
Abstract
The development of the musculoskeletal system is a complex process that involves very precise control of bone formation and growth as well as remodeling during postnatal life. Although the understanding of the transcriptional mechanisms of osteogenesis has increased considerably, the molecular regulatory basis, especially the gene regulatory network of osteogenic differentiation, is still poorly understood. This review provides the reader with an overview of the key transcription factors that govern bone formation, highlighting their function and regulation linked to Runt-related transcription factor 2 (Runx2). Runx2 as the master transcription factor of osteoblast differentiation, Twist, Msh homeobox 2 (Msx2), and promyelocytic leukemia zinc-finger protein (PLZF) acting upstream of Runx2, Osterix (Osx) acting downstream of Runx2, and activating transcription factor 4 (ATF4) and zinc-finger protein 521 (ZFP521) acting as cofactors of Runx2 are discussed, and their relevance for tissue engineering is presented. References are provided for more in-depth personal study.
Introduction
The process of intramembranous ossification begins with mesenchymal stem cells (MSCs), and the bone tissue is generated by the development of MSCs into osteoblasts and osteocytes. The osteocytes are the most numerous cells in mature bone, which reside in a mineralized extracellular matrix. 1 So far, the process of osteocytogenesis is largely unknown. A cartilage intermediate is not involved during intramembranous ossification. Bone formation by endochondral ossification is a complex phenomenon whereby recruitment, replication, and condensation of mesenchymal precursors occur at sites of future skeletal elements. Within these condensations, cells differentiate into chondrocytes followed by replacement by bone. 2 The process involves preosteoblasts, osteoblasts, mature osteoblasts, and ultimately the accumulation and mineralization of the extracellular matrix. A number of factors influence development, growth, and repair of bone. The mediators involved in osteogenesis include transcription factors, growth factors, cytokines, metabolites, hormones, mechanical loading, and aging. Hormones are involved in bone formation. Estrogen deficiency causes bone loss and osteoporosis by increasing the generation and activity of osteoclasts. 3 Excessive glucocorticoid also results in osteoporosis, which is a major cause for bone fractures in the elderly, by decreasing bone formation 4 and promoting bone resorption. 5 These data show that glucocorticoid signaling is required for normal bone formation. A recent report showed that Runt-related transcription factor 2 (Runx2) played a role in glucocorticoid-mediated BIM induction and apoptosis of leukemia cells. 6 Studies by Phillips et al. 7 also showed that the coordinated action of dexamethasone and the osteogenic transcription factor Runx2/Cbfa1 synergistically increased osteogenic gene expression, including osteocalcin (OC), bone sialoprotein (BSP), and alkaline phosphatase (ALP), and biological mineral deposition in primary dermal fibroblasts.
Major advances in the genetics of skeletogenesis have been occurred over past 15 years, especially in the identification of genetic factors regulating osteogenesis. However, the gene regulatory network (GRN) of osteoblast differentiation still remains poorly understood. Understanding the transcription factors affecting osteogenesis and related GRN is crucial to harness the inherent regenerative potential of skeletal tissues for possible use for gene therapy in bone repair and regeneration. This review provides the reader with an overview of the transcription factors linked to Runx2, the master transcription factor of osteoblast differentiation, highlighting the transcriptional cascade of osteogenesis involved in bone formation and remodeling. The master transcription factor of osteoblast differentiation Runx2, its upstream factors Twist, Msh homeobox 2 (Msx2), and promyelocytic leukemia zinc-finger protein (PLZF), downstream factor osterix (Osx), and coactivators activating transcription factor 4 (ATF4) and zinc-finger protein 521 (ZFP521) are discussed in the review. References are provided for more in-depth personal study.
