
Abstract
This study addresses a critical challenge in bioprinting for regenerative medicine, specifically the issue of hypoxia compromising cell viability in engineered tissues. To overcome this hurdle, a novel approach using a microfluidic bioprinter is used to create a two-layer structure resembling the human ovary. This structure incorporates a liposomal oxygen-releasing system to enhance cell viability. The bioprinting technique enables the simultaneous extrusion of two distinct bioinks, namely, bioink A (comprising alginate 1% and 5 mg/mL PEGylated fibrinogen in a 20:1 molar ratio) and bioink B (containing alginate 0.5%). In addition, liposomal catalase and hydrogen peroxide (H2O2) are synthesized and incorporated into bioinks A and B, respectively. The liposomes are prepared using thin film hydration with a monodisperse size (140–160 nm) and high encapsulation efficiency. To assess construct functionality, isolated human ovarian cells are added to bioink A. The bioprinted constructs, with or without liposomal oxygen-releasing systems, are cultured under hypoxic and normoxic conditions for 3 days. Live/Dead assay results demonstrate that liposomal oxygen-releasing systems effectively preserve cell viability in hypoxic conditions, resembling viability under normoxic conditions without liposomes. PrestoBlue assay reveals significantly higher mitochondrial activity in constructs with liposomal oxygen delivery systems under both hypoxic and normoxic conditions. The evaluation of apoptosis status through annexin V immunostaining shows that liposomal oxygen-releasing scaffolds successfully protect cells from hypoxic stress, exhibiting a proportion of apoptotic cells similar to normoxic conditions. In contrast, constructs lacking liposomes in hypoxic conditions exhibit a higher incidence of cells in early-stage apoptosis. In conclusion, the study demonstrates the promising potential of bioprinted oxygen-releasing liposomal scaffolds to protect ovarian stromal cells in hypoxic environments. These innovative scaffolds not only offer protection but also recapitulate the mechanical differences between the medulla and the cortex in the normal ovary structure. This opens new avenues for advanced ovary tissue engineering and transplantation strategies.
Impact Statement
Ovary tissue engineering is a strategy to preserve fertility in young women and prepubertal girls diagnosed with cancer, who cannot benefit from transplantation of cryopreserved ovarian tissue. In this study, we used bioprinting technology as a pioneering approach to investigate the fabrication of human ovarian tissue, incorporating oxygen-releasing liposomes to mitigate the adverse effects of hypoxia on transplanted engineered ovarian tissue. Bioprinting offers an innovative platform capable of mimicking the intricate architecture of the human ovary, including its mechanical nuances, while facilitating the precise extrusion of functional materials, such as nanocarriers. Therefore, this research holds significant promise for advancing the field of ovary tissue engineering and providing a ray of hope to those in need of fertility preservation under challenging clinical circumstances.
Introduction
Preserving fertility in patients with cancer, especially in prepubertal girls and women requiring immediate treatment, stands as a paramount concern. Although ovarian tissue cryopreservation and transplantation have shown promising results, there are still significant challenges to overcome, especially for patients diagnosed with tumors carrying a high risk of ovarian involvement, such as blood-borne and metastatic cancer. These cases pose significant challenges as the transplantation of cryopreserved ovarian tissue is not allowed. These cases pose significant hurdles to the transplantation of cryopreserved ovarian tissue because of the potential reintroduction of malignant cells, thereby heightening the risk of disease recurrence.1–5 It is important to note that chemotherapy and radiotherapy, while crucial in treating cancer, can lead to primary ovarian insufficiency, early menopause, and infertility. Moreover, they are associated with higher risks of osteoporosis and cardiovascular mortality, posing significant concerns for female patients with cancer. 6
In response to these challenges, the ongoing research on the development of an engineered ovary emerges as a promising avenue.7–13 This innovative approach aims to create bioengineered structures that mimic the function of the ovary, providing a means to restore fertility without the risk of reintroducing malignant cells. This transformative technology holds the potential to instill hope in patients with cancer facing fertility issues because of their treatment, presenting a safer alternative to traditional transplantation methods.
