
Abstract
Previously, chitosan reduces the senescence-related phenotypes in human foreskin fibroblasts through the transforming growth factor beta (TGF-β) pathway, and enhances the proliferation and migration capabilities of these cells are demonstrated. In this study, we examined whether the senescence-delaying effect of chitosan could be applied to primary knee-related fibroblasts, such as human synovial membrane derived cells (SCs) and anterior cruciate ligament fibroblasts (ACLs). These two types of cells were obtained from donors who needed ACL reconstruction or knee replacement. We found that chitosan treatment effectively reduced aging-associated β-galactosidase (SA-β-gal)-positive cells, downregulated the expression of senescence-related proteins pRB and p53, and enhanced the 5-bromo-2’-deoxyuridine (BrdU) incorporation ability of SCs and ACLs. Moreover, chitosan could make SCs secret more glycosaminoglycans (GAGs) and produce type I collagen. The ability of ACLs to close the wound was also enhanced, and the TGF-β and alpha smooth muscle actin (αSMA) protein expression decreased after chitosan treatment. In summary, chitosan not only delayed the senescence but also enhanced the functions of SCs and ACLs, which is beneficial to the application of chitosan in cell expansion in vitro and cell therapy.
Impact Statement
When human synovial membrane derived cells (SCs) and anterior cruciate ligament fibroblasts (ACLs) cultured in vitro, they cease dividing gradually. Chitosan treatment could significantly decrease the senescent phenotypes and enhance the proliferating potential of SCs and ACLs. Moreover, SCs secreted more GAGs and type I collagen after treatment, and chitosan treatment also avoided the ACL differentiation and enhanced the mobility of fibroblasts to accelerate wound closure.
Introduction
Cell therapy is used in the field of tissue healing engineering because cells can provide more growth factors and extracellular matrix to continuously induce a series of paracrine reactions of affected tissue and accelerate wound healing1,2 compared to just once administering drugs or growth factors. Although the allogeneic cell therapy solves the shortage of the cell amount, the immune response of the patients after implantation is concerned.3–5 Autologous cell therapy is still a prioritized choice for patients if enough cell amounts can be gotten. For example, mosaicplasty6–8 and autologous chondrocyte implantation (ACI)9,10 have been developed as mainstream cell therapies for knee cartilage repair. However, not only is the amount of natural cells limited, but also isolated cells become older and lost their ability to proliferate after being expanded in vitro. How to avoid cell senescence and keep the cellular function are issues for both the autologous and allogeneic cell therapy when cells are in vitro proliferated. In order to reach this goal, the methods for in vitro cell expansion such as bioreactors,11–13 hypoxic cultures14–16 and 3 dimensional cultures17–19 boost.
In addition to in vitro culturing aging, the cells from old and sick donors have bad quality 20 is another issue we need to focus on. Edward L. et al. proposed that the skin fibroblasts isolated from young and normal volunteers had longer in vitro lifespan, higher cell population replication rate and better cell migration ability than that isolated from old volunteers. 21 There are differences in synovial surface roughness, cell distribution uniformity, cell size, collagen secretion, etc. between the elderly and young people. 22 Aged tendon cells have lower proliferative capacity, and the expression of aging-associated β-galactosidase (SA-β-gal) is higher than that of young cells. 23 The cell activity and the extracellular matrix secretion of aging tenoblasts and tenocytes are also reduced.24,25 A significant negative correlation between the total population doublings (PDs) of skin fibroblasts and the ages of the diabetic and prediabetic donors is also found. 26 In other words, to enhance the proliferation and delay the senescence of the cells isolated from old and sick donors are more necessary than that from young and normal donors when cells expanded. Many senescence-delaying methods have been proposed through regulating the TGF-β 27 or oxygen 28 concentration in cell in vitro culture environment. Coculturing with younger cells is also used to enhance the proliferation and function of the aging knee-related cells in vitro.29,30 In this study, we used chitosan treatment on synovial membrane derived cells (SCs) and anterior cruciate ligament fibroblasts (ACLs) from donors with different ages to evaluate the effect of the senescence delaying.
Chitosan is a biodegradable and biocompatible polymer,31,32 and it has been used to upregulate the stemness capabilities of normal stem cells 33 and cancer stem cells. 34 Because fibroblasts loss their stemness, they mediate their self-renewal ability through ceasing the senescence and promoting the proliferation. In previous studies, the chitosan treatment not only decreases the senescence-related phenotypes but also increases the proliferating ability and the motility of foreskin fibroblasts 35 through the TGF-β pathway. 36 In this study, chitosan was used to treat on SCs and ACLs, and the senescence and proliferation-related phenotypes of these cells were examined. In addition, the cellular functions of SCs and ACLs were also analyzed. This study would provide some information for considering the application of chitosan treatment on ACLs and SCs when cell expansion.