Runx2, the Master Transcription Factor for Osteogenesis
Runx2 is a transcription factor that belongs to the runt homology domain protein family, which contains a glutamine-/alanine-rich domain at its N-terminal end (a runt domain) and a proline/serine/threonine (PST)-rich region at the C-terminus. Runx2 is also called core-binding factor-alpha (CBFA1), Acute Myeloid Leukemia 3 (AML3), PEBP2alphaA, and osteoblast-specific factor 2 (OSF2). To unify the naming system for this exciting class of transcription factors and facilitate cross-referencing of articles, the Nomenclature Committee of the Human Genome Organization (HUGO) adopted the use of the term RUNX to refer to genes encoding the runt-related proteins in November 1999. Runx2 is expressed in cells prefiguring the skeleton as early as E10.5, 8 at which stage cells still have the capacity to differentiate into osteoblasts or chondrocytes. Runx2 is expressed in the osteoblast lineage, 9 hypertrophic chondrocytes, 10 odontoblasts, and ameloblasts. 11 Runx2 decreases and eventually vanishes at E16.5 in differentiating chondrocytes. 10
In vitro and vivo studies show that Runx2 is an essential transcription factor for osteoblast differentiation, matrix production, and mineralization during bone formation. Runx2 controls the expression of major bone matrix protein genes through a direct binding site called osteoblast-specific cis-acting element (OSE2), which is present in the promoter of several osteoblast-specific genes such as OC, 9 osteopontin (OPN), 9 BSP, 9 and collagen, type I, alpha 1 (Col1A1).9,12 Runx2 also directly regulates cranisynostosis-associated gene NEL-like 1 (NELL1), 13 skeletal tissue-enriched gene Pannexin 3 (PANX3), 14 and zinc-dependent endopeptidases matrix metallopeptidase 9 (MMP9) 15 and MMP13. 16
Runx2 binds OSE2 in the promoter to initiate mesenchymal condensations of the developing skeleton and regulate osteogenesis. In immature stage, Runx2 enhances osteogenesis. However, Runx2 is not essential to maintain the expression of the major bone matrix protein genes in mature osteoblasts and needs to be suppressed to form mature bone 17 (Figs. 1 and 2). Overexpression of Runx2 in adipose tissue-derived MSCs inhibits adipogenesis as demonstrated by decreased lipoprotein lipase and peroxisome proliferator-activated receptor (PPARγ) expression and reduced lipid droplet formation. In osteogenesis, Runx2-overexpressing adipose tissue-derived MSCs undergo rapid and marked osteoblast differentiation as determined by osteoblastic gene expression, ALP activity, and mineral deposition. 18 Even forced expression of Runx2 in nonosteoblastic cells induces the expression of the major osteoblast-specific genes. 9 Runx2 is also a critical transcription factor that promotes chondrocyte maturation (Fig. 1). The formation of hypertrophic chondrocytes is severely impaired in some skeletal elements in Runx2-knockout mice. 19 Expression of Runx2 in nonhypertrophic Col2a1-expressing chondrocytes accelerates chondrocyte differentiation and partially rescues the chondrocyte phenotype in Runx2 knockout mice. 20
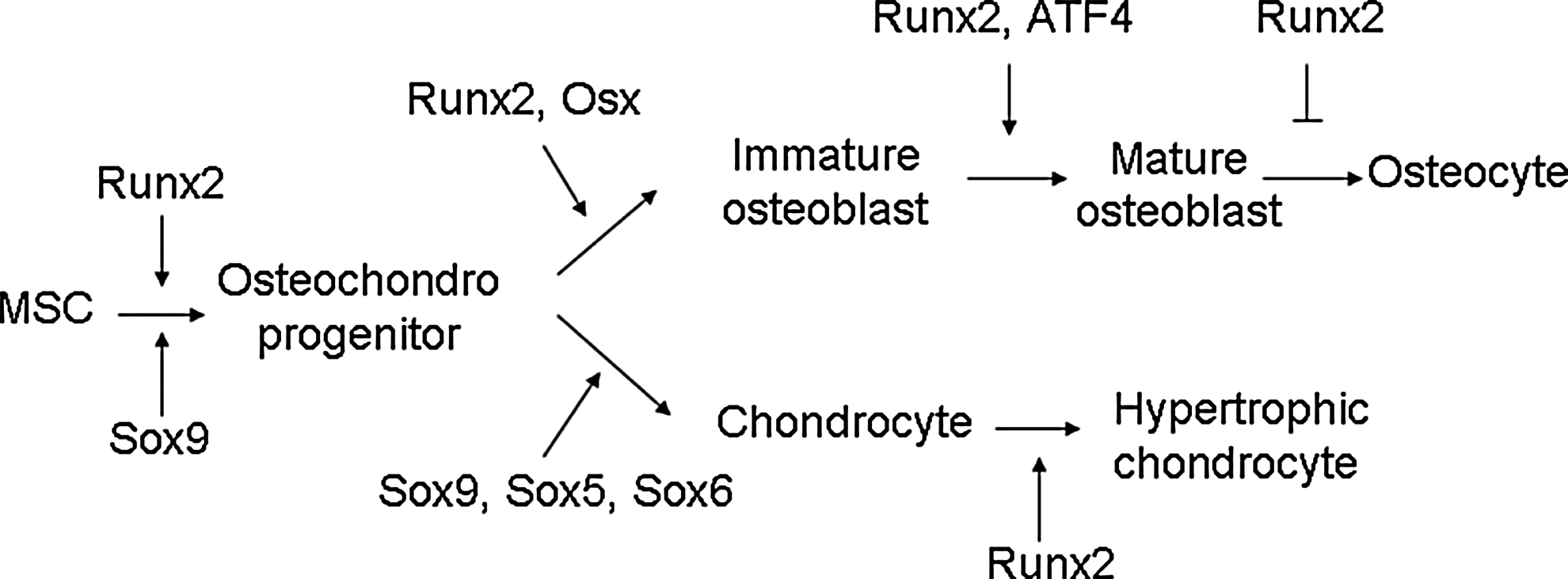
Role of transcription factors in cell differentiation along the osteoblast and chondrocyte lineage. Runt-related transcription factor 2 (Runx2) is an essential transcription factor of early osteoblast differentiation as the master gene of bone formation. However, Runx2 is not essential for late stage of osteoblast differentiation and needs to be suppressed for terminal differentiation into osteocytes. Runx2 is also required for chondrocyte maturation.

