
Abstract
Tissue engineering—part of regenerative medicine—is a promising technology that could potentially offer elegant solutions to urogenital defects, but so far, it has fallen short of its potential. Within experimental studies for bladder and urethra reconstructions, two clinical applications have been described, but extension of these techniques to the broader urological patient population has not happened so far. In this article, we aim to identify the ethical road blocks in the clinical evaluation of tissue-engineered products under the European Medicines Agency and Food and Drug Administration regulations for pediatric urological conditions and, ultimately, to recommend strategies to overcome them. The use of human tissue-engineered products (HTEPs) to treat children with congenital urogenital defects poses challenges in the clinical testing phase, connected to three features of the application of this treatment in this patient group: (1) those associated with the product, namely, the multifaceted complexity of the HTEP; (2) those connected to the procedure, namely, the lack of a randomized controlled trial (RCT)-tested gold standard to compare the new treatment to and difficulties surrounding standardization of the treatment protocol; and (3) the patient's young age and associated problems concerning possible long-term effects and the informed consent process. Due to these problems, a conventional RCT is not the methodology of choice to evaluate this treatment in this patient group. The unpredictability of HTEPs necessitates stringent and long-term surveillance and registry to ensure the safety of patients treated with these products.
Introduction
Tissue engineering is a promising technology that could potentially offer elegant solutions to structural defects. However, so far, the practical applications of the technology have fallen short of its potential. Currently, few clinical conditions are being addressed by tissue engineering, and commercial success has been difficult to attain.3,4 One of the clinical areas that has seen some promising clinical developments is urology.5–8
The EuroSTEC project (Soft tissue engineering for congenital birth defects in children: from biomatrix–cell interaction–model system to clinical trials), an integrated project funded by the European Commission under the Sixth Framework Program was aimed at the development of treatments for structural disorders present at birth.9,10 Two of EuroSTEC's target areas were the bladder and the urethra. In conditions affecting those areas, the success of conventional surgical reconstructions is often diminished by a shortage of native urothelium. Therefore, gastrointestinal tissues are currently used for reconstruction. However, these tissues are associated with complications, such as infection, metabolic disturbances, urolithiasis, perforation, increased mucus production, and malignancy.8,11–12
These complications have stimulated tissue engineering research into the creation of implantable bladder and urethral substitutes. There is a fair amount of animal studies examining tissue-engineered constructs for congenital urogenital defects,13,14 but as of yet, few clinical studies. The clinical studies that were conducted resulted in either an unsatisfactory outcome as compared to conventional treatment, or were halted because of a significant amount of adverse events.7,8,15,16 Therefore, we think that the development of HTEPs for the treatment of urogenital defects can serve as an example. Through surveying the problems that were encountered in the development process of these clinical products, we will highlight the challenges to clinical testing of tissue-engineered products in general.
Objective of the current article is to identify the ethical challenges in clinical testing of tissue-engineered products for pediatric urological conditions. To this purpose, we will first survey the regulatory environment created for HTEPs. Subsequently, we will look at the specific challenges associated with clinical testing of HTEPs for this clinical application, and ultimately, recommend strategies to overcome the identified challenges.
The Regulatory Environment of HTEPs
In the past, medical products were regulated as either medicinal products or medical devices. Tissue-engineered products fell between these two categories: in some ways, an HTEP is an active implantable medical device, but there is also a significant role for pharmaceuticals. 5 Additionally, an HTEP may contain viable cells. 17 In addition to tissue-engineered products, other cell therapy medicinal products fell between the existing categories as well. This prompted the European Union to create a new class of medical products: Advanced Therapy Medicinal Products or ATMPs (see Fig. 1).
