
Abstract
Self-assembled microvasculature from cocultures of endothelial cells (ECs) and stromal cells has significantly advanced efforts to vascularize engineered tissues by enhancing perfusion rates in vivo and producing investigative platforms for microvascular morphogenesis in vitro. However, to clinically translate prevascularized constructs, the issue of EC source must be resolved. Endothelial progenitor cells (EPCs) can be noninvasively supplied from the recipient through adult peripheral and umbilical cord blood, as well as derived from induced pluripotent stem cells, alleviating antigenicity issues. EPCs can also differentiate into all tissue endothelium, and have demonstrated potential for therapeutic vascularization. Yet, EPCs are not the standard EC choice to vascularize tissue constructs in vitro. Possible reasons include unresolved issues with EPC identity and characterization, as well as uncertainty in the selection of coculture, scaffold, and culture media combinations that promote EPC microvessel formation. This review addresses these issues through a summary of EPC vascular biology and the effects of tissue engineering design parameters upon EPC microvessel formation. Also included are perspectives to integrate EPCs with emerging technologies to produce functional, organotypic vascularized tissues.
Introduction
F
Incorporating cells that secrete angiocrine factors within the scaffold, such as endothelial cells (ECs) cocultured with mural cell precursors or fibroblasts, can induce de novo blood vessel development to create capillary networks and generate a prevascularized construct.14–17 These self-assembled capillary networks significantly reduce the time to anastomose with host vasculature and enhance construct viability in comparison to implants of nonvascularized tissue constructs.14,18,19 Despite these significant advances, the source of ECs remains an issue.3,20,21 Isolation of tissue-specific ECs requires additional, invasive surgeries for the patient. As well, fully differentiated cells have limited expansion potential and may lack responsiveness to growth factors and cytokines, impeding integration with vascular and parenchymal cells, which allows recapitulation of the intricate vascular patterns that result from intimate cell associations.22–24
The cell of greatest interest in vascularizing engineered tissues is the endothelial progenitor cell (EPC) because of its potential to differentiate into all capillary niches, such as the continuous capillaries in skeletal muscle, fenestrated capillaries in endocrine glands, and discontinuous capillaries in the liver.25,26 Furthermore, EPCs may be noninvasively isolated from recipients of tissue-engineering therapy through adult peripheral blood and umbilical cord blood, as well as derived from human-induced pluripotent stem cells (hiPSCs), avoiding immunogenicity concerns.27–30 EPCs form vascular structures in vitro and in vivo, possess permeability values similar to vessel-derived endothelium, and can outperform vascular-derived endothelium in forming vascular networks.15,31–36 In addition to their use in therapeutic angiogenesis, vascularized engineered tissues provide in vitro platforms for personalized medicine. 37 For example, utilizing autologous EPCs to develop vasculature in vitro can identify optimal drug combinations, tailored to the patient's unique genetic background.
Still, the identity and differentiation mechanisms of EPCs are not well understood, likely due to a lack of standardization in their isolation and characterization. 38 Heterogeneity is also present in the vasculogenic potential of EPC subsets and in the ability of tissue-specific stromal and parenchymal cells to promote EPC microvessel formation.39–44 As a result, wide variation exists in the reported engraftment potential of EPCs to host vasculature.45–48 This review aids the tissue engineers in their efforts to utilize EPCs by clarifying EPC identity and characterization, reviewing the current understanding of EPC vascular morphogenesis and differentiation, and summarizing key design parameters to generate EPC microvessels in vitro. Also provided are perspectives to incorporate existing knowledge of EPCs with advances in materials science and genetic engineering to enable the next generation of tissue vascularization research.