Materials and Methods
Preparation of chitosan substrates
1% (w/v) chitosan (Sigma-Aldrich C3646) solution was prepared by an acetic acid solution. The solution was added into tissue culture polystyrene (TCPS) plates and dried at 60°C to form thin membranes. Then, the plates were neutralized by sodium hydroxide and sterilized by ethanol and ultraviolet light for subsequent cell culture.
Cell culture
Anterior cruciate ligament and synovial membrane explants were obtained from donors who underwent ligament reconstruction or knee replacement. Twelve donors between the ages 25 and 81 provided their anterior cruciate ligament specimens, and 12 donors between the ages 25 and 79 provided part of their synovial membrane. The study protocol was approved by the Institutional Review Board of the National Taiwan University Hospital.
Anterior cruciate ligament specimens were cut into small fragments about 4–5 mm3 in size and cultured in medium. A few days later, ACLs migrated out from the fragments. In the other side, the synovial specimens were cut into small pieces and incubated for 2 h in a medium containing 2.5 mg/mL collagenase (Sigma-Aldrich). Then, SCs were filtered by sterile cell strainers (40 um mesh) and cultured on TCPS. SCs and ACLs were cultured on TCPS for 14 days without any passage, and the culture medium was renewed regularly. After 14 days culturing, the in vitro first-passage ACLs and SCs were harvested by 0.05% trypsin/ethylenediaminetetraacetic acid (Trypsin-EDTA, Gibco) for subsequent treatment. The culturing medium was Dulbecco's Modified Eagle Medium (Gibco), which contained 10% fetal bovine serum (Biological Industries), 100 U/mL penicillin G, 100 μg/mL streptomycin, and 250 μg/mL amphotericin B (Biological Industries). All cells were cultured in a 37°C humidified chamber with 5% CO2 and 95% air atmosphere. For chitosan treatments, in vitro first-passage cells were collected and seeded on chitosan coated plates at the density about 25,000/cm2 for 3 days. Seeding density and culture time follow our previous researches, 36 and we have proved that these parameters are ideal for chitosan treatment of fibroblasts. Cells were photographed under a microscope (Olympus IX71).
Viability assay
Cell viabilities were examined by the Live/Dead kit (Invitrogen 3224). 37 Spheroids were collected and stained by Live/Dead kit for 30 min. Live cells were labeled with calcein AM (green) and dead cells were stained with ethidium homodimer (red). They were observed under a confocal fluorescence microscope (LEICA SP5).
SA-β-gal staining
After fixed by formaldehyde, cells were incubated in SA-β-gal staining solution with X-gal at pH 6 and 37°C38–40
at the time points. These cells were photographed by the microscope, and the percentages of cells with blue sedimentation were counted. At least 400 cells were calculated from 15 randomly selected fields for each case. The senescence index refers to the SA-β-gal positive percentage of chitosan-treated cells divided by the percentage of untreated cells.
Western blotting
After cells were lysed, cell proteins were collected and quantitated by Bio-Rad total protein assay. Then, these proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The proteins were probed with human primary antibodies against pRB (Merck OP66; LM95.1; 1:100), p53 (Merck OP09; PAb1891; 1:200), collagen I (Epitomics S2307; polyclone; 1:3000), TGF-β (Cell signaling 3709; 56E4; 1:1000), αSMA (Abcam ab5694; polyclone; 1:4000), and GAPDH (Abcam ab22555; polyclone; 1:10000). The membranes were incubated in HRP-conjugated secondary antibodies (Goat antirabbit IgG (Abcam ab97051; 1:5000) and Goat antimouse IgG (Abcam ab97023; 1:5000)), and visualized by enhanced chemiluminescence (Millipore) following.
5-bromo-2’-deoxyuridine (BrdU) incorporation assay
Cells were cultured with BrdU for 24 h, and then the Cell Proliferation Enzyme-Link/′ed ImmunoSorbent Assay (ELISA) BrdU kit (Roche 11 647 229 001) was used to evaluate the cell amount incorporated by BrdU. Measure the absorbance of the samples in an ELISA reader at 450 nm (reference wavelength: 690 nm). The proliferation index was calculated by dividing the BrdU absorbance of the chitosan-treated cells by the BrdU absorbance of the untreated cells.
Safranin O staining
Safranin O is a compound which marks glycosaminoglycans (GAGs) so it is used to observe the glycoprotein secretion of cells. After fixed by 4% formaldehyde, SCs were washed by a 1% acetic acid solution for 10 s. Then, cells were stained by a 0.1% Safranin O (Sigma-Aldrich S2255) solution and photographed.
Scratch assay
ACLs were cultured on TCPS at the density about 20,000/cm2. The well surfaces were scratched gently to create scratches on the next day. After removal of detached cells, attaching cells were cultured in medium. The cell images were photographed under a Leica DMI600 microscope, and the closure degree of the wound was analyzed by using ImageJ software.