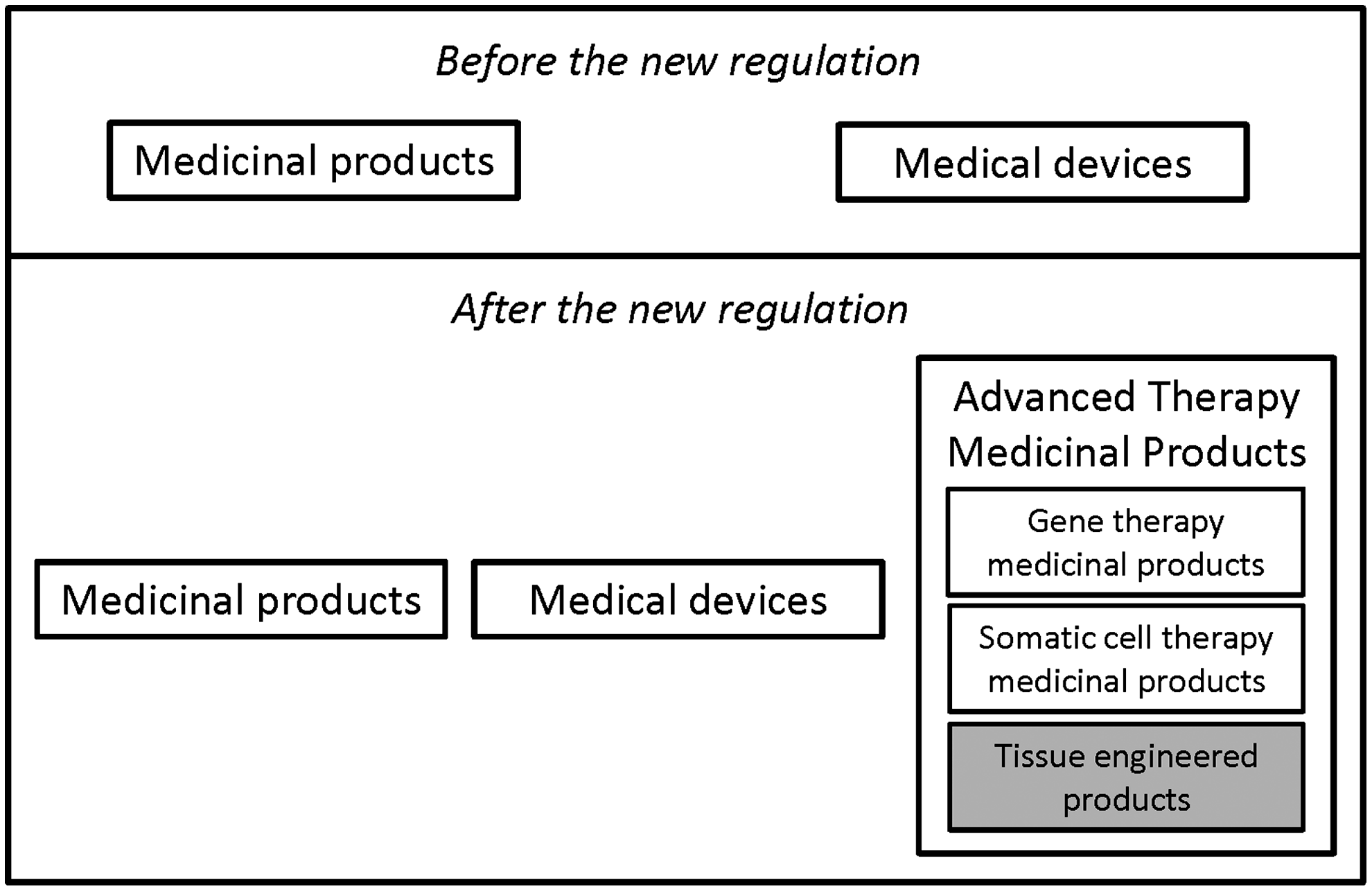
Before and after the new regulation.
An ATMP is either (1) a gene therapy medicinal product, (2) a somatic cell therapy medicinal product, or (3) a tissue-engineered product (see Table 1). 18 Regulation No 1394/2007 requires those planning to market an ATMP within the European Union to seek authorization from the European Medicines Agency (EMA).
