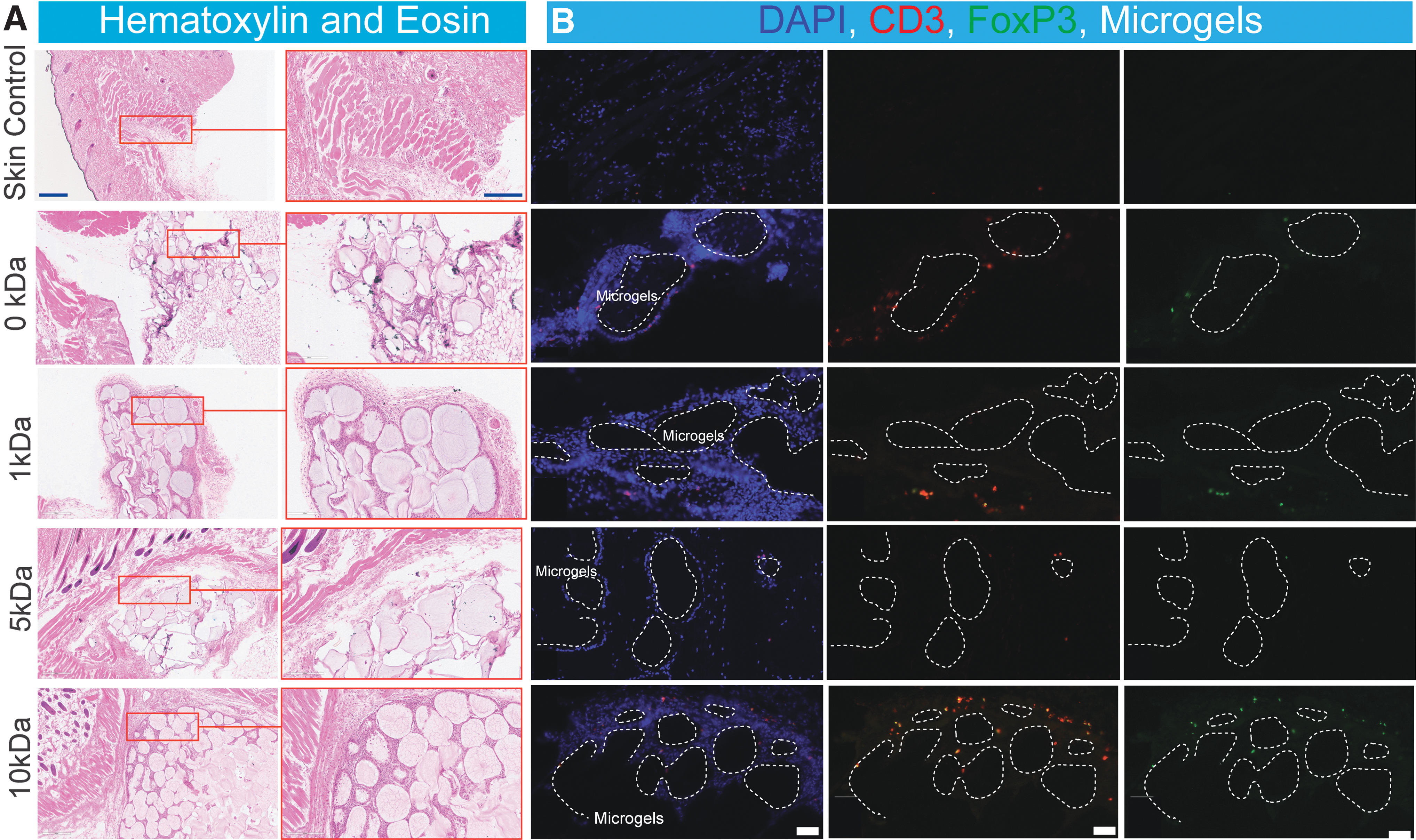
Effect of linker size on PD-L1 mediated cell infiltration and polarization following subcutaneous implantation.