Statistical analysis
All measurements were shown from at least 3 independent samples. Statistical significance was determined using one-way analysis of variance followed by Duncan’s test. p value of < 0.05 and 0.01 was considered significant.
Results
SCs and ACLs culturing on chitosan to form spheroids
Isolated SCs and ACLs were in vitro cultured on TCPS to observe their senescent phenotypes and PDs. Figure 1a shows more SCs and ACLs with enlarged and flatted morphology, typical aging features, were observed in the 28 DIV groups compared with the 0 DIV groups. These SCs and ACLs were also stained with SA-β-gal, a senescent biomarker [25]. Cells with enlarged and flattened morphology were stained by SA-β-gal in the cytoplasm. With the prolongation of in vitro culture time, the proportion of SA-β-gal positive SCs and ACLs increased (Fig. 1B and 1C; p < 0.01). Average in vitro PDs of SCs were 4.48 ± 1.12, and that of ACLs were 3.01 ± 0.91.
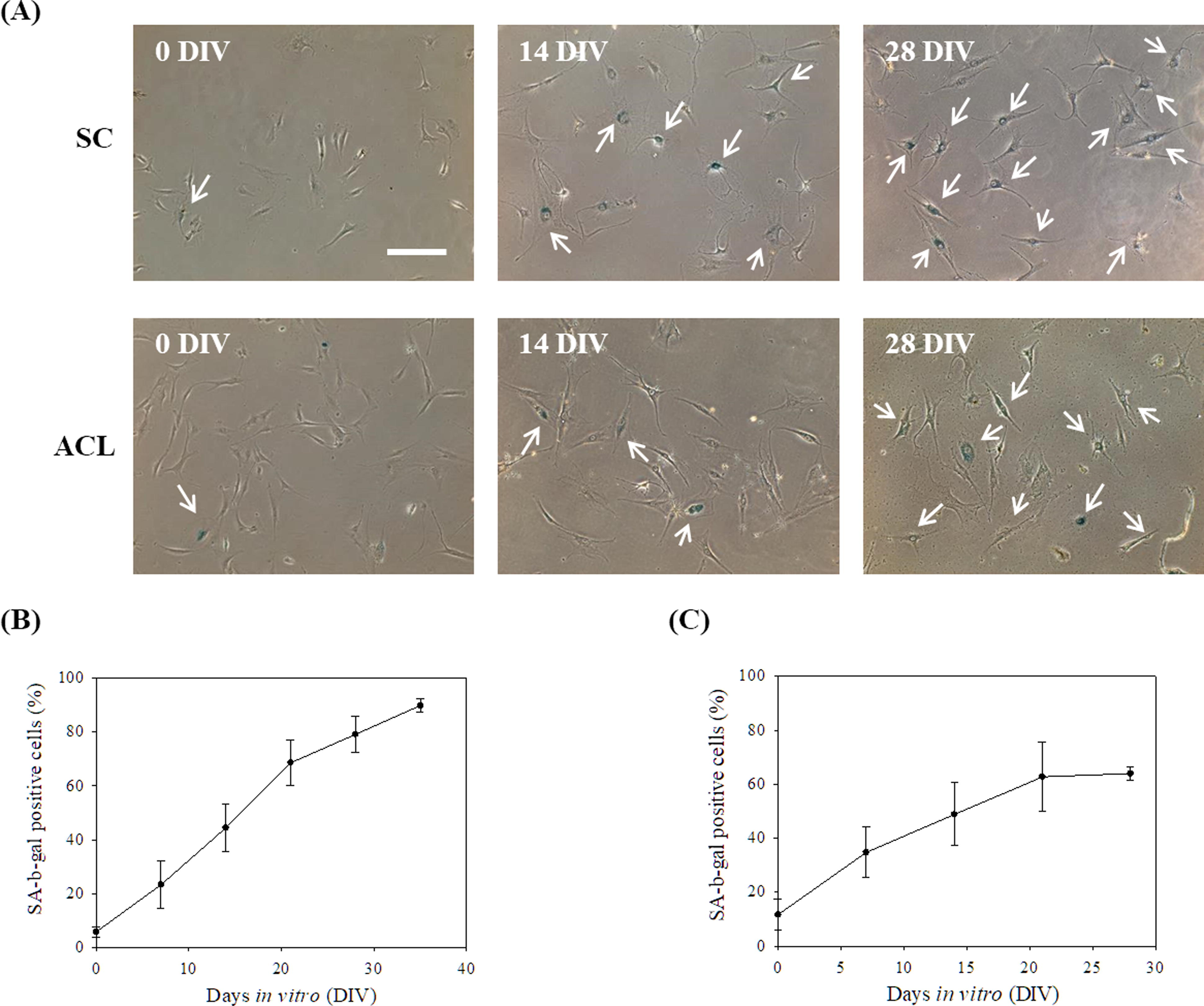
Human synovial membrane derived cells (SCs) and anterior cruciate ligament fibroblasts (ACLs) were serially passaged on TCPS in vitro.