Role and delicate regulation of Runx2 during osteoblast differentiation. Runx2 is essential for skeletal development. In osteoblast differentiation, Runx2 expression is detected in preosteoblasts expressing type I collagen weakly and is upregulated in immature osteoblasts expressing osteopontin. However, Runx2 expression is downregulated in mature osteoblasts expressing osteocalcin (OC). As the osteoblast transitions to an osteocyte expressing secreted proteins CD44, DMP1, and MEPE, alkaline phosphatase is reduced, whereas OC is elevated. The expression and transcriptional activity of Runx2 are tightly regulated by multiple proteins during osteoblast differentiation.
Runx2 expression level is important for normal bone development. Decreased Runx2 expression results in abnormal bone development. Mice with a homozygous mutation in Runx2 died just after birth without breathing, and analysis of their skeletons revealed lack of ossification of bone due to the maturational arrest of osteoblasts characterized by lack of expression of a later-stage marker OC, demonstrating that Runx2 plays an essential role in osteogenesis.21,22 Runx2 mutations cause cleidocranial dysplasia (CCD) in humans, an autosomal-dominant condition exhibiting defective endochondral and intramembranous bone formation, which is characterized by hypoplasia/aplasia of clavicles, patent fontanelles, supernumerary teeth, short stature, and other changes in skeletal patterning and growth. 23 Heterozygous loss of Runx2 function in mice is sufficient to produce CCD disorders similar to humans.21,23 Further data demonstrate that truncated Runx2 protein severely impaired Runx2 transactivation activity and failed to interact with and respond to Smads to induce osteogenesis and cause CCD. 24
Transgenic data show that increased Runx2 expression also cause bone dysplasia. By overexpression of Runx2 under the type I collagen promoter in transgenic mice, Runx2 transgenic mice end up with osteopenia with multiple fractures. Thin, porous, and OPN-enriched cortical bone is invaded by osteoclasts, despite the absence of acceleration of osteoclastogenesis. The number of neonatal osteoblasts increases, but matrix production and mineralization are impaired. Terminally differentiated osteoblasts and osteocytes decrease greatly, whereas less-mature osteoblasts accumulate in adult bone, demonstrating that Runx2 inhibits osteoblast differentiation at late stage by blocking maturation of osteoblasts. 17 Runx2-overexpressing mice by the MMP13 promoter show increased mRNA expression of bone-forming genes and decreased MMP13, which plays a role in recruiting osteoclasts to the bone surface in the tibiae of transgenic mice. Further histological analyses of the proximal tibiae show increased bone mineralization surface, mineral apposition rate, and bone formation rate, but decreased osteoblast number. 25 These data suggest that the Runx2 expression level is very important to maintain the balance between the bone formation–bone resorption process. Overall, Runx2 is the master transcription factor of bone formation.
Regulators of Runx2: Delicate Regulation of Osteogenesis
Bone development is a complex process, and three types of cells, including osteoblasts, osteoclasts, and chondrocytes, need be elaborately orchestrated to result in normal bone development. The expression and transcriptional activity of Runx2 are tightly regulated by upstream factors and cofactors to control downstream factors and function in skeletogenesis as the master transcription factor of osteogenesis.
