
Abstract
Bioscaffolds composed of extracellular matrix (ECM) have been shown to promote a profound transition in macrophages and T-cells from a proinflammatory to a prohealing phenotype with associated site-appropriate and constructive tissue remodeling rather than scar tissue formation. Matrix-bound nanovesicles (MBV) are a distinct class of extracellular vesicles that can be isolated from the ECM and can recapitulate these immunomodulatory effects on myeloid cells in vitro and in vivo, as shown in multiple preclinical models of inflammatory-driven diseases. However, the effect of this MBV-mediated immunomodulation upon the ability to mount an adaptive immune response following pathogenic challenge is unknown. The present study assessed the humoral immune response with and without repeated MBV administration in a mouse model of Streptococcus pneumoniae vaccination and infection. Mice were immunized on day 0, followed by an intraperitoneal MBV or methotrexate (MTRX) injection the next day and weekly thereafter for 5 weeks. Antipneumococcal polysaccharide immuglobulin G and immuglobulin M titers were no different between the vaccine + MBV and the vaccine-only groups, in contrast to the decreased titers in the MTRX-treatment group. Fifty percent of animals treated with MBV were protected from lethal septic infection with S. pneumoniae, and MBV treatment altered the population of immune cells within the lung following sublethal intranasal infection. Macrophages derived from bone marrow mononuclear cells harvested from MBV-treated mice showed persistent immunomodulatory effects following ex vivo challenge with bacterial antigens. The results of this study show that MBV treatment does not compromise the ability to mount an adaptive immune response and suggest that MBV induce sustained immunomodulation in cells of the myeloid lineage.
Impact Statement
The current study shows the immunomodulatory effect of matrix-bound nanovesicles (MBV) on vaccinated mice, while demonstrating their compatibility with the adaptative immune system. Furthermore, results of this study suggest a sustained MBV-mediated immunomodulation on myeloid lineages, which could be used in the development of future vaccines and immunomodulatory therapies.
Introduction
Biological scaffolds composed of mammalian extracellular matrix (ECM) have been used to repair injured tissues in the gastrointestinal tract, 1 musculoskeletal system, 2 cardiovascular,3–5 and integumentary tract,6,7 among others. The constructive tissue remodeling response induced by these biological scaffold materials has been associated with stem/progenitor cell recruitment,8,9 angiogenesis,10,11 the generation of antimicrobial peptides, 12 and perhaps most notably, the modulation of the host immune response from an M1-like, proinflammatory state to an M2-like, proreparative state. 13 The cell-signaling mediators of these favorable wound healing events include embedded cytokines and chemokines within the ECM,14–16 the generation of cryptic peptides from parent molecules within the ECM bioscaffold, 17 and the activity of recently described matrix-bound nanovesicles (MBV), a distinct subtype of extracellular vesicle (EV) embedded within the ECM.18–20 MBV have been shown to downregulate the proinflammatory phenotype of macrophages independent of all other ECM constituents.18,19
Preclinical studies show that MBV reduce symptoms of inflammation after local or systemic delivery in a rat model of pristane-induced rheumatoid arthritis 20 and after H1N1 influenza infection in mice. 21 However, this potent inhibition of inflammation and associated clinical symptoms raises the question of concomitant immunosuppression and increased risk of infection. A study by Allman et al. showed that implantation of decellularized porcine small intestinal submucosa ECM sheets did not alter the ability of Balb/c mice to respond to antigenic stimuli after implantation, 22 but this question has not been investigated following treatment with MBV.
The objective of the present study was to evaluate the effect of systemic delivery of MBV upon the ability of Balb/c mice to mount an antigen-specific humoral response after vaccination and determine the effect of MBV upon bone marrow-derived macrophages (BMDMs) from the same animals with Streptococcus pneumoniae antigen.
Materials and Methods
Study design
The overall study design is depicted in Figure 1. All mice were inoculated with S. pneumoniae vaccine on day 0 and received a weekly dose of MBV or the immunosuppressant methotrexate (MTRX). Antibody titers were determined at weeks 1 and 4 postimmunization. At week 6, animals were inoculated with viable S. pneumoniae bacteria either intraperitoneally (IP) with a lethal dose or intranasally (IN) with a sublethal dose. IP survival was followed for 14 days, whereas IN-inoculated animals were euthanized at day 10 and serum, bone marrow, and tissue specimens collected for further analysis.
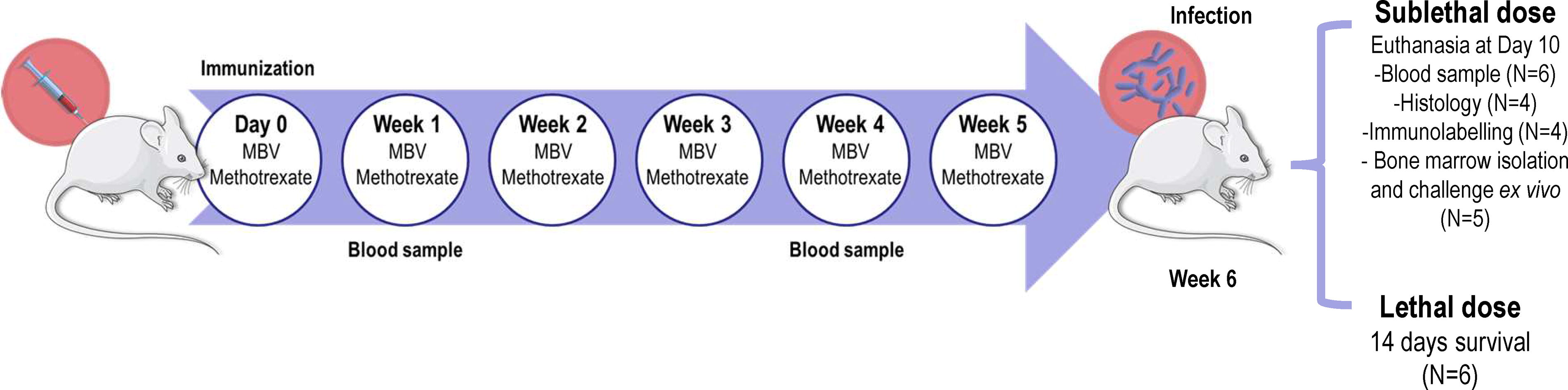
General study design.