Defining EPCs
The controversy surrounding EPC definition can be traced back to the 1917 observations of blood vessel development in the chick embryo by Florence Sabin.49,50 Sabin described angioblasts, which are cells derived through fibroblast growth factor (FGF-2 or bFGF) induction of the mesoderm, as giving rise to primitive erythroblasts enclosed by an EC border, known as blood islands, which formed primitive vascular networks that further developed into arteries and veins.49,51 The erythroblasts and ECs of the blood islands appeared to form from within the angioblasts, prompting Sabin to conclude that the angioblasts could produce blood plasma and red blood cells, as well as ECs. 49 This discovery led to the widely accepted hypothesis that hematopoietic stem cells and EPCs arose from a common precursor, the hemangioblast.52,53 Yet, subsequent observations utilizing fate mapping of murine embryonic cell populations in situ demonstrated that erythropoietic progenitors arose independently of ECs. 54 Similar findings from the developing vasculature within murine embryos are displacing the idea of a hemangioblast with the concept that while hematopoiesis and vasculogenesis occur almost simultaneously and in close proximity, the processes originate from distinct mesodermal precursors.25,50,55
Nonetheless, the hypothesis that hematopoietic stem cells and EPCs arose from a common hemangioblast precursor led to the seminal work of Asahara in 1997 and Rafii in 1998, which proposed that angioblasts, the endothelial precursor cells, could be isolated from human peripheral blood and bone marrow utilizing markers for hematopoietic stem cells, CD34, and ECs, CD34, and Flk-1/KDR.56,57 Specifically, Asahara showed that adherent CD34 antibody-captured mononuclear cells (CD34+ MNCs) possessed hematopoietic lineage through CD45 expression (27.2% ± 2.2%), as well as endothelial lineage through the expression of CD31 (71.5% ± 7.1%), Flk-1 (35.8% ± 8.8%), Tie-2 (54.6% ± 14.2%), E-selectin (9.0% ± 3.0%), and >80% uptake of DiI-labeled acetylated low-density lipoprotein (Fig. 1A). Furthermore, by demonstrating that CD34+ MNCs could assist in therapeutic angiogenesis on reinjection to ischemic tissue, Asahara catalyzed the idea of utilizing adult peripheral blood EPCs as an autologous cell therapy for patients afflicted with cardiovascular disease.56,58,59 Although the work of Asahara et al. was promising, it did not thoroughly confirm EPC identity of the examined CD34+ cell population. 56 As a result, studies attempting to replicate and compare Asahara's original findings to obtain a population of true EPCs, summarized in Table 1, have spanned nearly two decades of research. At present, a combination of surface antigen expression for EC markers and lack of expression for hematopoietic cell markers, as well as functional assays assessing proliferative and vasculogenic potential, is needed to define EPCs, summarized in Table 2. The rationale for these requirements is explained by a review and comparison of reported EPCs.
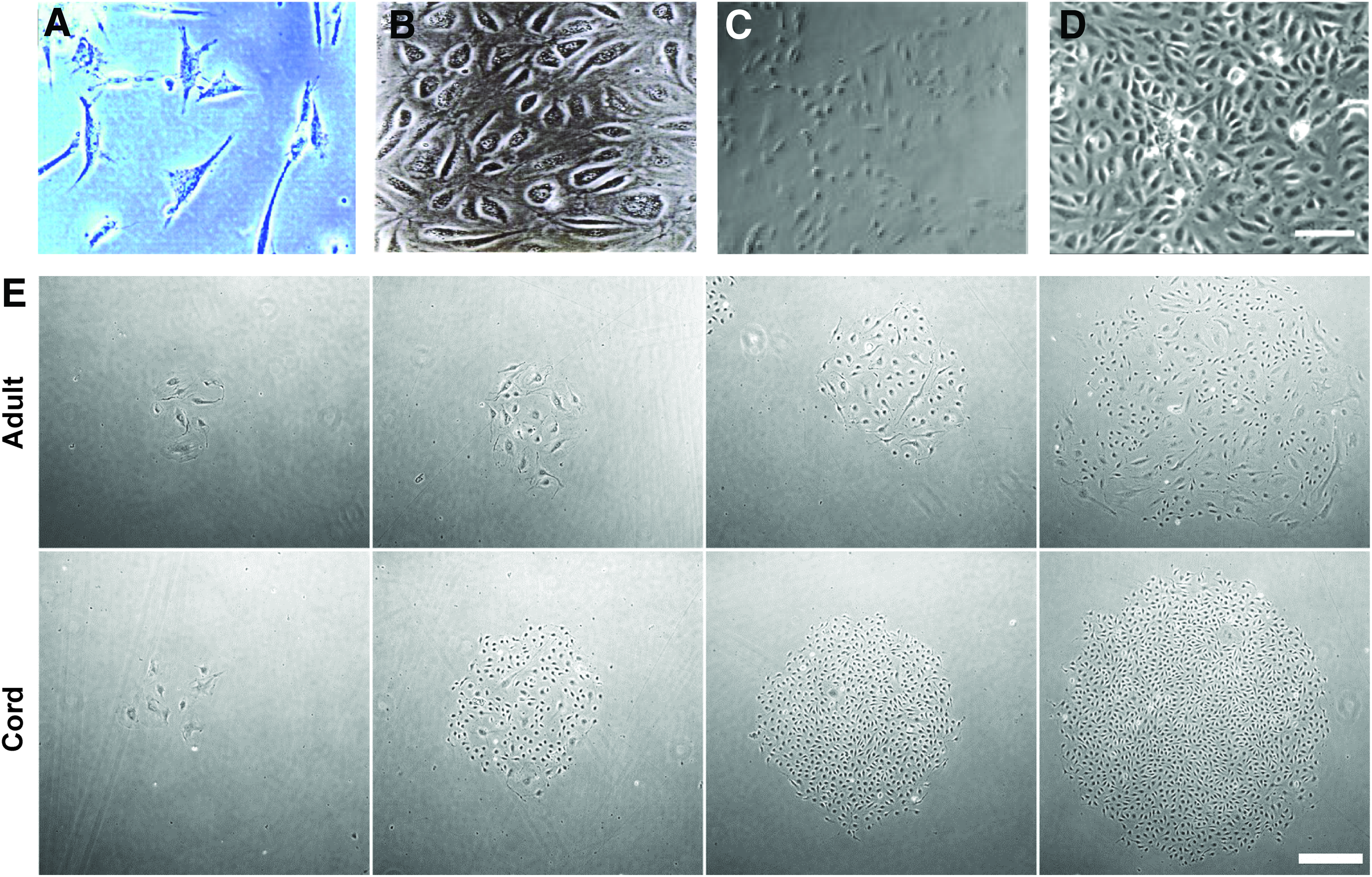
Photomicrograph comparisons of reported human EPCs.