Upstream factors of Runx2
Many factors such as Twist, Msx2, and PLZF have been identified to be involved in the regulation of Runx2 expression or transcription activity as upstream factors and therefore regulate osteogenesis (Fig. 3). It has also been shown that SRY (sex-determining region Y)-box 8 (Sox8) 26 and homeobox A2 (Hoxa2) 27 reduce expression of Runx2 to regulate bone formation. Sox9 directly interacts with Runx2 and represses its activity via their evolutionarily conserved high-mobility-group and runt domains. 28 p53-null osteoprogenitor cells show increased expression of Runx2. 29 Conversely, some transcriptional factors increase the expression of Runx2 to regulate osteogenesis. Forkhead box protein O1 (Foxo1) directly interacts with the promoter of Runx2 and regulates its expression. 30 SATB homeobox 2 (Satb2), 27 Hoxa10, 31 distal-less homeobox 3 (Dlx3), 32 and Dlx5 33 activate the expression of Runx2 and osteogenic genes. Very interestingly, Dlx3 exerts both positive and negative regulation of gene transcription by a different molecular mechanism. Dlx3 protein–DNA interactions increase OC promoter activity, whereas Dlx3–Runx2 protein–protein interactions decrease Runx2-mediated transcription. 34 Mice lacking Bapx1 die at birth showing dysplasia of the axial skeleton and strongly decreased Runx2, indicating that Bapx1 is required for Runx2 expression. 35 Wnt10b directs cell fate toward the osteoblast lineage by inducing osteoblastogenic transcription factors Runx2, Dlx5, and Osx. 36 Serum response factor (SRF) deficiency decreases the transcriptional activity of Runx2, whereas overexpression of SRF induces Runx2 transactivity, suggesting that SRF regulates bone formation through Runx2 (Fig. 2). 37
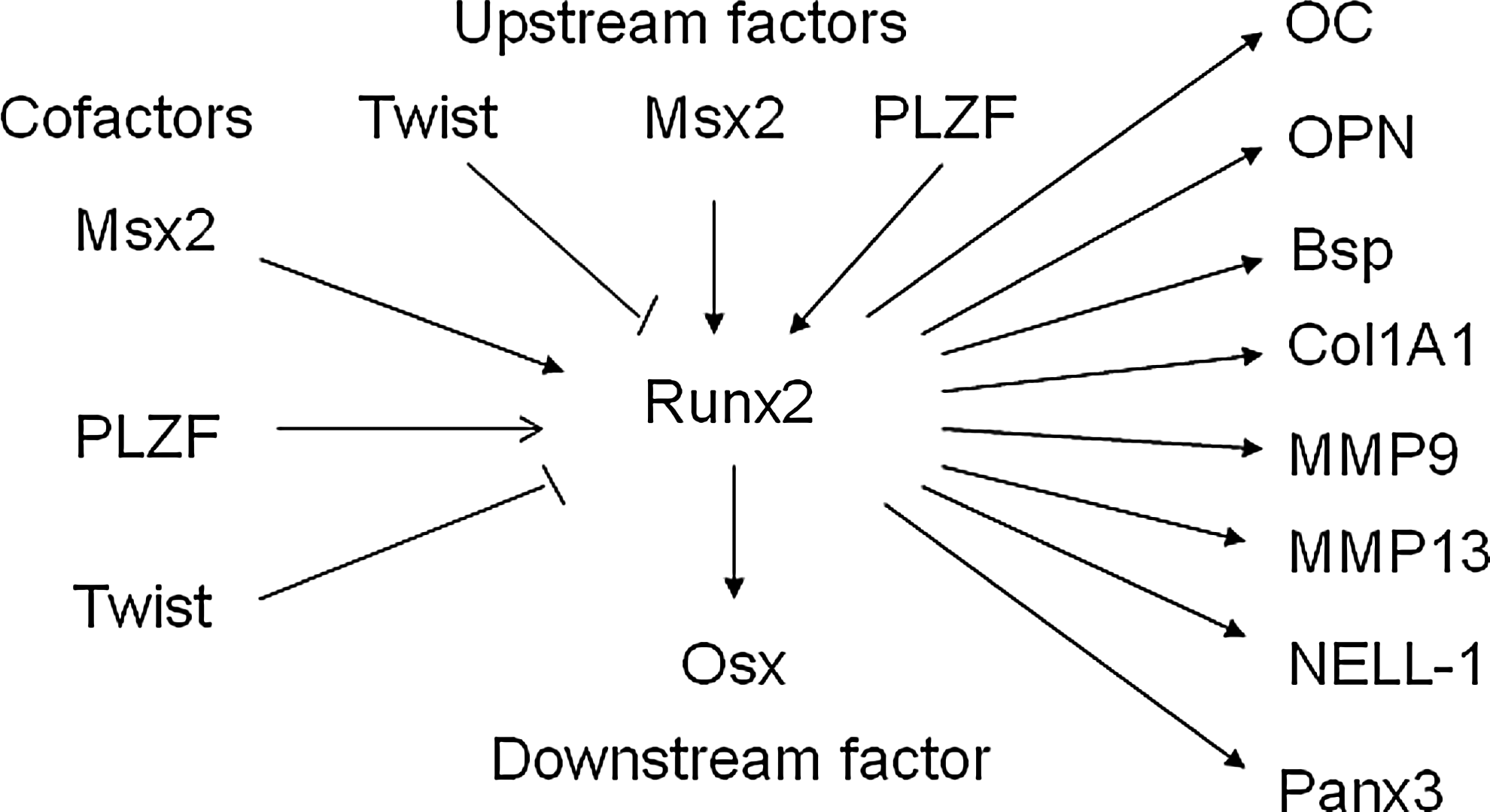
Runx2 as the master regulator of osteoblast differentiation is modulated by multiple transcription factors to regulate downstream factors and osteoblast differentiation. The expression and transcriptional activities of Runx2 are tightly modulated by upstream transcription factors and cofactors to regulate downstream transcription factors. As the master regulator of osteoblast differentiation, Runx2 directly binds and regulates the expression of multiple osteogenic genes that determine the osteoblast phenotype.