Isolation of MBV
The urinary bladders from pigs 6–8 months of age and ∼260 lbs weight were subjected to manual delamination of the urothelium and the submucosa, subjacent muscularis externa, and serosa. The remaining lamina propria and basement membrane were exposed to ethanol/peracetic acid incubation as previously described. 23 Urinary Bladder Matrix was then lyophilized, powdered, and digested by incubation with Liberase TL (highly purified collagenase I and collagenase II, Roche, Switzerland) in 50 mM Tris-HCl buffer overnight at 37°C with continuous rotation. MBV were then isolated by serial centrifugation steps at 500, 2500, and 10,000 g for 15, 15, and 30 min, respectively, and an ultracentrifugation step at 100,000 g for 70 min. As per the International Society for Extracellular Vesicles guidelines,24,25 MBV were characterized by measuring particle size and concentration with a NanoSight NS300 (Malvern Panalytical, US), EV markers were determined using an exosome antibody array (Exo-Check™, System Biosciences, US), EV were imaged with TEM, and RNA and protein levels were measured with a Nano-drop and BCA assay (ThermoFisher, US), respectively.
Immunization and antibody titer measurement
Six- to eight-week-old Balb/c mice (Jackson Laboratories, US) were distributed into four groups (N = 6): (1) control with no vaccine, (2) vaccine, (3) vaccine + 1 µg MTRX (Sigma-Aldrich, US), and (4) vaccine + 1 × 1011 MBV. IL-12 has been reported to augment the ability to mount a robust antibody response 26 and thus 1 µg of murine IL-12 (Biolegend, US) was administered IP on days −1, 0, and 1 in all groups. Mice were vaccinated with human 23-polyvalent S. pneumoniae vaccine (Pneumovax® 23, Merck & Co, US) on day 0. MBV and MTRX were administered IP once weekly beginning at day 0.
Three mice from each group were euthanized on days 7 and 28, serum was collected after cardiac puncture, and titers for immunoglobulin G (IgG) and immunoglobulin M (IgM) anti-S. pneumoniae serotype 3 capsular polysaccharide antibodies were determined by ELISA. Briefly, plates were coated with 100 μg/mL poly-L-lysine (Sigma-Aldrich, US) in phosphate-buffered saline (PBS) at 37°C for 2 h, washed with PBS, and further coated with serotype 3 pneumococcal polysaccharide (PPS) (ATCC, US) overnight at 4°C. Plates were washed with PBS and blocked with 3% bovine serum albumin for 1 h at room temperature (RT) under gentle agitation. Plates were then incubated with serial dilutions of mouse serum for 2 h at RT, washed, and incubated with antimouse IgG or IgM horseradish peroxidase (HRP)-conjugated antibodies (R&D Systems, US, and Abcam, US, respectively) for 1 h at RT. Plates were washed, signal was developed with ELISA TMB substrate (ThermoFisher Scientific, US), and absorbance was measured at 450 nm with a plate reader. Positive titers were determined by a signal twofold greater than the background signal.
Challenge inoculation protocol
S. pneumoniae (A66.1 strain, Public Health England, UK) was grown in Todd Hewitt Broth (THB, Sigma Aldrich, US) with 1% yeast extract (Sigma Aldrich, US) until bacteria reached logarithmic growth. The determination of colony-forming units (CFUs) per optical density (OD) reading was established by generating a standard curve for OD versus CFU counts in blood agar plates (ThermoFisher Scientific, US) after serial dilution plating. The CFU administered to animals were confirmed by blood agar plate CFU counts after serial dilutions plated immediately after inoculation, which also confirmed the viability of the bacteria.
Mice were randomly assigned to one of the following groups (N = 12): (1) control with no vaccine, (2) vaccine, (3) vaccine + 1 µg MTRX, and (4) vaccine + 1 × 1011 MBV. After the immunization, MTRX or MBV was administered for 5 weeks as per the protocol described above. Animals were inoculated at week 6 with a lethal IP dose exposure (N = 6) or a sublethal intranasal dose (N = 6) in separate experiments. For the lethal exposure, mice were injected with 1011 CFU of S. pneumoniae in sterile PBS, and animal survival was recorded for 14 days. For sublethal exposure, 1 × 107 CFU of S. pneumoniae were administered IN at week 6 postimmunization. After 10 days, serum was collected at the time of sacrifice by cardiac puncture and titers were measured for IgG and IgM anti-S. pneumoniae serotype 3 capsular polysaccharide antibodies with ELISA as described above. Lungs were collected for histological analysis and femurs were collected for BMDM isolation.
Histology and immunolabelling
Lung tissue from sublethal exposure IN inoculated mice was collected and fixed in buffered 10% formalin (ThermoFisher, US) for 1 week. Tissue sections were embedded in paraffin and cut into 5 µm sections and stained with hematoxylin and eosin and Picrosirius red. Quantification of cellularity and cell spatial distribution clustering were determined for the lung sections with QuPath® software. Collagen deposition was evaluated in Picrosirius red-stained sections by imaging under polarized light with a Zeiss Axio Observer Z1 (Zeiss, US) microscope. Five random fields of view of each section were imaged and measured with ImageJ software for deposition of mature and immature collagen by splitting color channels and calculating their area (green and orange = immature collagen, red = mature collagen).