Cell survival after transplantation of tissue-engineered scaffolds remains an important challenge, primarily because of the limited oxygen supply that results from a lack of vascular network. Moreover, it is well established that oxygen dissolved in a liquid can only diffuse a maximum distance of 200 µm in a scaffold.14,15 Based on Fick’s first law, the diffusion of oxygen in in vitro culture systems decreases as the medium volume increases. Although reducing the medium height in culture plates can mitigate the gradient decrease in oxygen, it introduces the risk of medium evaporation and cell dehydration. 16
Several strategies have been devised to overcome the limitations of oxygen diffusion within engineered scaffolds, such as using oxygen-releasing particles,17–19 vascularization-inducing growth factors,20,21 and endothelial cells. 22 Notably, hydrogen peroxide (H2O2) can be used as an oxygen-generating source, breaking down into water and oxygen in the presence of catalase.23–25 Abdi et al. 26 demonstrated that encapsulation of H2O2 offers precise control over its decomposition, thereby protecting cells from direct exposure of H2O2 and its potential harmful effects.
Another limitation of conventional tissue-engineered constructs is their inability to provide the complex architecture found in natural tissues. 27 However, advanced technologies, such as bioprinting and microfluidics, can be used to some degree to recapitulate the heterogeneity observed in normal tissues. For instance, in an effort to mimic the diverse mechanical properties inherent to ovarian tissue’s cortex and medulla, Choi et al. 28 encapsulated ovarian follicles in two-layered microcapsules using microfluidic technology. In this innovative approach, the outer layer exhibited greater rigidity, whereas the inner layer demonstrated a softer composition, thus effectively replicating the contrasting mechanical characteristics present in the ovarian tissue.
The bioprinting technology uses robotic additive manufacturing techniques that dispense biomaterials precisely in X, Y, and Z directions to control cell distribution for fabricating 3D complex tissue constructs that mimic natural tissues.29–31 Three distinct types of bioprinting approaches have been developed, which include vat photopolymerization-based (including stereolithography, digital light processing, and volumetric printing), jetting-based (including inkjet, microvalve, acoustic, and laser assisted), and extrusion-based bioprinting.32–35 Light-based vat-polymerization bioprinting creates 3D structures using photoactivatable bioresins in vats, initiated by patterned light doses for precise patterning in 2D and 3D. Inkjet bioprinters can be divided into two categories, namely, thermal and piezoelectric setups. The thermal printers generate bubbles from heated bioink and force the liquid to exit, whereas piezoelectric devices use mechanical stimulation to produce droplets.
The droplets created by both types of inkjet printers are deposited accurately. Laser-based bioprinters, as a nozzle-less method, use a laser beam to print bioink. The laser pulse is focused on the donor slide coated with an absorbing layer, such as titanium or gold, and induces a vapor bubble that ejects the bioink droplet onto the collection substrate. Extrusion-based bioprinters, in contrast, extrude bioink using pneumatic, piston, or screw dispensing systems to create 3D structures.30,36–39 Microfluidic extrusion represents an advanced methodology for generating printable hydrogels by simultaneously coextruding bioink and cross-linker through microchannels. This technology enables printing with multiple cell types while protecting them from shear stress during extrusion. The printhead has a microfluidic design that allows for effortless integration with various precise fluid and cell manipulation modules before fiber extrusion, ensuring versatility and efficacy.40–42
The goal of this study was to address two critical aspects. First, we aimed to assess the capabilities of microfluidic bioprinting technology to fabricate a bioprinted scaffold for ovarian tissue engineering, which replicates the intricate mechanical properties observed in natural ovarian tissue. Second, our study sought to assess the impact of an oxygen-generating bioprinted scaffold, enriched with liposomal catalase and liposomal H2O2, on the viability of ovarian stromal cells.