Twist, antagonist of Runx2
Runx2 directly binds and regulates the expression of OC. However, OC is expressed 4–5 days later than Runx2 expression during mouse development. It has been proposed that Runx2 function is transiently inhibited by other genes. Twist is a basic helix-loop-helix transcription factor. Twist1 is highly expressed in freshly purified bone marrow-derived MSCs and decreased during ex vivo expansion. 38 During early development, Twist-1 is coexpressed with Runx2 in cells destined to become osteoblasts and disappear when osteogenesis is initiated. Twist-1 inactivation alters Runx2 expression and Runx2-binding ability to the OC promoter. 39 Highly expressing Twist1 increases the expression of MSC markers, but decreases osteogenesis, implicating that Twist mediates self-renewal and lineage commitment of MSCs. 38 Haploinsufficiency of Twist1 causes Saethre-Chotzen syndrome, a form of craniosynostosis.40,41 Removing one allele of Twist1 is sufficient to correct the skull defects from heterozygous mutation of Runx2, and Twist-1 or 2 deficiency results in premature osteoblast differentiation. Conversely, Twist-1 overexpression inhibits osteogenesis without affecting Runx2 expression, 42 whereas Twist-1 silencing enhances osteogenesis of MSCs. 43 Further molecular analysis shows that Twist directly binds to the Runt DNA-binding domain of Runx2 to decrease binding of Runx2 to DNA. 38 These data show that Twist determines the onset of osteogenesis as an antagonist of Runx2. 42
Msx2, a regulator of bone development
Msx2 is a homeodomain transcription factor first identified in craniofacial bone and human femoral osteoblasts.44–46 Mice deficient in Msx2 manifest defects in skull ossification and a marked reduction in bone formation associated with decrease in osteoblast numbers, thus suggesting that Msx2 is involved in bone formation.45,46 Tracing the origin of the calvarial foramen defects shows that mesenchymal cell populations decreased due to defects in differentiation and proliferation, and not because of apoptosis or deficient migration of neural crest-derived precursor cells. 47 Msx2 also affects tooth development. Msx2−/− homozygous mice develop compound amelogenesis imperfecta, dentinogenesis imperfecta, and periodontal osteopetrosis. 48 The expression of Runx2 is strongly reduced in Msx2-deficient mice, suggesting that Msx2 acts as upstream factor of Runx2 to regulate osteoblast differentiation (Fig. 3). Overexpression of Msx2 affects bone formation. In vitro, Msx2 promotes osteogenesis of MSCs. 49 However, a controversial result was reported in chick calvarial osteoblasts, showing that ectopic Msx2 overexpression prevents osteoblastic differentiation and mineralization, whereas Msx2 knockdown by antisense decreases proliferation and accelerates differentiation. 50 Overexpression of Msx2 in the Runx2-deficient MSCs induces Osx expression, whereas knockdown of Msx2 inhibits Osx by BMP2. 51 Msx2 transgenic mice increase bone formation by increasing osteoblast numbers. Further molecular studies reveal that the Msx2 transgene promotes osteogenic differentiation in part by reducing Dkk1 (an antagonist of Wnt receptors LRP5 and LRP6) expression and enhancing Wnt signaling. 52 These data show that Msx2 plays an important role in regulating bone development.
Plzf, an upstream factor of Runx2
PLZF encodes a zinc-finger-type transcription factor, comprising 9 Kruppel-like C2H2 zinc fingers located in the C-terminus of the protein 53 and a BTB (bric-a-brac, tram track, broad complex)/POZ (poxvirus, zinc finger) domain in the N-terminal end. PLZF is shown to be one of the highly upregulated genes during osteogenesis and is functionally involved in osteogenesis of MSCs. PLZF knockdown results in decreased expression of osteoblast-specific genes, whereas PLZF overexpression improves osteogenesis.54,55 Similar effects of PLZF overexpression on immortalized MSCs are also observed. 55 Deletion of the BTB domain abrogates the effect of PLZF on osteogenesis, showing that BTB plays a crucial role in osteogenesis. Further analyses show that PLZF affects Runx2, whereas Runx2 does not affect the expression of PLZF, suggesting that PLZF regulates osteogenesis as an upstream factor of Runx2. 54 PLZF is also shown to regulate chondrogenesis of MSCs as an upstream factor of Sox9, the master regulator of chondrocyte differentiation. PLZF-overexpressing MSCs repaired cartilage defects better and earlier than control MSCs. 55 It is proposed that PLZF regulates the osteogenic master regulator Runx2, which directly regulates osteoblast-specific genes, including OC, 9 OPN, 9 BSP, 9 and Col1A19,12 during osteogenesis (Fig. 3). Therefore, PLZF represents a very promising strategy for bone regeneration and repair.