For immunolabelling, following deparaffinization and rehydration in xylene and decreasing ethanol dilutions, sections were subjected to antigen retrieval in 10 mM citric acid (pH 6) for 1 min, and endogenous peroxidase activity was quenched by incubating sections with 3% H2O2 in PBS for 30 min at RT. After rinsing in PBS, sections were blocked with 5% normal goat serum (NGS) + 0.1% Triton-X100 in PBS for 1 h at RT and in primary antibody (anti-CD11b ab128797, Abcam, US; anti-CD4 bs-0766R and anti-CD8 bs-4791r, Bioss Antibodies, US) solution diluted 1:50 in blocking buffer for 1 h at RT. Sections were then washed three times with PBS, incubated with HRP-conjugated secondary antibodies (antirabbit HRP-conjugated, Dako, US) diluted 1:200 in blocking buffer for 45 min at RT, washed three more times in PBS, and developed with VECTASTAIN® DAB substrate (Vector Laboratories, US). Signal development was stopped by immersing slides in PBS three times. Sections were counterstained with hematoxylin, dehydrated in increasing ethanol solutions, cleared in xylene, and coverslipped. Finally, images were obtained with a slide scanner (Motic ScanPro6, Motic, US), and full sections were analyzed with QuPath Software to determine the percentage of cells positive for CD11b, CD4, and CD8, and the CD4/CD8 ratio.
BMDM isolation and ex vivo inflammatory challenge
BMDM derived from the different groups inoculated IN (control; vaccine; vaccine + 1 µg MTRX; vaccine + 1 × 1011 MBV) were isolated at day 10 after inoculation. Briefly, using aseptic technique, the skin from the proximal hind limb to the tarsus was removed, the coxofemoral joint was disarticulated, and muscle was excised for isolation of the femur. Harvested femurs were kept on ice and rinsed in a sterile dish containing macrophage complete medium, which consisted of DMEM (Gibco, US), 10% fetal bovine serum, 10% L929 fibroblast supernatant, 0.1% beta-mercaptoethanol (Sigma-Aldrich, US), 1% penicillin/streptomycin, 10 mM wnon-essential amino acids (Gibco, US), and 10 mM HEPES buffer. Using aseptic technique, the ends of each femur were transected with sterile scissors and the marrow cavity flushed with complete macrophage medium using a 30G needle. Collected cells were washed and plated at 106 cell/mL and allowed to differentiate into macrophages for 7 days at 37°C and 5% CO2, changing medium every 48 h. At day 7, BMDM from each group were treated with either normal medium, Escherichia coli lipopolysaccharide (LPS) at 100 ng/mL or PPS at 10 μg/mL for 24 h at 37°C and 5% CO2. Medium was collected for cytokine analysis, and RNA was isolated with Trizol® (Fisher Scientific, US) for gene expression assessment by RT-qPCR, or cells were fixed in 4% paraformaldehyde (Sigma Aldrich, US) for 20 min at RT for further immunocytochemistry analysis.
For immunolabelling studies, fixed cells were washed three times in PBS and blocked with 5% NGS + 0.1% Triton-X100 in PBS for 1 h at RT, incubated with primary antibodies (anti-F4/80 NB600-404, Novus Biologics, US; anti-CD11b ab128797, Abcam, US) diluted 1:100 in blocking buffer. Following three washes with PBS, fixed cells were incubated with secondary antibodies (Alexa Fluor® 488 goat antirabbit and Alexa Fluor® 594 goat antimouse, Invitrogen, US) diluted 1:200 in blocking buffer, washed three further times in PBS, incubated with DAPI for 5 min for nuclei staining, and washed once more in PBS. Immunolabeled cells were imaged with a Zeiss Axio Observer Z1 (Zeiss, US) microscope, and images were quantified for the fluorescence intensity of each marker per cell number ratio.
Cytokines released into the medium were identified and quantified by ELISA according to the manufacturer’s protocols (IFNγ, TNF-α, and IL-6 Biogelend, US; IL-23 R&D Systems, US), while RNA was isolated with Trizol® RNA isolation protocol, reverse transcribed to cDNA with high capacity cDNA reverse transcription kit (Fisher Scientific, US), and mRNA expression measured by TaqMan® RT-qPCR using primers for TNF-α (Mm00443258_m1), IL-6 (Mm00446190_m1), IL-23 (Mm00518984_m1), IFNV (Mm01168134_m1), MR1 (Mm00468487_m1), CD74 (Mm00658576_m1), CSF2 (Mm00436450_m1), and CXCL2 (Mm00468487_m1) primers, with HPRT (Mm03024075_m1) as housekeeping primer (all primers were purchased from ThermoFisher, US).
Statistical analysis
Statistical analysis was performed with GraphPad Prism 9 (GraphPad Holdings LLC, California, US). Normal distribution of data was assessed using the Shapiro–Wilk test. One-way ANOVA and Fisher’s least significant difference post hoc analysis were used to assess statistical significance for normally distributed data, which was assumed when p < 0.05. Kruskal–Wallis and Dunn’s post hoc nonparametric tests were used for non-normally distributed data. For survival analysis, a simple survival Kaplan–Meier estimate was performed.
Results
MBV do not compromise humoral immunity
The isolation of MBV yielded a homogeneous population of vesicles ranging from 100 to 200 nm in size, as also observed by TEM. In addition, MBV showcased EV markers and contained approximately 15 µg of protein and 2 µg of RNA per 1011 particles (Supplementary Fig. S1). On days 7 and 28, blood was collected, and specific antiserotype 3 anti-PPS IgG and IgM titers were quantified. At day 7, IgG and IgM titers were significantly higher in the vaccine (p < 0.05) and vaccine + MBV groups (p < 0.05) compared with the sham group (Fig. 2A and B). At day 28, IgG titers (Fig. 2C) in all groups were slightly higher compared with sham (p < 0.01, p < 0.01 and p < 0.05 for vaccine, vaccine + MTRX, and vaccine + MBV, respectively), while IgM titers (Fig. 2D) showed no significant differences (p > 0.05). Overall, antibody production in animals treated with vaccine + MBV was similar to animals treated with vaccine alone, demonstrating that MBV did not compromise the ability of mice to produce antigen-specific antibodies after immunization.