Materials and Methods
Ovarian tissue collection
For this study, ovaries from deceased multiorgan donors were collected following approval from the Université Catholique de Louvain’s Institutional Review Board for the use of human ovaries on May 25, 2019 (IRB reference 2018/19DEC/475). After transferring ovaries to the laboratory and the removal of the medullar section, ovarian cortexes were frozen based on our standard method. 43
Cell isolation and culture
The stromal cells were isolated after thawing cryopreserved ovarian fragments at 37°C, as previously described. 44 Briefly, tissues that were mechanically minced by a tissue chopper (Mickel Laboratory) were enzymatically digested using Liberase DH (05401054001; Sigma-Aldrich) and DNAse I (10,104,159,001; Sigma-Aldrich) at 37°C for 75 min. Then, the activity of enzymes was stopped by adding an equal volume of Dulbecco’s phosphate-buffered saline (DPBS; 14,190,144; Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS; 16,140–071; Gibco).
The final suspension was consecutively filtered through 80 (NY8002500; Millipore, Sigma-Aldrich) and 30 μm (NY3002500; Millipore) nylon net filters and centrifuged at 500 g for 10 min. Afterward, the cells were counted and cultured in a culture medium containing Dulbecco’s modified Eagle’s medium F-12 nutrient mixture (DMEM/F12; 21,041–025; Gibco), 10% FBS, and 1% antibiotic and antimycotic (Anti-Anti; A5955; Gibco) in a humidified incubator (37°C, 5% carbon dioxide [CO2]). Cells were subcultured after they had reached confluence, and the culture medium was replaced every other day.
Liposomal catalase and liposomal H2O2 syntheses
Liposomal catalase and liposomal H2O2 were prepared by the thin-film hydration method and followed the optimized formulation described previously. 45 Briefly, DSPC (1,2-Distearoyl-sn-glycero-3-phosphocholine; P1138; Sigma-Aldrich), DOPE (2-Dioleoyl-sn-glycero-3-phosphoethanolamine; 76548; Sigma-Aldrich), and cholesterol (C8667; Sigma-Aldrich) at a molar ratio of 1:1:1 were dissolved in chloroform, and the solvent was then evaporated at 60°C using a rotary evaporator. The film was then dried in a vacuum desiccator overnight to get rid of any remaining solvent. After hydrating the film with a 1 mL solution of PBS containing 800 mg/L catalase or 1M H2O2, the mixture was vortexed, heated to 40°C for 15 min in an ultrasonic bath (Powersonic 405, 40 kHz), and then centrifuged at 20,000 g for 20 min at 4°C to separate the liposomes from the aqueous solution. The final samples were sterilized by filtering through a 0.22 µm sterile filter before being kept at 4°C.
Liposomal catalase and liposomal H2O2 characterization
Dynamic light scattering technique using the Zetasizer Nano ZS (Malvern Instruments Limited) was used for the evaluation of the size, polydispersity index, and zeta potential of liposomal catalase and liposomal H2O2. The ultraviolet spectrophotometer method was used to measure the catalase concentration (NanoDrop 2000/2000c, Thermo Fisher Scientific) and the fluorometric Amplex UltraRed/horseradish peroxidase (HRP) was used for the quantitative assessment of H2O2.46,47 Briefly, a mixture of Amplex UltraRed (100 µM) and HRP (1U/mL) was added, and after a 15-min incubation, the fluorescent reaction products were quantified using a fluorometer (Multilabel reader, Victor X4). The total amount of catalase or H2O2 (WT) and their concentration in the supernatant following centrifugation (WF) were measured to compute the entrapment efficiency (EE %) using the following equation:48,49
Bioink preparation
Alginate (9842; Aspect Biosystems) was dissolved in PBS at 4°C overnight to prepare a concentration of 1.18%. PEGylated fibrinogen (PF) was also prepared as described previously 50 with a molar ratio of PEG: Fib 20:1. Briefly, syringe-filtered human fibrinogen (F3879; Sigma-Aldrich) at a concentration of 85 mg/mL was incubated at 37°C before PEGylation. A total of 10 mg/mL of O,O’-Bis[2-(N-Succinimidyl-succinylamino)ethyl]polyethylene glycol (NHS-PEG-NHS; 2,000 Da; 713783; Sigma-Aldrich) in PBS was prepared, incubated at 37°C, and syringe filtered. Then, an equal volume of NHS-PEG-NHS was added to the fibrinogen solution and incubated at 37°C for 2 h.