SCs and ACLs were collected after 14 days in vitro culture from TCPS, and cells were seeded on chitosan and TCPS separately for 3 days. In contrast to cells cultured on TCPS, cells could suspend in culture medium to form spheroids on chitosan within 3 days (Fig. 2A). Live/Dead staining data shows that the majority of cells in spheroids were viable (Fig. 2B).
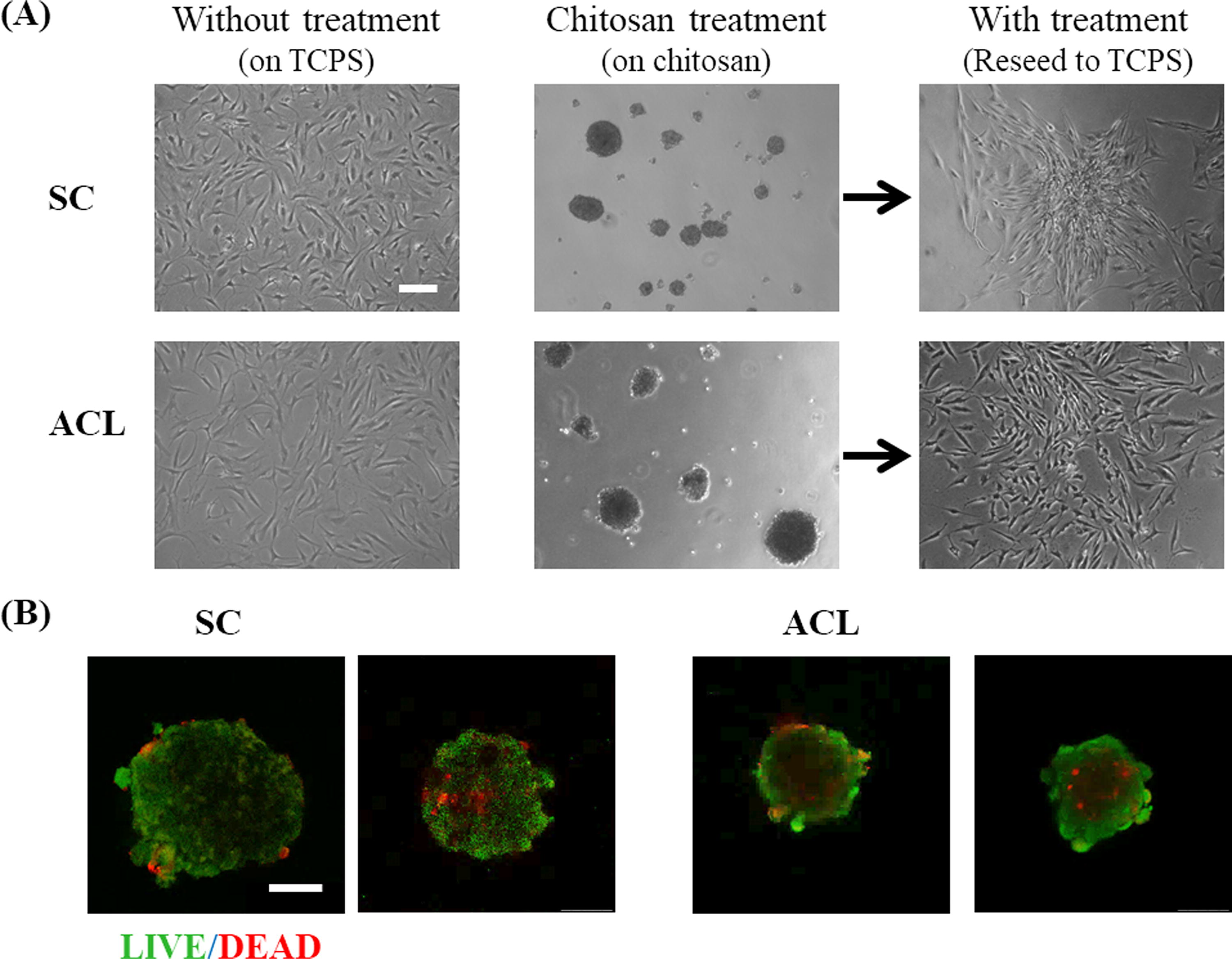
SCs and ACLs were cultured on chitosan to form multicellular spheroids for 3 days.