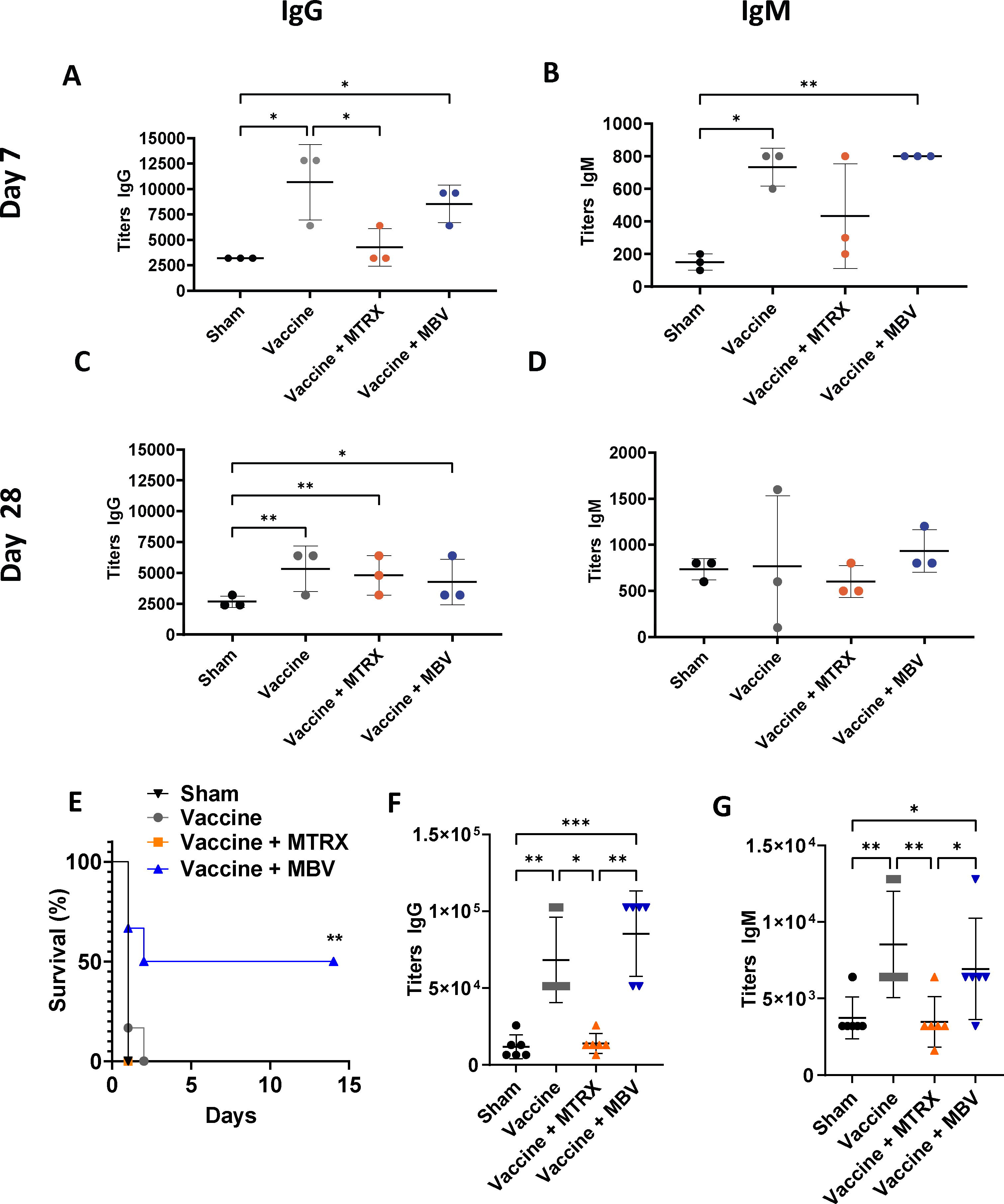
MBV treatment does not compromise the humoral immune response and increases survival of immunized mice after lethal dose inoculation. IgG
The studies to evaluate the protective effects of acquired immunity against S. pneumoniae following vaccination showed that all animals succumbed to sepsis 2 days after IP inoculation with a lethal dose of bacteria except for 50% (n = 3/6) of mice in the vaccine + MBV group (Fig. 2E; p < 0.01). These results suggest that treatment with MBV may augment the cellular immune response above that of vaccination alone.
Mice inoculated IN with a sublethal dose of S. pneumoniae (1 × 107 CFU) showed only mild symptoms after 10 days, matching previous studies that reported high survival of Balb/c mice after IN inoculation with S. pneumoniae. 27 Blood titers of antipneumococcal-specific IgG were higher in the vaccine and vaccine + MBV groups (Fig. 2F) when compared with the sham and MTRX groups (p 0.01), whereas no differences were observed between the sham and vaccine + MTRX groups (p > 0.05). IgM levels in the vaccine group and the vaccine + MBV group were higher than the sham and vaccine + MTRX groups (Fig. 2G; p < 0.05). These results further show that MBV treatment did not compromise the ability of immunized mice to generate a specific humoral immune response.