Osx, downstream factors of Runx2
Runx2 regulates downstream genes that determine the osteoblast phenotype and function in skeletogenesis. Besides major osteoblast-specific genes such as OC, Col1A1, BSP, and OPN, 9 Runx2 also regulates Osx, which is essential for osteoblast differentiation. Osx is a zinc-finger-containing transcription factor expressed in osteoblasts 56 and chondrocytes. 57 Inactivation of Osx in mice results in perinatal lethality owing to a complete absence of bone formation and completely arrested osteoblast differentiation displaying lack of early and late markers of osteoblast differentiation. 56 A homozygous single-base-pair deletion in Osx resulted in recessive osteogenesis imperfecta in an Egyptian child. 58 Osx-deficient mice lack a mineralized matrix in bones formed by intramembraneous ossification, which is different from Runx2-deficient mice, which show an entirely nonmineralized skeleton. Osx-deficient bones formed by endochondral ossification contain some mineralized matrix, which resembles calcified cartilage, not mineralized bone matrix, showing that Osx is not required for chondrocyte hypertrophy. Osx is not expressed in Runx2-deficient embryos, whereas Runx2 is normally expressed in Osx-deficient embryos, showing that Osx is a downstream factor of Runx2 during osteoblast differentiation. Further data show that Runx2 directly binds to Osx at a responsive element in the promoter of the Osx gene. 59 These data demonstrate that Osx specifically induces osteoblast differentiation and bone formation in vivo. Further molecular data show that the nuclear factor of activated T cells (NFAT) and Osx form a complex that binds to the osteoblast-specific Col1a1 promoter and synergistically stimulates its activity. 60 All these data suggest that Runx2 and Osx belong to two independent pathways; Osx is a Runx2-dependent transcription factor and is absolutely required for bone formation.
Cofactors of Runx2-regulating osteogenesis
Runx2 is a transcription factor essential for osteoblast commitment and early stages of osteoblast differentiation, whose activity is tightly controlled by transcriptional factors through protein–DNA or protein–protein interactions. It was shown that Stat1 interacts with Runx2 to inhibit its nuclear localization. 61 Yes-associated protein (YAP) suppresses Runx2 transcriptional activity in a dose-dependent manner. 62 AJ18 modulates Runx2 activity and osteogenic differentiation by suppressing Runx2-mediated transactivation of an OC promoter. 63 Myeloid Elf-1 like factor (MEF) forms a complex with Runx2 to interfere with binding of Runx2 to the OC promoter at the OSE2 site. 64 Nrf2 interacts with Runx2 to inhibit Runx2-dependent stimulation of OC promoter activity and recruitment of Runx2 on the OC promoter without affecting the expression of Runx2 mRNA. 65 pRb serves as a linker connecting p204 and Runx2 by forming a ternary complex to stimulate Runx2 activity. 66 Schnurri 3 (Shn3) controls the protein level of Runx2 by mediating recruitment of E3 ubiquitin ligase WW domain-containing protein 1 (WWP1) to alter the availability of Runx2 in the nucleus. 67 Core-binding factor-beta (CBFbeta) 68 and Smads 24 interact with Runx2 to regulate skeletal development. Smad does not directly induce Runx2 expression, and Runx2 is induced by the receptor-activated Smad through Smad-induced junB functions. 69 TAZ (transcriptional coactivator with the PDZ-binding motif) coactivates Runx2-dependent gene transcription while repressing PPAR-gamma-dependent gene. 70 Id4 promotes osteoblast differentiation by releasing Hes1 from Hes1–Hey2 complexes to increase the stability and transcriptional activity of Runx2. 71 GLI family zinc-finger 2 (GLI2) interacts with Runx2 to enhance osteogenesis. 72 Chicken ovalbumin upstream promoter–transcription factor II (COUP-TFII) physically interacts with Runx2 to impair the Runx2-dependent activation of the OC promoter 73 (Table 1 and Fig. 2).
ATF4, activating transcription factor 4; ZFP521, zinc-finger protein 521; OC, osteocalcin; Satb2, SATB homeobox 2; YAP, yes-associated protein; MEF, myeloid Elf-1-like factor; Rb, retinoblastoma; Shn3, schnurri 3; CBFbeta, core-binding factor-beta; COUP-TFII, chicken ovalbumin upstream promoter–transcription factor II.