The delaying senescence and enhancing proliferation of SCs and ACLs after chitosan treatment
The spheroids were harvested from chitosan substrates and reseeded on TCPS for 5 days, to which the spheroids could attach and cells then migrated out of the spheroids, as shown in Figure 2A. The migrating cells were harvested for the following tests without extra passages. Chitosan treatment cells could reduce the SA-β-gal positive ratios of SCs and ACLs cells significantly (Fig. 3A). Cells from different donors were examined to evaluate the effect of chitosan treatment on SA-β-gal expression. If the treatment could reduce the SA-β-gal positive expression after the treatment, the senescent index should be below 1. Table 1 shows that SC and ACL cell populations isolated from different donors could reduce the amount of SA-β-gal positive cells after chitosan treatment. The expressions of senescence-associated molecular markers pRB and p53 of cells were tested. Figure 3B and 3C show that chitosan-treated cells had lower protein expression of senescence-related markers, which happened on both SCs and ACLs (p < 0.5 for p53 and p < 0.01 for pRB). BrdU cell proliferation assay was used to evaluate the cell proliferation ability. If the treatment could enhance the cell proliferation, the proliferating index should be higher than 1. BrdU incorporation data shows significant increase in the proliferation of chitosan-treated SCs and ACLs (Table 2).
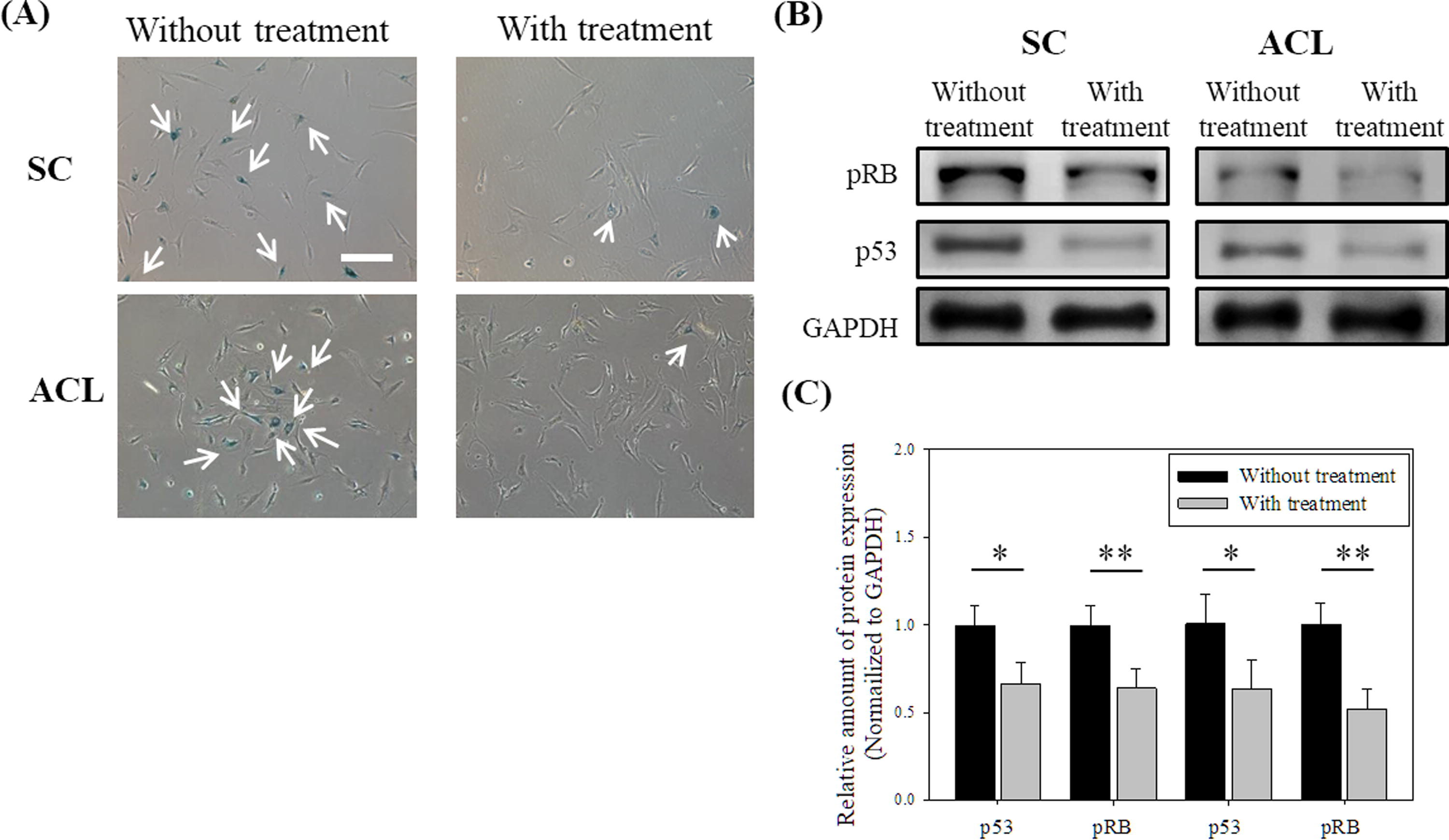
Effect of chitosan treatment on senescent phenotypes of SCs and ACLs.