Two types of bioinks (shell A and shell B) were prepared to print the oxygen-releasing scaffold. Shell A was composed of alginate, PF 20:1, ovarian stromal cells at passage 7, and liposomal catalase with a final concentration of 1%, 5 mg/mL, 1 × 106 cells/mL, and 40 mg/L, respectively. Shell B contained alginate and liposomal H2O2 with a final concentration of 0.5% and 40 mM, respectively.
Bioprinting and in vitro culture of the scaffolds
The extrusion-based RX1 bioprinter (Aspect Biosystems) was used to print parallel filaments of shell A and shell B. This bioprinter is operated by air pressure and contains microfluidic printheads that allow the printing of different materials at the same time. The channel specified for the cross-linker initiates cross-linking of bioinks as well as minimizes shear stress on the cells during printing.
A rectangular print structure with a volume of 1 × 1 × 10 mm3 was designed in the bioprinter software (Aspect Studio). The CENTRATM printhead was used to print the desired structure with optimized printing pressures of 105, 43, 200, and 150 mbar for shell A, shell B, cross-linker, and buffer, respectively. CAT-2™ cross-linker (0458; Aspect Biosystems) and saline buffer solution (flushing solution; 0460; Aspect Biosystems) were used to cross-link filaments during printing and flush the channels of the bioprinter printhead after printing, respectively. Moreover, 40 mM calcium chloride (CaCl2; C5080; Sigma-Aldrich) and 5 IU/mL of human thrombin (T7009; Sigma-Aldrich), which was reconstituted and diluted in 40 mM CaCl2, were used as postprinting cross-linkers. Three repetitions were bioprinted for each condition of hypoxia (1% oxygen [O2], 5% CO2) and normoxia (20% O2, 5% CO2) with or without liposomes and cultured for three days in humidified incubators at 37°C (Fig. 1).
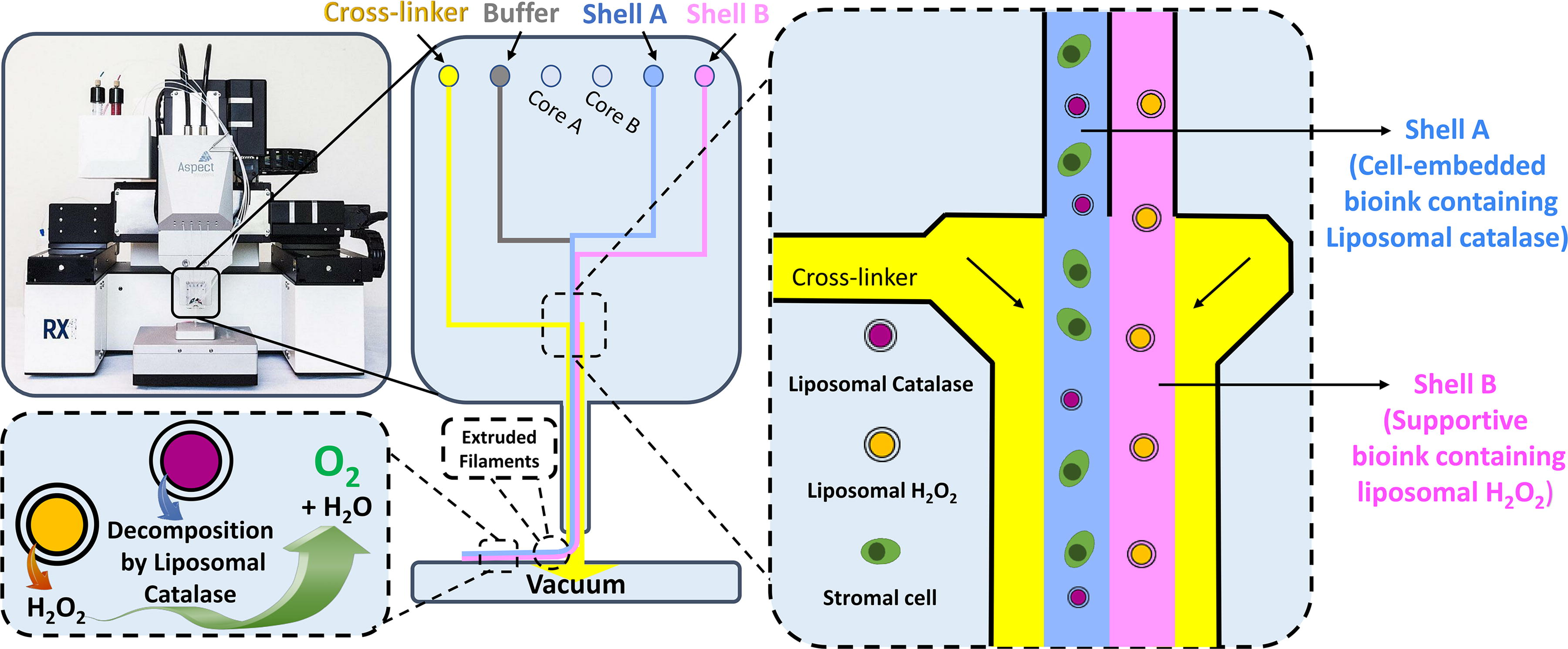
A schematic of bioink compositions in a microfluidic printhead, RX1 bioprinter, and a representative bioprinted scaffold.
Live/Dead assay