Activating transcription factor 4
ATF4 encodes a transcription factor that plays a crucial role in osteoblast differentiation and function. In vitro kinase assay shows that AFT4 is strongly phosphorylated by ribosomal S6 serine/threonine kinase 2 (RSK2) as a critical substrate, and this phosphorylation is undetectable in osteoblasts from mice lacking RSK2, encoding a growth factor-regulated kinase, in which the mutation results in DuCoffin-Lowry syndrome (CLS). CLS is an X-linked mental retardation condition associated with skeletal abnormalities. In addition, ATF4 knockout mice displayed delayed skeletal development, decreased bone formation, and thereafter develop a severe low-bone-mass phenotype. 74 Molecular studies reveal that ATF4 binds to an osteoblast-specific element in the OC promoter and directly activates its transcription. 74 ATF4 is also required for proper synthesis of Col1A1. Col1A1 synthesis is decreased in ATF4-decifient osteoblasts compared with wild-type osteoblasts. This decrease in Col1A1 synthesis is not due to decreased gene expression of Col1A1, Runx2, and Osx, as their expression is normal in mutant osteoblasts compared with wild type osteoblasts, suggesting that ATF4 affects synthesis of Col1A1 through a post-transcriptional mechanism. Addition of nonessential amino acids in a culture medium rescues the defect in collagen synthesis in osteoblasts lacking ATF4, suggesting that ATF4 is required for efficient amino acid import into osteoblasts. 74 Forced expression of a constitutively active form of RSK2 (RSK2-T707A) 75 enhances the activity of the pOG2-luc reporter in COS cells, whereas RSK2-T707A is not able to increase the activity of the ATF4 mutant that cannot be phosphorylated by RSK2, 74 suggesting that phosphorylation of ATF4 by RSK2 enhances its transactivation ability. Further studies showed that ATF4 interacts with Runx2 through Satb2 to synergize their activities by binding proximal binding sites at the OC promoter. 27
ATF4 also regulates osteoclast differentiation and ultimately bone resorption through its expression in osteoblasts.76,77 This can be explained by the binding of ATF4 to the promoter of the receptor activator of nuclear factor-KB ligand (RANKL) gene, 78 which encodes a factor secreted by osteoblasts and binds to its receptor RANK on osteoclasts to trigger intricate and distinct signaling cascades that control lineage commitment and activation of osteoclasts. RANKL binding to RANK is negatively regulated by OPG to inhibit bone turnover by osteoclasts. 79 ATF4-deficient mice have decreased osteoclast numbers owing to reduced RANKL expression, demonstrating that ATF4 is involved in the control of bone resorption. 72 These data demonstrate that ATF4 is a major regulator of osteoblast differentiation.
Zfp521, a binding partner of Runx2
Zfp521, a 30-zinc-finger protein, is a new player in bone formation. Zfp521 is a nuclear protein highly expressed at the periphery of mesenchymal condensations, and plays an important role in developing bones, including the perichondrium and periosteum, osteoblast precursors, osteoblasts, ostocytes, chondroblasts, and prehypertrophic chondrocytes. Zfp521 is also highly expressed at the periphery of mesenchymal condensations and calvarial osteoblasts.80,81 Zfp521 strongly represses activation of the Runx2-mediated reporter gene. Zfp521 calvarial cells from Zfp521-transgenic mice show decreased osteogenesis as determined by less nodule formation and mineralization, and decreased early osteoblast marker genes, 82 suggesting that Zfp521 antagonizes early stages of osteoblast differentiation. Conversely, depletion of Zfp521 increases expression of Runx2 and Runx2 target genes. 83 Forced expression of Zfp521 in osteoblasts in vivo leads to an increase in bone mass due to a marked increase in the osteoblast number and bone-forming activity. 81 Removing one copy of Zfp521 can rescue the CCD phenotype due to lack of one copy of Runx2 in mice, whereas overexpressing Zfp521 exacerbates the bone defects. 83 Zfp521 is an important PTHrP target gene.82,84 Further molecular data have shown that Zfp521 blocks Runx2 in the presence of HDAC3 by recruiting the histone deacetylase HDAC3 to repress its transcriptional activity. 83 Zfp521 coimmunoprecipitates with Runx2 in the same complex to repress its transcriptional activity, demonstrating that Zfp521 is a binding partner of Runx2. These data show that Zfp521 restricts early stages of osteoblast differentiation, but promotes late stages of osteoblast maturation by antagonizing Runx2 transcriptional activity.
HDACs, cofactors of Runx2
Runx2 is the master regulator of osteogenesis and is crucial for regulating the expression of bone-specific genes. Runx2 either activates or represses transcription of tissue-specific genes by binding specific DNA sequences or interacting with transcriptional coactivators and corepressors to regulate bone formation. It has been shown that transcriptional activity of Runx2 is inhibited by HDACs. HDACs are a class of enzymes that remove acetyl groups from an ɛ-N-acetyl lysine amino acid on a histone, allowing the histones to wrap the DNA more tightly to regulate DNA expression by acetylation and deacetylation. HDAC activity plays important roles in the development of bone formation by post-translational modification. HDAC4 and HDAC5 inhibit transcriptional activity of Runx2 by deacetylating. 85 HDAC4 also inhibits Runx2 transcriptional activity by binding the Runt domain and interfering with DNA binding. 86 In a different manner, HDAC3 and HDAC6 repress Runx2-mediated transcription by binding the aminoterminal domain 87 and the carboxy terminus 88 of Runx2 to deacetylate lysines in the Runx2 protein, respectively. In a deacetylase-independent manner, HDAC7 represses Runx2 transcriptional activity by indirectly binding Runx2 through the aminoterminus of HDAC. 89
Identification of Genetic Factors Regulating Osteogenesis
Great progress has been made over the past 15 years in identification of genetic factors involved in skeletogenesis and the understanding of the molecular events of bone development. To take these studies further, it is important to make use of human disease models, animal models, and well-designed in vitro studies.