In the United States, articles containing or consisting of human cells or tissues intended for transplantation, implantation, infusion, or transfer to a human recipient are regulated by the Food and Drug Administration (FDA) as human cells, tissues, and cellular and tissue-based products (or HCT/Ps).19–22 The level of regulation depends on the characteristics of the product (see Fig. 2), but good tissue practices (or GTPs) are required regardless of the specific characteristics. GTPs describe the methods, facilities, and controls used in the manufacturing of HTEPs to prevent the infectious disease transmission and cross-contamination.19–22
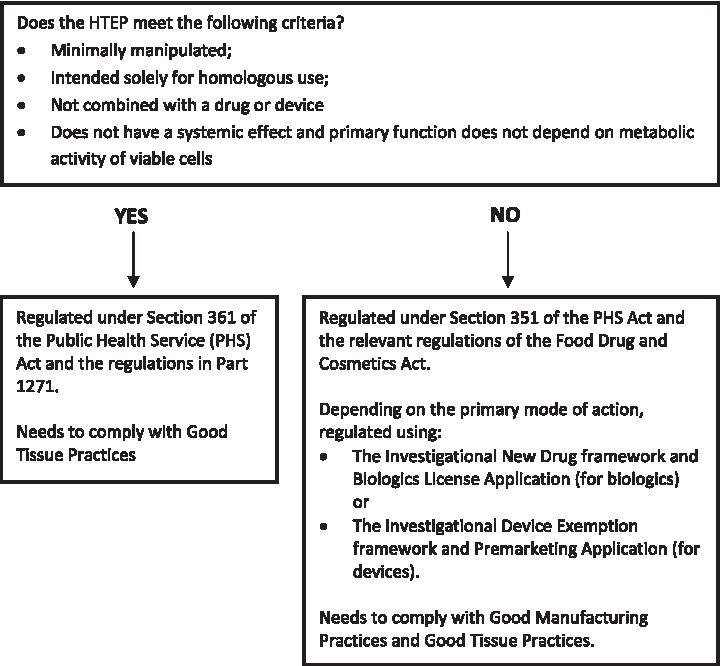
Criteria to determine level of Food and Drug Administration regulation for human tissue-engineered products.22,23
From preclinical to clinical practice
Before any HTEP can be considered for market authorization, information needs to be collected about its quality, as well as about its behavior in animals and, ultimately, humans. A common step in the process of proceeding to clinical trials are animal studies. In tissue engineering, there is some debate on the adequacy of animals as models for the human situation.23–25 It appears to be difficult to gather information about regenerative activity and safety of HTEPs in humans from animal trials. 26
For clinical research to be justified, three conditions need to be met: (1) minimization of potential risks to the trial participant, (2) enhancement of the potential benefits to individual subjects, and (3) the potential benefits to individual subjects and society are proportionate to or outweigh the risks.27,28 Additionally, for a clinical trial of an HTEP to be successful, it should not only confirm therapeutic value, but also provide insight in regenerative activity, safety, and long-term effects of the HTEP.
The risks that are anticipated (see Table 2) need to be weighed against the potential benefits, and against the risk–benefit ratio of existing therapies. Because the process of tissue engineering is complex, risk assessment is of vital importance and should be rigorous.
HTEP, human tissue-engineered products.
The last step in proceeding from clinical research to clinical practice is market authorization. HTEPs are expected to show greater variability in composition and performance as compared to traditional medicinal products or medical devices, which has consequences for, among other things, labeling and product descriptions. The risk potential varies between different types of HTEPs, which has led both the EMA and the FDA to install the so-called risk-based approach in the market authorization process. This approach is used to determine the amount of data needed in the market authorization application, and is based on the identification of risk factors associated with the quality, safety, and efficacy inherent to the nature of the HTEP in question.20,21
Challenges to Clinical Testing
A clinical application of an HTEP that was researched in the EuroSTEC project is the use of tissue-engineered constructs in children with congenital urological defects. The use of HTEPs to treat children with these conditions creates an ethically complex situation because of a combination of factors, which have to do with (1) the product (an HTEP), (2) the procedure (a surgical intervention), and (3) the patient (a child). Separately, these three factors complicate the design and conduct of a clinical trial. When combined, they pose numerous challenges to the clinical evaluation of this new treatment.