The SA-β-Gal Positive Ratio and the Senescent Index of SCs and ACLs
At least 400 cells were calculated from 15 randomly selected fields for each case. The senescence index refers to the SA-β-gal positive percentage of chitosan-treated cells divided by the percentage of untreated cells.
p < 0.05.
p < 0.01.
The BrdU Absorbance Value and the Proliferating Index of SCs and ACLs
The proliferation index was calculated by dividing the BrdU absorbance of the chitosan-treated cells by the BrdU absorbance of the untreated cells. (n = 4).
p < 0.05.
p < 0.01.
The cellular functions of SCs after chitosan treatment
Synovial membrane mesenchymal stem cells have been proved, and these cells have stem cell characteristics and the in vitro multidirectional differentiation potential. 41 Synovial membrane mesenchymal stem cells also have powerful chondrogenic differentiation potential compared with mesenchymal stem cells harvested from other sources. 42 We examined the GAGs expression of SCs by Safranin O staining. 43 With and without treatment SC spheroids were both seed on TCPS for 3 days to migrate out and stained with Safranin O. Figure 4A shows that more cells treated with chitosan were stained by Safranin O than untreated cells. On the other hand, the fibrous cartilage contains a high level of type I collagen, so we analyzed the amount of the type I collagen of chitosan-treated SCs. As shown in Figure 4B and 4C, chitosan treatment stimulated SCs to produce more type I collagen (p < 0.5).
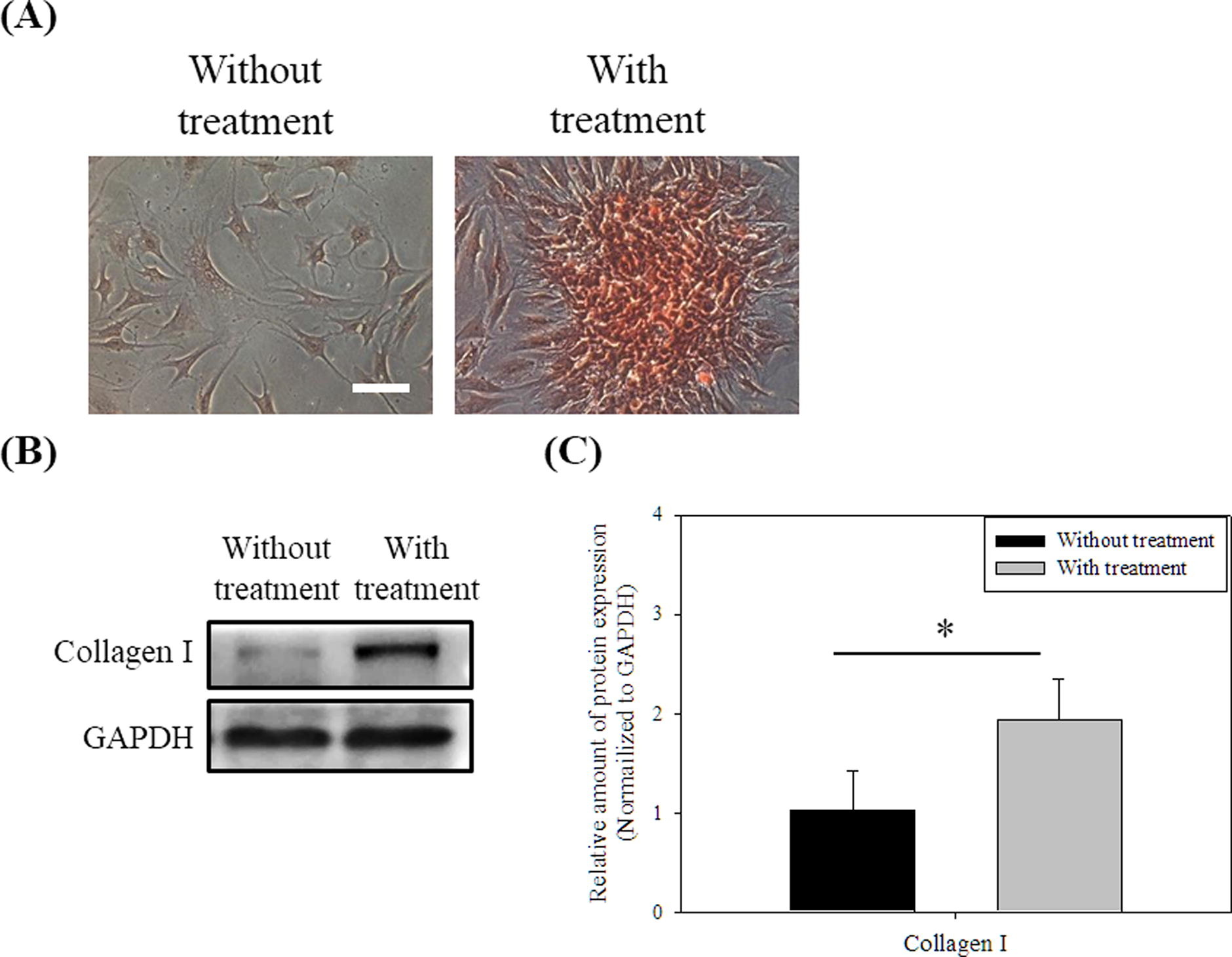
Effect of chitosan treatment on cellular characteristics of SCs.