Discussion
T cells are the driving force mediating transplant rejection, as well as transplant side effects, such as graft versus host disease.48,49 While modulating T cell suppression is key for optimal treatment, the complexity in immune activation makes achieving this dynamic balance a nontrivial task. Currently, clinical approaches involve the use of drugs that systemically inhibit T cell proliferation (Tacrolimus or Sirolimus) and corticosteroids (prednisone), 50 which leave recipients exposed to opportunistic infections and long-term malignancies. 51 While biological therapies targeting costimulation pathways (i.e., abatacept and belatacept which inhibit CD80/CD86, CTLA-4 targeting CD28)52,53 have been tested, overall complications from viral and fungal infection, as well as remaining toxicity issues, have limited and scrutinized their role in clinical applications. 54
Our previous studies on protein delivery for transplant tolerance have demonstrated that immobilization and presentation of PD-L1 through synthetic microgels lead to local immunomodulation sufficient for sustained allograft function. This effect has been attributed to the enhanced protein retention at the transplant site provided by the immobilization of the protein on the synthetic nondegradable microgel carrier surface.30,31,46 This localized approach is highly desirable as it can circumvent issues of dosing, systemic toxicity, and potential long-term malignancies by controlling regulatory cell localization and functions. Control of this cell phenotype does not affect systemic responses, as blood circulating cells from tolerant mice can still amount a strong response to matched antigens in ex-vivo assays. 30 Thus, providing this localized approach can aid in overcoming limited long-term therapeutic effects, while minimizing the risks of long-term immunosuppression.
Beyond their use for localization, these biomaterial platforms grant the opportunity to discriminate the effect of immunotherapies based on tissue-specific immune environments. This provides a unique advantage to tailor immunotherapy regimens, opening the door for precision medicine approaches. While the authors and others have explored multiple bioengineering platforms for the spatial localization of immunomodulatory molecules,55,56 the role of the mechanics of immunotherapeutic presentation on the recipient host cells has remained largely underexplored.
In this study, we demonstrate an important role for linker length in modulating downstream effects of PD-L1 mediated immunotherapies in the implant microenvironment. PD-L1 has been shown in the past to have a pronounced role in the development and sustenance of function of induced Tregs. 33 This role has been correlated to PD-L1 effect on FoxP3 expression, which while not acting as the master regulator of Treg cell fate it contributes essential signals that drive their development. 57
Consistent with this observation, PD-L1 presented in PEG linkers of length 6, 30, and 60 nm in vitro promoted the conversion of T conventional into Tregs to extents higher than control conditions only in the group where the ligand was presented through a longer more flexible linker (60 nm). As the amount of grafted protein was the same among all sample groups, as evaluated by imaging and spectrophotometer analysis, we suggest that this difference is attributed to an effect of the linker length on either ligand engagement or signaling. Future studies will aim to dissect if linker length has an effect on Akt-mTOR activation, 33 which has been shown to be critical for Treg conversion.
We further proceed to validate the role of the linker on immunomodulatory function of the protein in vivo by implementing a subcutaneous transplant model in which previously developed microgel technology was used as a carrier for delivery. 30 To immobilize the protein ligand on the surface, we implemented linkers of different lengths which did not affect protein capturing efficiency. Our results demonstrated a dynamic immune microenvironment which peaked at day 7 postimplantation and resulted in elevated immune infiltration in the T cell compartment only in the group with the longer flexible linker. Notably, just a 10-fold change in linker size was sufficient to boost PD-L1 role in regulating the dynamic balance between T effector and Tregs in vivo.
We showed that a longer linker could enhance the presence of Tregs at the implant site by 15-fold, with the majority of these cells expressing the coreceptor PD-1. The majority of these cells had a CCR7-dependent migratory phenotype and was modulated locally as no differences were observed in local draining lymph nodes to the graft. Previous literature has demonstrated that migrating Tregs from the periphery express and retain a higher number of PD-1 receptors than other CD4 subsets. 58 Moreover, they have determined a direct role for PD-1 in preferentially modulating migration in induced Tregs but not on non-Tregs through their interaction with lymphatic endothelial cells. 58 Thus, it remains to be determined if PD-1 expression based on linker length can aid in the preferential migration from draining lymph nodes through PD-1/PD-L1 mediated pathways.