Methods to identify genetic factors regulating osteogenesis
Human disease cases with skeletal dysplasia
Naturally occurring human disease cases with bone dysplasia offer an excellent platform to explore the underlying genes causing the various bone abnormalities and their phenotypes. However, the incidence is rare. Studies of extreme phenotypes will yield very useful data that can be used for studies that may result in therapeutic intervention in the form of gene therapy.
Animal models
Animal models, especially mouse genetics models, allow researchers to investigate the role of genetic factors involved in skeletogenesis and their phenotypes, which would otherwise not be possible in humans. Producing genetically engineered animals by modulating in vivo gene expression is widely used to study the underlying molecular alterations and screen genes related to bone development, including mutated animals with homozygous or heterozygous mutations and transgenic animals. Conditional gene knockout by the Cre/loxP or FLP/FRT recombination system is widely used to study gene function in adult mice and selected cell types based on a tissue-specific inactivation of the gene of interest. This approach is advantageous over the conventional type in that conditional mice survive longer, especially when conventional knockout mice exhibit embryonic or early postnatal lethality, or when a gene alteration exerts its effects in multiple different cell and tissue types, which make it difficult to distinguish direct function in a particular tissue or secondary effects resulting from altered gene function in other tissues. The technique is also much cleaner and scientifically more precise for elimination of a specific target gene from a single organ in the body. Animal models can provide insights into the biological role in human disease and permit the identification of the genetic defect in humans. It was shown that mice with mutations in GMAP-210 were similar to patients with Achondrogenesis type 1A in humans, a neonatal lethal form of skeletal dysplasia, suggesting that Achondrogenesis type 1A may be caused by GMAP-210 deficiency. Further sequence analysis revealed mutations in the unrelated patients with Achondrogenesis type 1A. 90
In vitro study
Microarrays and high-throughput sequencing technology are being employed to screen and identify novel molecular signature related to osteogenesis. Screened candidate genes will then be validated by gain or loss of function in cells in vitro. This will be very useful in studying human genes regulating osteogenesis without human disease cases.
Challenges of study in genetic factors regulating osteogenesis
Although extraordinary progress has been made in the identification of transcription factors involved in osteogenesis, detailed GRN of osteogenesis remains poorly understood. So far, the repair of massive bone defects still represents a major clinical orthopedic challenge. Bone is a highly vascularized tissue dependent on the close spatial and temporal connection between endothelial cells and bone cells to maintain skeletal integrity. Bone tissue engineering is a complex undertaking that involves the combination of growth factors (such as osteogenic and angiogenic factors), cells, and scaffolds. To repair the massive bone defects by tissue engineering, only study of the genetic factors in osteogenesis is not sufficient. To improve bone regeneration and repair, it is necessary to identify novel genetic factors regulating osteogenesis and decipher GRNs of osteogenesis. At the same time, it is also crucial to understand how osteogenic and angiogenic factors interact with each other and with cells during the multistem processes of bone development and repair. Combined strategies of osteogenic and angiogenic factors will enhance the regenerative capacity of bone. It is important to establish how osteogenic and angiogenic factors can be delivered in a spatial and temporal manner in biomimetic scaffolds so that bone regeneration can occur.
Transcription factor-targeted gene therapy for bone regeneration
Gene therapy represents one the most promising approaches for orthopedic regenerative medicine. The use of transcription factors for bone tissue engineering has been examined. It has been shown that Runx2 overexpression significantly upregulates osteoblastic differentiation and enhances mineralization of bone marrow stromal cells (BMSCs) in vitro and in vivo.
91
Similar results are obtained in rat BMSCs.
92
Further data show that Runx2-modified BMSCs are able to accelerate healing of critical-sized bone defects.
93
In fibroblasts, effects of Runx2 overexpression on mineralization depend on the scaffold used; collagen foams exhibit 10-fold higher mineral volume compared to PCL and PLGA matrices in vitro,
94
suggesting that the choice of scaffold will affect the reparative effects. Runx2-modified dermal fibroblasts form mineralized templates in vivo after subcutaneous implantation.
95
Graded tissue interface is engineered by gradients of immobilized Runx2 retrovirus via deposition of controlled poly(
Conclusions
We have provided a detailed overview of the identity of the important mediators and the molecular mechanisms of the genetic factors linked to Runx2 as the master transcription factor of osteoblast differentiation. The information presented can form the basis for many researchers in the stem cell arena to embark on further studies to enhance bone healing and bone repair. Bone development and repair is a very complex process, and many related issues remain to be elucidated. Further scientific endeavors need be undertaken to deepen our understanding of osteogenesis, so that it can lead to better solutions for bone repair and regeneration.
Footnotes
Acknowledgment
This work is supported by a BMRC grant (R-175-000-085-305).
Disclosure Statement
The authors have declared that no competing interests exist.