The viability of cells was analyzed using a Live/DeadTM viability/cytotoxicity kit (L3224; Invitrogen), which is based on the membrane integrity and intracellular esterase activity. To perform this analysis, the medium of constructs was removed and washed with DPBS before adding the Live/Dead solution containing 0.5 μL of calcein-AM and 2.5 μL of ethidium homodimer-1 in 997 μL DPBS. Then, the samples were incubated for 45 min at 37°C. The cell survival was characterized under a fluorescent microscope, in which green and red colors indicate live and dead cells, respectively. The viable and dead cells were counted on a surface of 200 × 200 µm2 using Image J software (https://imagej.nih.gov/ij/).
Mitochondrial activity assay
The mitochondrial activity of cells in each condition was assessed by using a resazurin-based solution of PrestoBlue™ HS (P50200; Invitrogen) following our published protocol. 50 PrestoBlue solution was prepared in the culture medium with a proportion of 1:10 PrestoBlue in the medium. Then, the PrestoBlue solution was added to each well after aspirating the medium of samples. After an incubation time of 3 h at 37°C, the medium in each well was pipetted a few times to be homogenized, and 100 µL of the medium was transferred to a 96-well plate to measure the fluorescent values by fluorescence spectroscopy (Multilabel reader; Victor X4). The emission and excitation wavelengths were set at 620 nm and 560 nm. The average fluorescent value of hydrogels on day 0 and normoxic samples without liposomes on day 3 were used for normalization at 0% and 100% viability, respectively.
Immunofluorescence staining
After 3 days of in vitro culture of bioprinted samples, the samples were put in HistoGel (HG-4000-012; Thermo Fisher Scientific) and fixed with a combination of 75% of paraformaldehyde and 4% and 25% of CaCl2 200 mM for 1 h at room temperature. Then, the constructs were dehydrated and clarified before embedding in paraffin. The paraffin-embedded samples were cut, and the slides were prepared for an immunohistochemical study. Given that early apoptosis in cells can be identified through the detection of membrane phospholipid phosphatidylserine (PS) exposed on the cell’s outer membrane, we conducted annexin V immunostaining to detect cells in the early apoptosis state. Briefly, after deparaffinizing and rehydrating different sample sections on slides, the nonspecific antigens were blocked using 10% NGS in 1× Tris-buffered saline (TBS) for 1 h at room temperature.