MBV have an immunomodulatory effect after infection in immunized mice
Histological evaluation of lung biopsies from the 10-day IN-inoculated mice showed extensive cellular infiltration of the alveoli (Fig. 3A). Quantification revealed no significant differences in total cellularity between infected groups (Supplementary Fig. S2), and likewise no differences in clustering (Fig. 3B). These results indicate local inflammation of the lungs in infected mice, matching previous reports. 28 Picrosirius red staining of lung sections revealed lower percent areas of mature collagen deposition around bronchi in the vaccine + MBV group compared with sham (Fig. 3C), which was confirmed after quantification of area of mature collagen fibrils by polarized light microscopy (p < 0.01, Fig. 3D). A similar decrease in lung fibrosis in the MBV-treated groups after pathogenic infection has been previously reported in H1N1-infected mice. 21 Since fibrosis is closely tied to the effect of macrophages on tissue-resident fibroblasts, the findings of the present work together with previous studies suggest a change in the phenotype of innate immune cells following systemic MBV treatment.20,21,29–31

Histopathological analysis revealed lower collagen deposition in MBV-treated mice after infection. Lung sections from intranasally infected mice
The different immune cell subpopulations most relevant in pulmonary inflammation (i.e., CD11b+ myeloid cells, CD4+ and CD8+ T-cells) were determined by immunolabelling (Fig. 4A) of lung sections and quantified with a machine learning-based classification model in QuPath after immunolabeling. No differences in percentage of CD11b+ cells were observed (Fig. 4B). However, the percentage of CD4+ cells was significantly lower in both the vaccine + MBV and vaccine + MTRX groups when compared with the vaccine-only group (Fig. 4C; p < 0.05). There were no significant differences in the percentage of CD8+ cells between any groups (Fig. 4D). The CD4+/CD8+ ratio was higher in the vaccine-only group compared with the sham (p < 0.01) and vaccine + MTRX groups (p < 0.05), and not significantly different from the vaccine + MBV group (Fig. 4E). These results demonstrate that systemic MBV treatment during immunization modulated the immune response, resulting in a lower recruitment of CD4+ cells in the locally infected area, decreasing the acute inflammatory response without compromising the adaptive immune response as previously assessed.
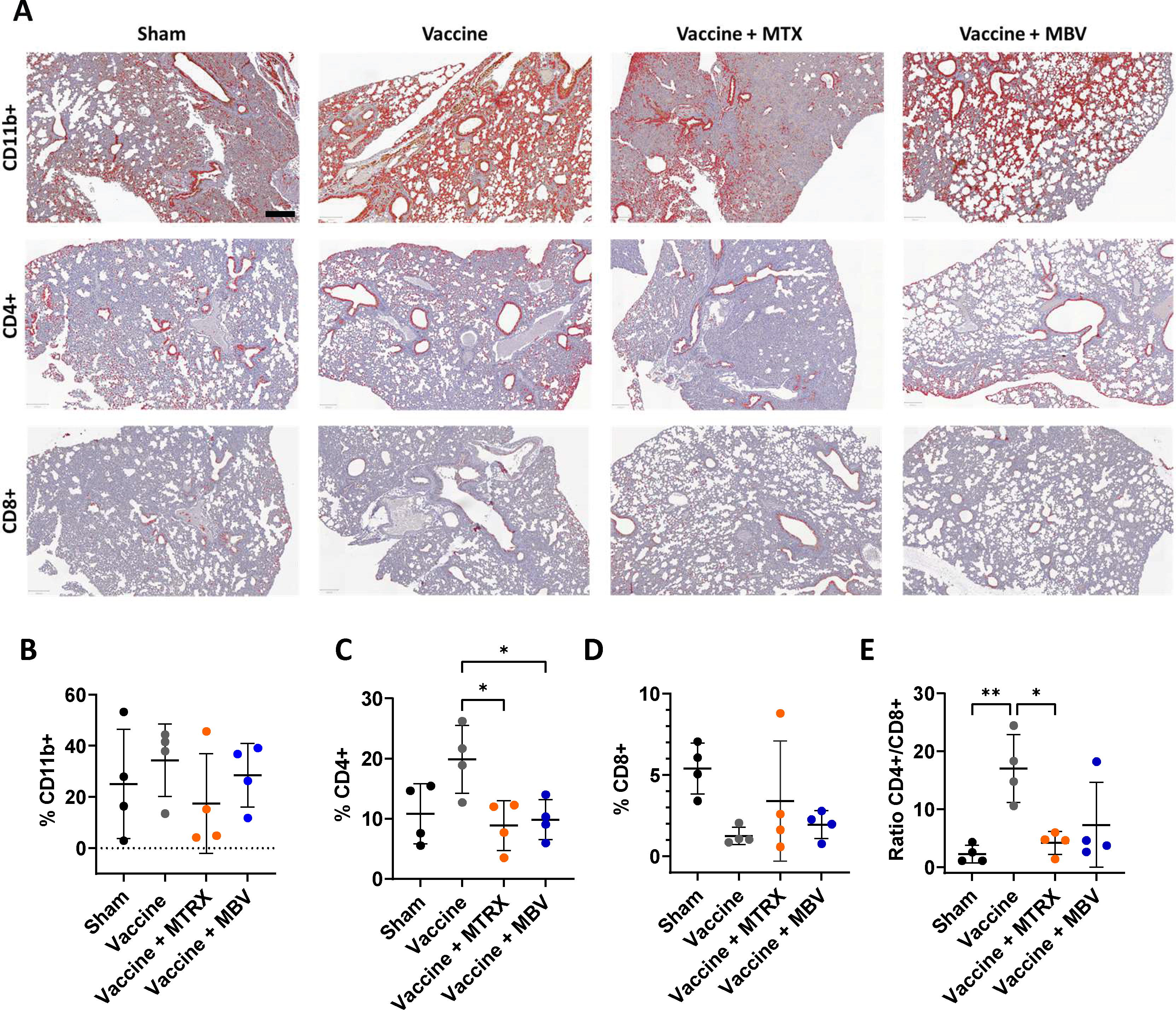
MBV treatment during immunization modulates immune cell infiltration after infection. Immunolabeling and machine learning-based classification (red) of CD11b+ (myeloid cells), CD4+ (T-cells), and CD8+ (cytotoxic cells) in infected mice lungs sections
MBV modulate myeloid progenitor cell phenotypes without compromising specific immune memory
To determine if the immunomodulatory effects observed in vivo were associated with phenotypic changes in cells of the myeloid lineage, bone marrow cells were isolated from the femurs of the vaccinated and bacterially infected mice at 10 days after IN infection. Bone marrow mononuclear cells were cultured in vitro in macrophage maturation medium for 7 days, followed by challenge with either LPS from E. coli or serotype 3 PPS. BMDM were positive for the pan-macrophage marker F4/80 as expected (Fig. 5A, red channel), with quantification showing no differences in fluorescence intensity between groups after treatment with LPS or PPS (Fig. 5B). Treatment with PPS induced an increase in expression of the phagocytic marker CD11b similarly across all groups (Fig. 5A, green channel, and C).