We can attribute our observed results to the modulation of the linker used for ligand presentation, as extensive evaluation of the delivery microgel carrier physicochemical and mechanical properties demonstrated no differences in protein capturing or stiffness, all properties which could indirectly affect the polarization of the immune cell phenotype observed. 26 In addition, retention times at the implant site were comparable among linkers and similar to clearance rates observed after subcutaneous delivery of therapeutic proteins. 30
However, while not significant in terms of protein half-life, protein presentation using the 10 kDa linker was lower at day 3 postimplantation compared to the intermediate and shorter sizes implemented. This accelerated clearance could be related to interference from biotin present in plasma, 59 which through competitive unbinding of the grafted chimeric SA-PD-L1 could be modifying presentation of the ligand only in circumstances where protein is more accessible (such as when it is presented through a longer linker). This interference cannot be easily predicted or quantified; however, immunoblotting and in vivo trafficking through positron emission tomography 60 studies could help shed some light on the role of protein clearance on immune trafficking.
Notably, linker length modulated the expression of adhesion marker, CD69, which has been shown to correlate to levels of TCR signaling both at population and single cell resolution levels. 42 Future studies seek to validate if the long-term maintenance of the observed biased phenotypic distribution is through TCR signaling mediated phenotypic differentiation of CCR7-dependent migrating CD4 cells into tissue resident skin cells capable of maintaining a skewed tolerogenic immune environment.
While our study indicates that ligand length can promote ligand engagement, it is important to note that we did not evaluate changes in molecular mechanism of interaction between the ligand, PD-L1, and its receptor, PD-1. Previous studies have demonstrated that parameters such as linker length, hydrophobicity, and conjugation chemistry can affect protein presentation due to steric hindrance.61,62 Linker length is associated with increased hydrophilicity, which translates to better end exposure in an aqueous environment. Indeed, amino acid based flexible linkers which have been used to promote movement in engineered proteins are mostly made from connecting functional domains to polar amino acids (Lys and Glu) which improve functional domain solubility. 61 However, there have several drawbacks in terms of affecting protein folding and optimal biological activity. Thus, future studies should focus on investigating the effects of linker length on PD-1-PD-L1 interactions and its potential for implementation in the treatment of autoimmune diseases and tolerance induction.
Conclusion
In conclusion, these studies show preliminary evidence of the use of linker mobility on enhancing the presentation of PD-L1 for dampening effector T cell response and promoting local immunoregulatory phenotypes. Through both in vitro and in vivo studies, we demonstrated that increased length of immobilized linker correlated to an increase in regulatory and exhausted phenotypes at the implant site. This approach provides a promising strategy to harness the therapeutic potential of PD-L1 expression for tolerance induction that can be highly translational without exacerbating immunogenicity or attenuating protein bioactivity.
Footnotes
Acknowledgments
The authors thank the University of Michigan In Vivo Animal Core for imaging and histology resources. The authors thank the University of Michigan Pathology Department Molecular Pathology Research Laboratory for immunofluorescence staining and imaging resources. The authors also thank the Biointerfaces core for training and access to equipment for rheological evaluation. The authors thank Robert Heizelman for his assistance in animal handling during imaging.
Authors' Contributions
N.M.R. synthesized and characterized SA-PD-L1 presenting microgels, performed in vitro assays, transplants, and the collection of all data for article. A.D. performed rheological assessments and immunoblotting. K.R. synthesized microgels. E.S.Y. and H.S. fabricated SA-PD-L1 proteins and provided input on immunological experiments and edited the article. N.M.R. and M.M.C. conceived and designed all experiments and wrote the article.
Data and Material Availability
All data needed to evaluate the conclusions in the article are present in the article and/or the Supplementary Materials. Additional data related to this article may be requested from the authors.
Disclosure Statement
H.S. and E.S.Y. hold equity in FasCure Therapeutics, LLC, which has an option to license the SA-PD-L1 technology from the University of Louisville. M.M.C., H.S., and E.S.Y. are inventors on a provisional patent related to materials for PD-L1 presentation. All other authors declare that they have no competing interests.
Funding Information
This work was supported, in part, by the Juvenile Diabetes Research Foundation (JDRF; Grant No. 2-SRA-2022-1274-S-B).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.