Conventionally, clinical testing is conducted through several phases. So-called phase I trials, aimed at safety and (in pharmaceuticals) dose-ranging, generally include a small group of healthy volunteers. Once initial safety has been confirmed, the process moves on to phase II trials. In these studies, efficacy of the intervention is tested in healthy volunteers. Phase III studies are large, randomized controlled multicenter trials in which the intervention is applied in patients and compared to the gold standard treatment. After an intervention is approved for the market, phase IV trials are often started. These studies are aimed at continued safety surveillance. 30
Large randomized controlled trials (RCTs) have become a staple of pharmaceutical research, and medical research in general, and as such there is a well-defined, internationally agree-upon set of rules and regulations for clinical research of drugs. Additionally, there has been some history of trials with implantable medical devices, their performance being evaluated in clinical trials and postmarket surveillance procedures. 31 With clinical testing of HTEPs, however, there is substantially less experience.
Product
One of the most important characteristics of the tissue engineering process is its complexity. This complexity has consequences for clinical trial design and evaluation.
First, HTEPs show a certain amount of variability, because they contain metabolically active cells in a dynamic extracellular environment. Second, HTEPs are intended to integrate with the body and eventually achieve regeneration of the tissue. The HTEP itself evolves, the dynamic in the body of the recipient influences the incorporation of the HTEP and, finally, the recipient's body and the HTEP interact with each other. Third, because of this interaction, once the process of regeneration is started, it is impossible to fully reverse it. The HTEP itself can be removed, but cells or biomolecules from the HTEP may have influenced surrounding tissues in a way that cannot be undone. This may lead to problems for the trial participant when the results of a trial with an HTEP turn out to be unfavorable.17,27
Like any clinical study, those focusing on HTEPs will have to confirm their therapeutic value. Additionally, they will have to show whether regeneration takes place, whether the product is safe, and what its long-term effects are. 27 A trial of a regenerative medicine treatment is not simply a test of a new medicinal product, but should be considered a trial of a complete process (construction, surgical implantation, and regenerative action of the HTEP in the body of the patient, and the final functional outcome). 32
Procedure
Some challenges in clinical testing of HTEPs for pediatric urological conditions are due to the surgical procedure through which the HTEP is implanted.
In evidence-based medicine, the RCT is the gold standard for assessing the effectiveness of clinical interventions. The RCT is considered the only clinical study design that allows for truly valid inferences considering cause and effect. 33 Therefore, when appropriate, practical, and ethical, a randomized trial design should be used. 34 In spite of this, studies featuring RCTs on surgical interventions in children are rarely reported. 33 This means that, as many new surgical treatments are not tested in classic clinical trials, there may not be a true gold standard to compare the new treatment with the HTEP.
When evaluating a new treatment, a certain uniformity of protocol is necessary to limit the amount of variation in the treatment being studied. However, complete standardization of a surgical intervention is difficult; the way a surgeon conducts an HTEP implantation may be refined over time due to the surgical learning curve and the advance in HTEP construction and the development of instruments. Additionally, different surgeons may refine a technique in different ways. In multicenter studies, the differences in skills and experiences of the operating teams may introduce further variation. 35
Patient and parents
The fact that the prospective patient is a child leads to two kinds of challenges: those associated with the patient's life span, and those connected to the informed consent process.
The prospective patients of HTEP-based treatments in question, in this article, are small children with, in the best case scenario, a long lifespan ahead of them. Some of the risks of HTEP-based treatments, if they occur, are expected to happen in the far future. In some tissue engineering-based treatments (like cartilage tissue engineering for elderly patients), the expected length of time for these adverse events to occur exceeds the average lifespan of the patients in question. In HTEP-based treatments for pediatric urological conditions, therefore, something like tumorgenicity may be a very real concern. As a consequence, follow-up to trials of HTEPs would ideally allow for these long-term negative effects to be taken into account. However, as these adverse events may only occur 20, 30 years into the future, arranging for such a long surveillance period may prove to be a challenge.