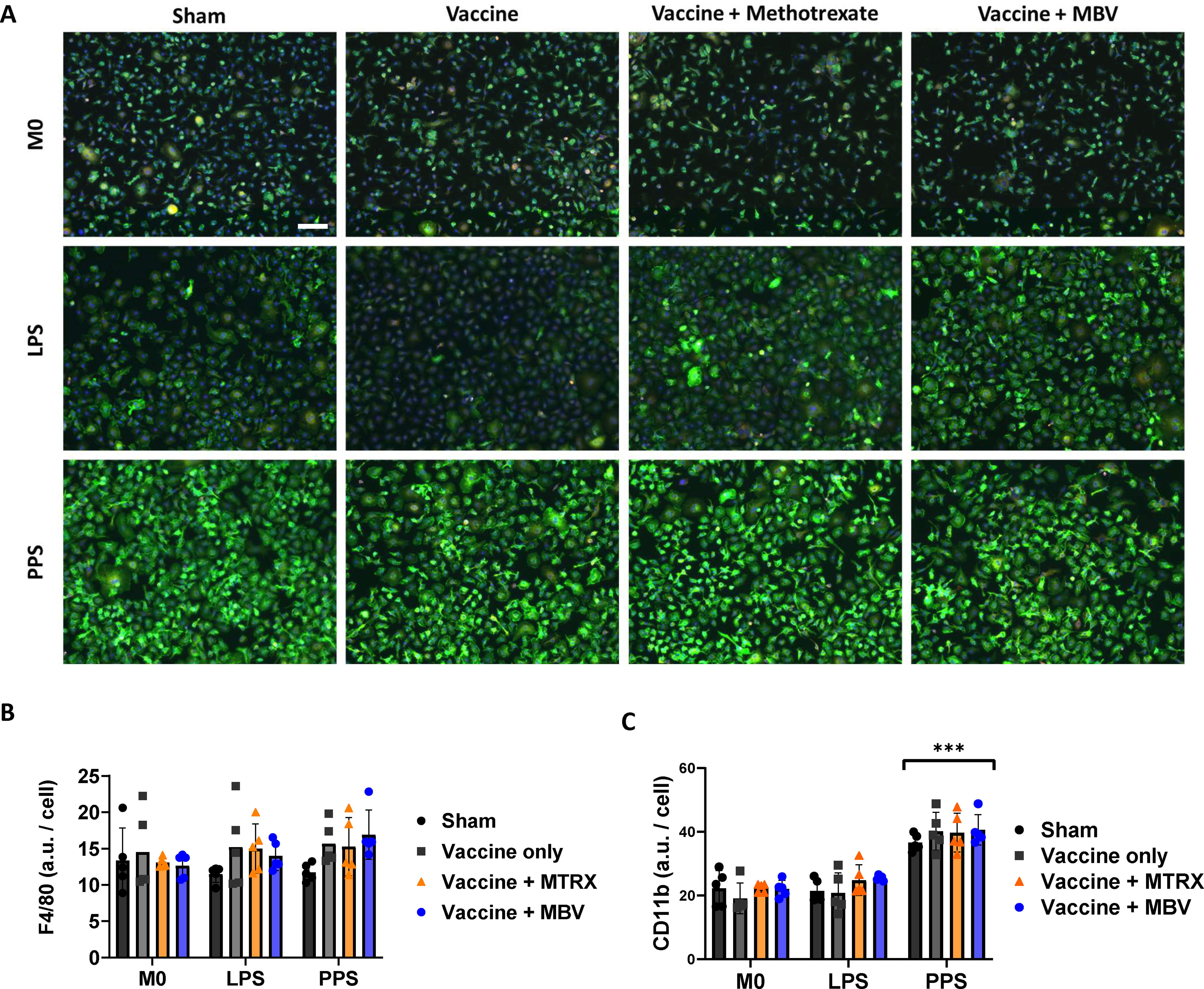
Macrophages isolated from MBV-treated mice maintain innate immune responsiveness to pathogens. Immunolabelling of BMDM from immunized and infected mice showed that cells were positive for the pan macrophage marker F4/80 (red) and the myeloid marker CD11b (green), scale bar (valid for all images) 50 µm
The production of multiple inflammatory cytokines in LPS- or PPS-challenged BMDM was quantified by ELISA and RT-qPCR. Secretion of TNF-α did not differ among groups after treatment with either PPS or LPS (Fig. 6A), whereas IL-6 secretion was significantly increased in the vaccine group compared with sham (Fig. 6B; p < 0.05) when macrophages were exposed to LPS.