Afterward, the slides were incubated with annexin V polyclonal primary antibody (11060-1-AP; Proteintech), diluted 1:400 in normal goat serum (NGS) 1%, for 1.5 h at room temperature. After washing slides in 1× TBS, they were incubated with Alexa Fluor 488 goat anti-rabbit secondary antibody, diluted 1:250 in NGS 1%, for 40 min at room temperature. Then, the nuclei were stained with 1:500 Hoechst 33342 (H3570; Thermo Fisher Scientific) for 5 min followed by washing in 1× TBS and distilled water. Finally, the slides were mounted with a fluorescence mounting medium (S3023; Dako) and scanned by a fluorescent scanner (ZEISS Axio Scan.Z1). The pictures were analyzed by ZEN 3.5 (blue edition), and the annexin V-positive cells were counted in a square with dimensions of 160 × 160 µm2.
Statistical analysis
One-way analysis of variance was performed to statistically analyze the data, and a p-value of 0.05 was used to define statistical significance. The quantitative data were reported as mean ± standard deviation (SD), and error bars in graphs reflect one sample SD.
Results
Liposomal catalase and liposomal H2O2 characterization
The size, polydispersity, and zeta potential of liposomal catalase and liposomal H2O2 were measured using dynamic light scattering technique (Table 1). Results indicated that the sizes of both formulations are in the range of 140–160 nm with a polydispersity index of 0.2–0.4, which is classified as relatively monodisperse nanoparticles (Fig. 2a). 51 In addition to being a crucial factor in the stability and biocompatibility of nanoparticles, zeta potential is an effective way for determining the strength of colloidal particle interactions. 52 The zeta potential of liposomal catalase and liposomal H2O2 was −7.74 ± 2.22 and −10.19 ± 0.96, respectively, which demonstrate relative stability, which is important in ensuring that aggregation does not occur during the bioprinting process (Fig. 2, b).

Characterization of liposomal catalase and liposomal H2O2 after preparation: size distribution
Characterization of Liposomal Catalase and Liposomal H2O2 After Preparation
Cell survival and mitochondrial activity of bioprinted scaffolds under hypoxic and normoxic conditions
The viability of cells bioprinted along with liposomes and cultured under hypoxic conditions exhibited no significant difference when compared with the viability of cells cultured in normoxia without liposomes (78.2 ± 5.3% and 83.9 ± 8.7%, respectively). Conversely, the viability of cells in hypoxia without liposomes (74.7 ± 8.4%) was significantly lower compared with the other experimental groups (p < 0.05).
In contrast, samples containing liposomal catalase and liposomal H2O2 and cultured in normoxia demonstrated the highest cell viability (90.1 ± 5.2%) compared with all other groups (vs. normoxia, p < 0.05; vs. hypoxia, p < 0.0001; vs. hypoxia contained liposomes, p < 0.01) (Fig. 3a–e). Moreover, mitochondrial activity was significantly higher in bioprinted oxygen-releasing constructs in both hypoxia and normoxia culture conditions when compared with bioprinted constructs without liposomes (normoxia–liposomes [Normoxia-LIP] vs. normoxia, 161.8 ± 23.8% vs. 100 ± 6.2%, p < 0.001; hypoxia–liposomes [Hypoxia-LIP] vs. hypoxia, 110.1 ± 7.9% vs. 89.3 ± 4.9%, p < 0.05). Notably, the group cultured in normoxia containing liposomes showed the highest mitochondrial activity compared with all other groups (vs. all groups, p < 0.0001) (Fig. 3f).