The cellular functions of ACLs after chitosan treatment
For ligament healing, ACLs play a role in migrating to the affected area so a scratch wound model was used to evaluate the cell mobility. Figure 5A shows that the wounds were more rapidly filled by the chitosan treatment ACLs than by the untreated ACLs. Quantification analysis shows that the wounds of the chitosan treatment ACLs almost completely repaired after 24 h and exhibited a significant difference to the untreated cells (Fig. 5B; p < 0.01).
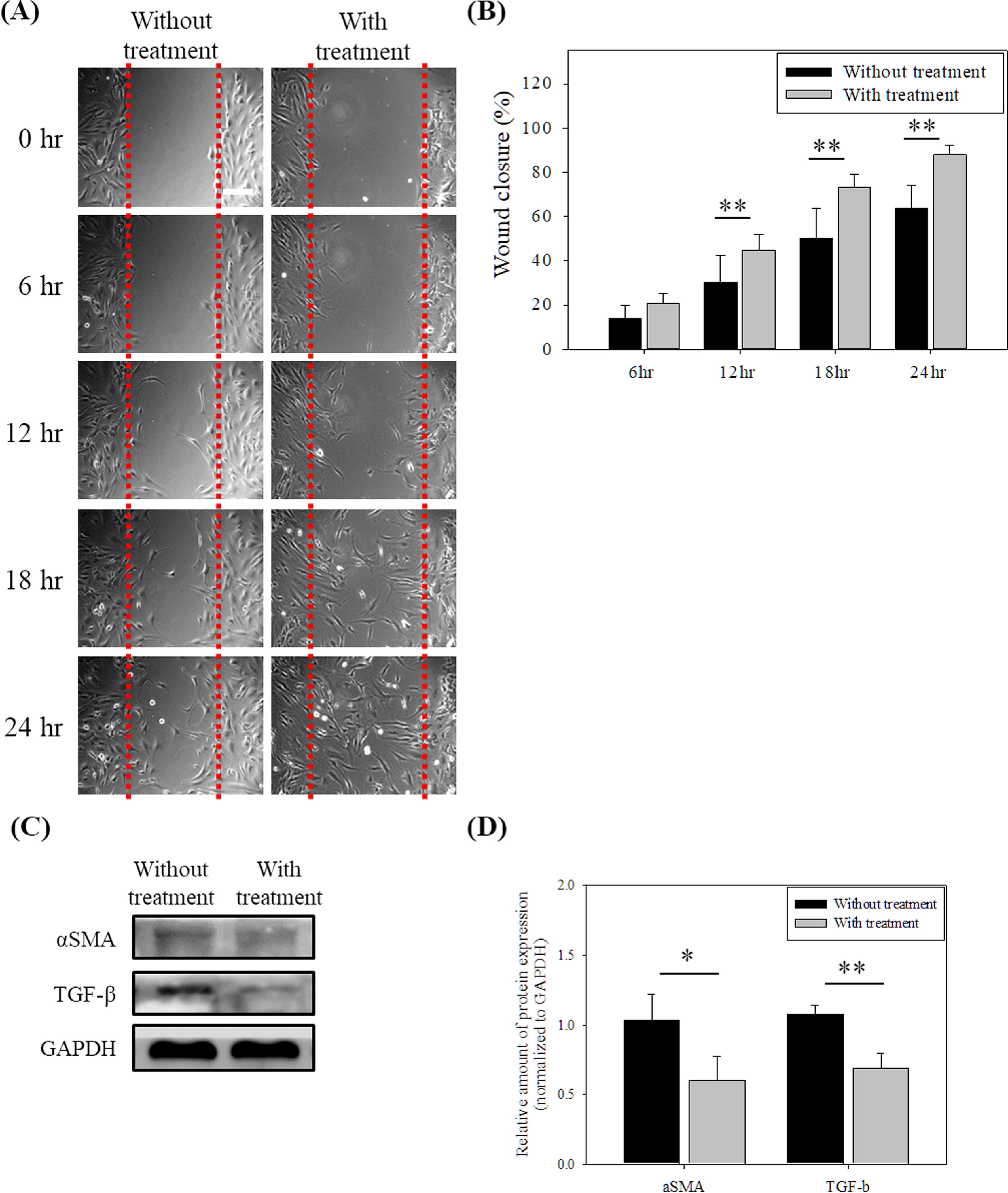
Effect of chitosan treatment on cellular characteristics of ACLs.