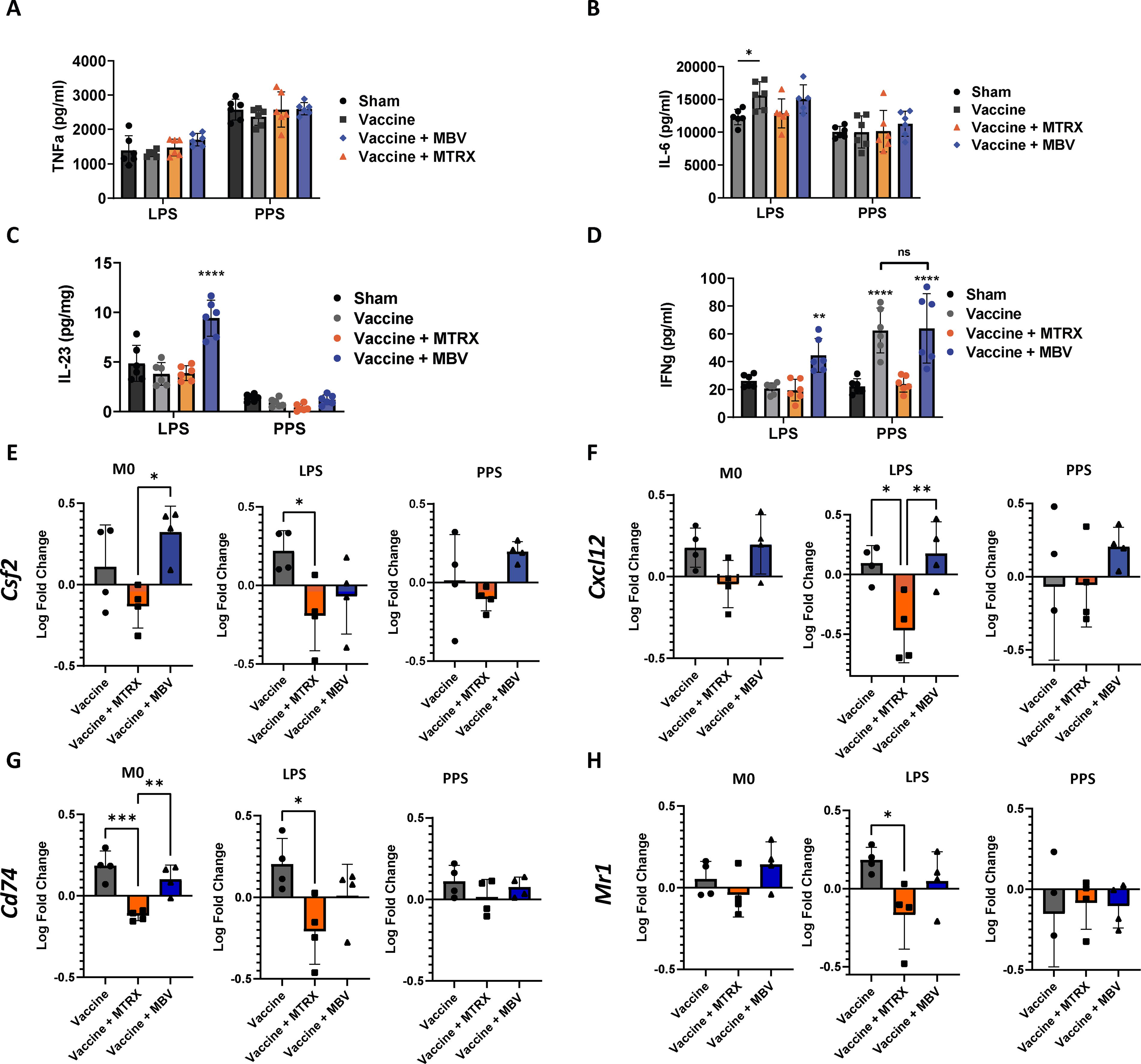
MBV treatment results in long-term modulation of innate memory of BMDM. Ten days after IN infection with 1 × 107 CFU S. pneumoniae, bone marrow from infected mice was isolated and mononuclear cells were differentiated ex vivo to macrophages over 7 days, followed by challenge with pneumococcal polysaccharide (of the same serotype as the infecting strain, PPS) or lipopolysaccharide from E. coli (LPS, a previously unencountered antigen). Secretion of TNF-α
Interestingly, IL-23 (Fig. 6C) and IFNγ (Fig. 6D), two cytokines involved in the response to pathogens in vivo, showed a pattern of regulation in the MBV-treated group that was different from the other groups. BMDM from mice exposed to MBV in vivo showed increased secretion of IL-23 (p < 0.0001) and IFNγ (p < 0.05 compared with sham and p < 0.001 compared with the vaccine-only and MTRX groups) after in vitro challenge with LPS, an antigen to which they had no prior exposure. In response to PPS challenge, however, IFNγ production was higher in both the vaccine-only and vaccine + MBV groups (p < 0.0001). The modulation of IFNγ was confirmed at the transcript level by RT-qPCR (Supplementary Fig. S3); however, the mRNA and protein expression did not correlate for IL-23 (Supplementary Fig.S3), which suggests additional layers of regulation of these pathways. These results show that exposure to MBV in vivo triggered phenotypic changes in myeloid progenitor cells which persisted after differentiation to macrophages, and conditioned the response to antigen challenge ex vivo. Furthermore, these changes resulted in a stronger response to pathogens (with and without previous exposure) than those observed in the vaccine-only group and could indicate a synergistic effect with vaccination.
Other relevant markers involved in immune cell chemoattraction and antigen presentation, including granulocyte colony formation stimulating factor (csf2), chemokine cxcl12, the major histocompatibility complex (MHC) type II marker cd74, and the MHC type I marker mr1 (Fig. 6E, F, G, and H, respectively), were unaffected by MBV treatment when compared with the vaccine-only group. On the contrary, treatment with MTRX lowered the expression of csf2 (p < 0.05 vs. the MBV group in M0 and LPS conditions; Fig. 6E), cxcl2 (p < 0.05 and p < 0.01 vs. the vaccine-only and MBV groups, respectively, in LPS condition, Fig. 6F), cd74 (p < 0.01 and p < 0.001 vs. vaccine-only and MBV in M0 condition, p < 0.05 vs. vaccine-only in LPS condition, Fig. 6G), and mr1 (p < 0.05 vs. vaccine-only in M0 condition, Fig. 6H). No difference in any of these markers was found between groups for the PPS condition. Overall, these results further confirm that MBV did not affect the ability of the innate immune myeloid cells to initiate an adaptive immune response.