Cell viability and mitochondrial activity of bioprinted cells under hypoxic and normoxic conditions. Representative Live/Dead cytotoxicity test pictures of samples in normoxia
Immunofluorescent staining on bioprinted scaffolds
Cells cultured under both normoxic and hypoxic conditions were examined for early apoptosis using annexin V antibody. The results demonstrated that cells in hypoxia exhibited a higher degree of translocation of PS to the outer cell membrane layer (39.5 ± 19.8%) compared with the bioprinted cells in normoxia (20.3 ± 15.7, p < 0.01) and hypoxia with the incorporation of liposomal catalase and liposomal H2O2 (17.9 ± 11.4%, p < 0.01). Interestingly, the number of annexin V-positive cells in the normoxia group did not significantly differ from the number observed in bioprinted constructs containing liposomal catalase and liposomal H2O2 cultured in hypoxia (Fig. 4).

Early apoptosis analysis of bioprinted ovarian stromal cells cultured under hypoxic and normoxic conditions for 3 days. Representative images of annexin V bound to translocated PS in different constructs and conditions
Discussion
Catalase is an antioxidant enzyme known for its ability to break down H2O2 into water and oxygen.53,54 In this study, liposomal catalase was used to produce oxygen from H2O2 molecules, which cross the lipid bilayer of liposomal H2O2 at a gradual rate.55,56
The encapsulation of liposomal H2O2 was executed in a separate bioink, deliberately excluding cells to avoid any potential adverse effect during the printing process. Following the bioprinting process, liposomal encapsulation yielded a sustained release of H2O2, thereby protecting the surrounding cells from high concentrations of free H2O2 and minimizing potential cellular damage. Therefore, this approach serves as a dual barrier for the controlled release of H2O2 before it undergoes decomposition by catalase. Furthermore, liposomal catalase was incorporated into the cell-laden bioink.
Recent literature has demonstrated sustained activity of liposomal catalase over a 24-day period, whereas free catalase exhibited a loss of 90% of its original activity within the same time frame. 57 Encapsulating catalase within liposomes enhances the enzyme’s short lifetime and provides a controlled, gradual release pattern.58–60 Notably, the usage of liposomal catalase and liposomal H2O2 has found application in enhancing cancer therapy. For instance, Song et al. 56 injected liposomal catalase and H2O2 into 4T1 tumor-bearing mice and reported effective oxygenation of tumors. In our study, we use catalase and H2O2-loaded liposomes for the oxygenation of scaffolds in a hypoxic environment.
Two distinct alginate-based bioinks were formulated: shell A (bioink A) and shell B (bioink B). Shell A contained a higher concentration of alginate (1%), PF, isolated human ovarian stromal cells from the cortex, and liposomal catalase. Shell B included liposomal H2O2 in a lower alginate concentration (0.5%). PF has been successfully used for encapsulation of either isolated human ovarian stromal cells 50 or follicles 44 and has been reported to have positive effects on preserving the survival of both ovarian stromal cells and follicles in vitro. Moreover, Dadashzadeh et al. 50 reported the positive impact of thrombin on ovarian cell proliferation.
In this study, the in vitro culture medium of bioprinted constructs was enriched by thrombin to polymerize the PF incorporated in bioink A. The selection of varying alginate concentrations aimed to replicate the mechanical differences between the ovarian cortex and the medulla. The mechanical properties of alginate hydrogels depend on several factors, including the molecular structure of alginate, concentration of divalent cations, cross-linking time, and alginate concentration.61–65 Here, CaCl2 and cross-linking time were kept constant, whereas alginate concentration was varied to form a stiffer alginate hydrogel in shell A (1%) than in shell B (0.5%). The microfluidic printhead of the RX1 bioprinter was used to bioprint oxygen-releasing scaffolds. This was achieved by simultaneously extruding a bioink containing liposomal catalase along with cells side by side with a bioink containing liposomal H2O2.