In the later stage of wounding healing, excess myofibroblasts would make scarring or fibrosis formation. 44 Myofibroblasts are differentiated from fibroblasts, and they express TGF-β and αSMA protein. Therefore, western blot assay was used to assess these proteins. After chitosan treatment, the TGF-β and αSMA protein expressions of ACLs were decreased compared to untreated ACLs (Fig. 5C–D; p < 0.5 for αSMA and p < 0.01 for TGF-β).
Discussion
We have demonstrated that chitosan decreased the senescence-related phenotypes, and increased the proliferation and migration abilities of human foreskin fibroblasts 35 through the TGF-β pathway 36 previously. In this study, we studied whether the senescence delaying effect of chitosan can be applied to other fibroblasts such as SCs and ACLs for the application of the cell therapy. From the data of the live/dead staining (Fig. 2B), SCs and ACLs were viable when they suspended on chitosan to form spheroids within 3-day culture even though anchorage-dependence is an important feature for normal cells to retain their proliferating ability. 45 Short-term chitosan treatment for SCs and ACLs would not make these cells accelerate apoptosis or necrosis. Chitosan could also significantly decrease the senescent phenotypes (Fig. 3) of SCs and ACLs. In order to evaluate the effectiveness and applicability of chitosan treatment, SCs and ACLs isolated from different donors were treated. Senescent index was identified by SA-β-gal expression of cells and proliferating index was calculated by the absorbance of BrdU assay of cells. Table 1 and Table 2 show that the chitosan treatment was effective in different SC populations and ACL populations isolated from different donors. Chitosan treatment system could be a suitable method for in vitro culture SCs and ACLs, which delayed the senescence and enhanced the proliferation of cells for the cell expansion.
In addition to the senescence and proliferation, the cellular functions of cells were also important for the cell therapy. Synovial membrane is a good source of mesenchymal stem cells applied on the chondrogenesis induction and the cartilage production in vitro 46 from the perspective of cell therapy. Two different types of articular cartilage exist in the knee joint, the hyaline cartilage and the fibrous cartilage. The hyaline cartilage covers the end of the bones, and the fibrous cartilage is mainly distributed in meniscus. 47 The hyaline cartilage is rich in water, GAGs and type II collagen to reduce the friction of bone to bone. 48 The type I collagen of cartilage provides the knee with a high degree of compression resistance.49,50 Compared with untreated SCs, there are more GAGs (Fig. 4A) and type I collagen (Fig. 4B and 4C) in chitosan-treated SCs, which is beneficial for cartilage regenerative engineering. In addition to the effect of chitosan, the structure of the spheroids would be another factor for the chitosan-treated SCs to express more GAGs and collagen. The structure of the spheroids is a critical feature of chondrogenesis differentiation in vitro.51,52
For ligament fibroblasts, the motility is a function when wound healing. Figure 5 indicates the chitosan treatment ACLs migrated faster to the wound area than the without treatment cells. ACLs not only were enhanced proliferation but also possessed better motility after chitosan treatment. On the other hand, the chitosan treatment also lowered the TGF-β and αSMA protein expression (Fig. 5C–D) which are markers of the myofibroblasts.53–55 Myofibroblasts are differentiated from fibroblasts, and they are responsible for wound contraction and extracellular matrix remodeling. In the later stage of wounding healing, it is not ideal to produce excessive myofibroblasts. 44 In this study, the chitosan treatment would avoid the ACL differentiation to form scarring and enhance the mobility of fibroblasts to accelerate the wound closure. These phenomena are beneficial for the future use on cell therapy field.
In summary, chitosan not only delayed the senescence but also enhanced the functions of SCs and ACLs, which is beneficial to the application of chitosan in cell expansion in vitro and cell therapy. However, whether chitosan can be applied to other human primary fibroblasts or directly to in vivo culture to reach a senescence-delaying effect requires further research.
Conclusion
A method to delay cell senescence and enhance proliferation is necessary for the cell expansion in vitro. Chitosan not only delays the cellular senescence but also enhances the proliferation and functions of SCs and ACLs, which benefits for the use of chitosan in the cell therapy field.
Footnotes
Acknowledgments
The authors thank the staff at the Third and Eighth Core Lab, National Taiwan University Hospital, and the staff of the Imaging Core and the Flow Cytometric Analyzing and Sorting Core at the First Core Labs, National Taiwan University College of Medicine, for technical assistance.
Authors’ Contributions
All authors contributed to the study conception and design. Tsai CW: Experiment execution, data analysis (equal) and original draft writing (lead). Chen TY: Experiment execution and data analysis (equal). Wang JH: Methodology (lead), writing review and editing (equal). Young TH: Conceptualization (lead), original draft (supporting); writing review and editing (equal). All authors read and approved the final article.
Author Disclosure Statement
The authors have declared that no competing interests exist.
Funding Information
The authors thank the