Because the prospective trial participant is a legally incompetent child, proxy consent needs to be obtained from the parent(s). In that case, the patient's proxy weighs the risks and benefits of the treatment in question (a decision that may have consequences in the far future, when the patient is no longer a child). The fact that the treatment in question is highly complex, as mentioned before, adds even more difficulty, since the risk of inadequate informed consent is present. The implantation of an HTEP has different goals and risks than a conventional surgical procedure, and is therefore difficult to explain to lay people. This endangers the ideal of a patient's proxy consenting to a treatment based on a clear understanding of the relevant facts.27,36
Discussion
The use of HTEPs to treat children with congenital urogenital defects poses challenges in the clinical testing phase, connected to three features of the application of this treatment in this patient group. First, those associated with the product, namely, the multifaceted complexity of the HTEP. Second, those connected to the procedure, namely, the lack of an RCT-tested gold standard to compare the new treatment to and difficulties surrounding standardization of the treatment protocol. Lastly, the patient's young age and associated problems concerning possible long-term effects and the informed consent process. Due to these problems, a conventional RCT will be difficult to conduct and is not the methodology of choice to evaluate this treatment in this patient group.
How should clinical evaluation of HTEPs in these patients take place? Implantation of an HTEP is quite a significant intervention. As is the case in, for instance, research on treatments for cancer or HIV infection, this intervention is too great a burden to test in healthy volunteers. Therefore, conventional phase I trials with healthy volunteers are unethical. Therefore, the next step following animal studies will most likely be small-scale expert case series with actual patients. For the transition from animal experiments to use in humans to be justified, three conditions need to be met. First, the animal models used need to be optimal representations of the situation in humans. This may prove to be problematic, as the adequacy of the animal models used in tissue engineering has been questioned.23–25 Research on the predictive value of animal experiments in tissue engineering-based treatments is therefore of vital importance. Recent developments in the field of systematic reviews of animal models, for the optimization of animal testing, are a step in the right direction.37,38 Second, minimization of the potential risks and maximization of the potential benefits to the individual trial subject. The tissue engineering process consists of an ex vivo part (manufacturing the HTEP in the laboratory) and an in vivo part (implantation of the construct and regeneration in the body). This necessitates the establishment of Good Manufacturing Practice guidelines pertaining to the ex vivo manufacturing stage, as well as the establishment of Good Clinical Practice guidelines for clinical trials and clinical practice for HTEPs. Third, the informed consent procedure should consist of a comprehensible explanation of the process, the possible benefits and possible risks (both short- and long-term).
If the results of the small case series are positive, the next step should be the evaluation of the HTEP-based treatment on a larger scale, where different teams in several expert centers treat patients based on a uniform protocol. Reliable therapies for the conditions in question are currently available—although they do come with complications in certain patients. Ultimately, for introduction of HTEPs in clinical practice to be justified, large clinical trials should prove superiority over conventional treatment. A newly developed treatment should have a more beneficial cost–effectiveness ratio than the previously available treatments. However, there is often no gold standard, RCT-tested conventional treatment with proven cost–effectiveness. Additionally, it seems difficult to demarcate what to include in the calculation of costs of treatment, and therefore cost–effectiveness studies in this field may prove to be very complex.
A treatment in the field of regenerative medicine needs to accomplish an important act: regeneration. HTEPs for pediatric urological conditions are designed not just to patch up a structural defect, but to ultimately, fully function as native urological tissue. Therefore, it is important that clinical studies prove that regeneration takes place. This means that ways to monitor (and possibly control) the behavior and development of the HTEP in the body after implantation need to be devised. Monitoring is also of vital importance to gather data on the behavior of the HTEP and/or the regenerated tissue in the long-term. Recently, the importance of postmarket surveillance and patient registries to track outcomes was stressed in a public hearing held by the FDA concerning complications of a medical device (a synthetic surgical mesh) for gynecological problems.39,40 Since an HTEP contains biologically active molecules and/or cells, its behavior in the body is even less predictable than that of a medical device. Therefore, stringent and long-term surveillance and registry are essential to ensure the safety of patients treated with these products. (see Fig. 3).