Discussion
The results of the present study show that the systemic administration of MBV has no detectable inhibition upon the ability to mount an antigen-specific adaptive immune response following vaccination with S. pneumoniae. Furthermore, the immunomodulatory effects of MBV appear to affect macrophage progenitor cells allowing for prolonged in vivo activity. Previous studies have shown that MBV have potent in vivo anti-inflammatory effects in a rat model of pristane-induced rheumatoid arthritis 20 and murine model of H1N1 influenza. 21 The promotion of transition from a proinflammatory M1-like to an anti-inflammatory M2-like macrophage phenotype following exposure to bioscaffolds composed of acellular ECM has been shown in both in vitro and in vivo studies with associated favorable tissue remodeling outcomes.32–34 It has also been shown that MBV that represent a single component of such ECM-based scaffolds can recapitulate these favorable immunomodulatory effects.18,29 The potential clinical implications of an anti-inflammatory immunomodulatory therapeutic that has no associated detectable immunosuppression is significant.
The innate immune system plays an essential role in the initial stages of the adaptive immune response. Antigen-presenting cells (APCs) such as neutrophils, dendritic cells, and macrophages are responsible for phagocytic uptake and lysis of pathogens and presentation of their antigens to B lymphocytes, stimulating production of specific antibodies against these pathogens. These events establish an immunological memory that facilitates a rapid and targeted response to future exposures. Such protective and clearly beneficial processes are associated with the classical signs and symptoms of inflammation mediated primarily by cells responsible for the innate immunity, especially APC.35,36 Therefore, anti-inflammatory immunosuppressive therapeutics such as MTRX in the present study can have the undesirable side effect of mitigating an otherwise protective humoral immune response. 36
The therapeutic use of ECM-based biomaterials has been shown to reduce proinflammatory activities and to promote a pro-remodeling or “M2-like” phenotype in innate immune cells, such as macrophages,34,37 and subsequently to promote a Th2 and Treg phenotype in T cells.38,39 Similarly, recent studies show that MBV 40 can reproduce these immunomodulatory effects. Furthermore, these effects persist for >70 days following systemic delivery of MBV. 20 It is plausible therefore that MBV could impair events necessary for the initiation of the adaptive immune response. The results of the present study, however, show that adaptive immunity remains intact and functional following a therapeutic dose of MBV. IgG and IgM titers after vaccination alone or vaccination followed by exposure to S. pneumoniae were unaffected by MBV in contrast to MTRX that markedly impaired the humoral response to S. pneumoniae at day 7. Recovery of antibody production was observed at day 28 as the effects of MTRX gradually diminished. The results were further supported by the contrasting histopathology and survival outcomes between the MTRX- and MBV-treatment groups.
The ability of MBV to mitigate the number and phenotype of inflammatory cells following intranasal exposure to H1N1 virus, similar to that observed in the present study, has been recently shown. 21 MBV treatment was associated with a decreased accumulation of the proinflammatory cytokines IFNγ, IL-6, IL1b, GCSF, and CXCL1, which in turn was associated with decreased pulmonary fibrosis. 41 In the present study, the CD4+ cell population in lungs was lower in the MTRX and MBV groups compared with the vaccination-only group. While CD4+ T cells are not required for protection against S. pneumoniae with 42 and without 43 vaccination. CD8+ cells showed no differences between groups in the present study and are required for protection against S. pneumoniae. ECM-based biomaterials, which all contain MBV as an integral component, have been associated with the promotion of a Th2 phenotype in CD4+ cells22,38,39 and the downregulation of inflammatory cytokines in myeloid cells. Such a decrease in inflammatory cytokines would logically reduce T cell recruitment, 44 thus leading to the lower percentage of CD4+ cells in lungs of mice from the MTRX and MBV groups; however, this mitigation of inflammatory cells occurred without immunosuppression in the MBV-treated group.
The 50% survival (3/6) of mice that received both vaccination and MBV followed by a lethal dose exposure to S. pneumoniae, in contrast to all other groups that showed 100% mortality (6/6), suggests an adjuvant effect of MBV. Of note, only BMDM isolated from the vaccination + MBV group showed an increased production of IFNγ and IL-23 in response to inflammatory challenges with either PPS or LPS. This finding is relevant because IFNγ is an activator of CD8+ cells and clearance of pneumococcal infections, 42 and monocyte-mediated activation of Th1 and Th17 T cells via IL-23 is crucial to an effective host response to S. pneumoniae.45,46 The fact that this cellular response occurred more than 2 weeks after the last administration of MBV in vivo, long after the lifespan of circulating monocytes and their differentiated macrophages,47,48 suggests a mechanism by which not only circulating macrophages are affected but also their progenitor cells.
The present study showed that the immunomodulatory effects of MBV treatment do not impede the ability to mount a humoral response and result in sustained modulation of innate immune cells. There are limitations to the present study that should be considered. The study was conducted with a single pathogen, S. pneumoniae, and the effects with other pathogens such as gram-negative bacteria and viruses would require dedicated singular studies. In addition, expansion of the present work to other innate APC such as dendritic cells and neutrophils would clearly add to further understanding the effect of MBV on the innate immune system. The biodistribution of MBV following systemic delivery, the specific cell types that are most responsive to MBV, and the transcriptional and proteomic effects of MBV upon innate immune cells are also of great interest but beyond the scope of the present study. However, considering the potential therapeutic effects of MBV, such studies are worthy of further investigation.
Conclusions
The present study shows that MBV treatment does not compromise the ability of the immune system to mount a protective humoral response after vaccination against S. pneumoniae and suggests a persistent modulation of innate immunity cells that should be investigated in future studies.
Footnotes
Acknowledgments
The authors would like to acknowledge the contributing assistance of the University of Pittsburgh Division of Laboratory Animal Research staff for animal care and support.
Authors’ Contributions
H.C.-M., R.J.C., and S.F.B. conceived the study, whereas H.C.-M. materialized the experimental design. H.C.-M. and R.J.C. carried out the animal work, and H.C.-M. performed the data analysis. H.C.-M. wrote the article. R.J.C., W.D.A., G.S.H., and S.F.B. edited the text.
Disclosure Statement
H.C.-M. is employed by Viscus Biologics LLC. G.S.H. is VP at ECM Therapeutics. S.F.B. is cofounder and CSO at ECM Therapeutics. R.J.C. and W.D.A. declare no interests.
Funding Information
The present study did not receive funding from any third party nor organizations.