The bioprinted oxygen-releasing scaffolds effectively preserved the viability of ovarian stromal cells in hypoxia, resulting in a viability similar to that observed in bioprinted scaffolds without liposomes under normoxic conditions (control). Notably, the oxygen-releasing liposomes improved cell viability in normoxia samples compared with the control (Fig. 3a–e). Moreover, the results of the mitochondrial activity in different samples indicated higher activity in scaffolds containing oxygen-releasing liposomes compared with samples without liposomal catalase and H2O2 (Fig. 3f). These results demonstrate the suitability of using oxygen-releasing systems not only in hypoxic conditions but also for culturing tissue-engineered scaffolds under normoxia. This is especially relevant owing to the limitations in oxygen diffusion imposed by medium height and scaffold thickness, where cells placed at the bottom of scaffolds experience the lowest oxygen pressure. 16 Indeed, oxygen diffusion in the tissue exhibits a direct correlation with surface area and an inverse relationship with thickness. 66
Hypoxia is known to induce apoptosis.67–69 Once apoptosis is initiated, PS translocates, becoming exposed on the outer surface of cells. 70 Annexin V has a high affinity to interact with PS, 71 and annexin V labeling of PS on the cell surface is indicative of cells in the early apoptosis stage. 70 Annexin V immunostaining analyses demonstrated that the bioprinted oxygen-releasing scaffold provides a suitable system for mitigating hypoxic stress on ovarian stromal cells, with the number of annexin V-positive cells being comparable with those cultured under normoxic conditions (control).
Conversely, bioprinted cells without a liposomal oxygen-releasing system experienced more apoptosis due to the hypoxic stress on the ovarian stromal cells. Kim et al. 18 reported that oxygen delivery under hypoxic conditions moderated cell apoptosis compared with samples exposed to hypoxia without oxygen delivery, which had a higher rate of apoptotic activity. Moreover, Suvarnapathaki et al. 72 provided oxygen-releasing scaffolds by synthesizing microparticles containing different concentrations of calcium peroxide (CaO2) in polycaprolactone and encapsulated them in gelatin methacrylate. Their results showed that low levels of oxygen provided by low concentrations of CaO2 led to an increase in apoptosis levels. Therefore, incorporating an appropriate oxygen-releasing system into tissue-engineered scaffolds holds the potential to safeguard cell survival.
Conclusion
In summary, our study effectively used bioprinting technology to replicate the intricate architecture of natural ovarian tissue, specifically its two-layered structure comprising the cortex and the medulla. This was achieved by simultaneously extruding two different bioinks using a microfluidic RX1 bioprinter, resulting in the creation of parallel filaments with different mechanical properties, achieved through the usage of high and low concentrations of alginate. Moreover, the persistent challenge of hypoxia in tissue-engineered scaffolds was addressed through the incorporation of an oxygen-releasing system. Liposomal catalase and liposomal H2O2 were synthesized and incorporated into the two bioinks. Our observations underscored the positive impact of the bioprinted oxygen-releasing scaffold on the viability of ovarian stromal cells, both in hypoxic and in normoxic environments. Notably, the liposomal oxygen-releasing system demonstrated the ability to protect cells from apoptosis induced by low oxygen levels and hypoxic stress as well as support cellular metabolism according to the mitochondrial activity.
In conclusion, our study illustrates the potential for bioprinting technology to replicate the complex architecture of natural tissues and offers a promising solution to mitigate hypoxia-related challenges in tissue engineering. The liposomal oxygen-releasing system shows promise as a valuable tool for enhancing cell viability under adverse conditions, providing new avenues for the development of functional tissue-engineered constructs.
Footnotes
Acknowledgments
We thank the Kidney and Pancreatic Transplantation Unit of UCLouvain’s Saint-Luc Hospital for donating ovaries for this study. We would like to express our gratitude to Aspect Biosystems for providing us with the opportunity to use a microfluidic RX1 Bioprinter and for their support in supplying materials and engaging in scientific communication.
Authors’ Contributions
A.D. and S.M.: Conceptualization, methodology, software, validation, formal analysis, investigation, writing original draft, and visualization; C.A.A.: Resources, review and editing, supervision, project administration, and funding acquisition.
Disclosure Statement
The authors declare no competing interests.
Funding Information
This study was supported by grants from the Fondation Louvain (PhD scholarship awarded to S.M., as part of a legacy from Mr. Frans Heyes, and PhD scholarship awarded to A.D. as part of a legacy from Mrs. Ilse Schirmer).