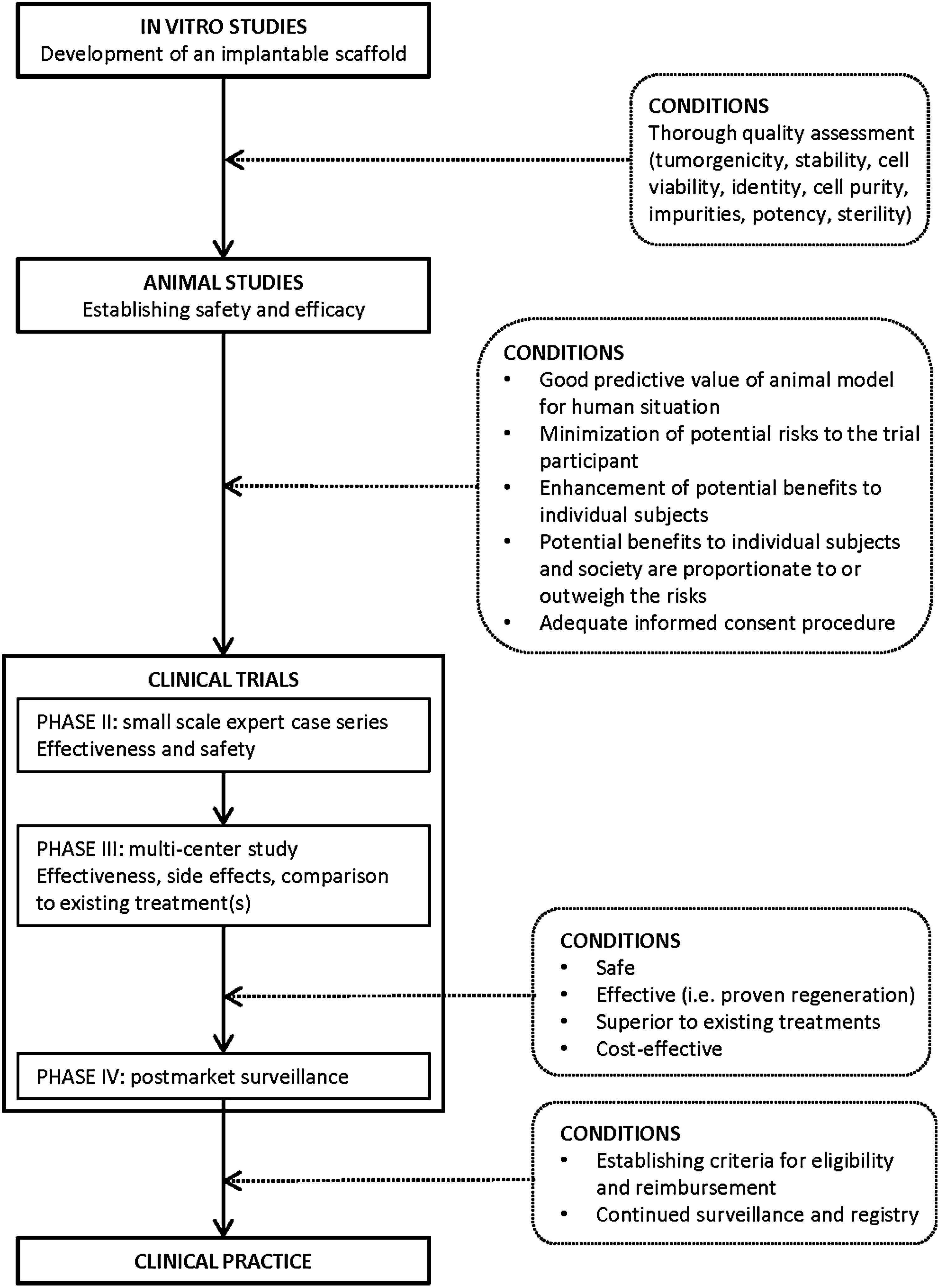
From bench to bedside.
Footnotes
Acknowledgment
The research for this contribution was funded by the European Commission (EuroSTEC: EU contract LSHB-CT-2006-037409).
Disclosure Statement
No competing financial interests exist.