α-SMA, alpha-smooth muscle actin; BBE, bovine brain extract; bFGF, basic fibroblast growth factor; BSA, bovine serum albumin; cAMP, cyclic adenosine monophosphate; DiI acLDL, DiI acetylated low-density lipoprotein; G-CSF, granulocyte colony-stimulating factor; EC, endothelial cell; EPC, endothelial progenitor cell; ecNOS, endothelial constitutive nitric oxide synthase; ECFCs, endothelial colony-forming cells; EDTA, ethylenediaminetetraacetic acid; FACS, fluorescence-activated cell sorting; FBS, fetal bovine serum; HBSS, Hank's balanced salt solution; hEGF, human epidermal growth factor; hiPSC, human-induced pluripotent stem cell; ICAM-1, intercellular adhesion molecule-1; IGF-1, insulin-like growth factor-1; IL-1, interleukin-1; LPS, lipopolysaccharides; MNCs, mononuclear cells; NRP-1, neuropilin-1; o-Me, 8-(4-chlorophenylthio)-2′-O-Me-cAMP; PB-MNC, peripheral blood mononuclear cells; ROCK, Rho-associating coiled kinase; SCID, severe combined immunodeficient; SCGF, stem cell growth factor; TCP, tissue culture plastic; TNF-α, tumor necrosis factor alpha; UEA-1, ulex europaeus agglutinin-1; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor.
Bone marrow-derived EPCs
Beginning in 1998, Rafii and colleagues expanded on Asahara's work by demonstrating that vascular endothelial growth factor (VEGF or VEGF-A) is critical to developing EC colonies from CD34+ MNCs derived from peripheral blood, bone marrow, and cord blood based on von Willebrand factor (vWF) expression and EC monolayer formation (Fig. 1B). 57 Yet, CD34 cannot serve as a unique EPC marker because it is expressed on both hematopoietic progenitor cells and mature ECs, as well as fibroblasts and epithelial cells. 60 To overcome this lack of specificity, in 2000, Gehling et al. tried to isolate peripheral blood EPCs using antibodies for CD133/AC133, a selective marker for CD34+ hematopoietic stem cells that does not recognize mature, differentiated CD34+ ECs.61,62 Gehling et al. observed that 2 weeks of CD133+ MNC treatment with VEGF and stem cell growth factor produced EC-like colonies containing Weibel-Palade bodies and expressing 30–35% CD34, 16–32% CD31, 48–56% VE-cadherin, >99.5% KDR/Flk-1, Tie-2, ulex europaeus agglutinin-1 (UEA-1), and vWF. 61
Around the same time of Gehling's work, however, Schmeisser et al. conducted a study that showed peripheral blood-derived monocytes, a potential progeny of CD133+ hematopoietic stem cells, could express vWF (45.2% ± 5%), VE-cadherin (12.4% ± 2%), endothelial constitutive nitric oxide synthase (ecNOS, 9.8% ± 3%), and form capillary-like network structures in vitro after 2 weeks of VEGF treatment. 63 The EC-like characteristics of monocytes continued to increase after 4 weeks of VEGF treatment, resulting in similar expression levels in EC markers vWF (94.2% ± 13%), VE-cadherin (89.7% ± 15%), ecNOS (58.8% ± 10%), as vascular EC controls, and decreased expression for hematopoietic markers CD45 (92.2% ± 6%) and CD14 (12.3% ± 4%). 63 Taken together with the knowledge that monocytes and macrophages can uptake acetylated low-density lipoprotein and express CD31, these findings suggest that instead of CD133+/CD34+ MNCs existing as hemangioblast progenitors as suggested by Asahara, Rafii, and Gehling, they could simply exist as monocytes, induced from hematopoietic progenitors to exhibit EC attributes on continuous VEGF treatment.25,56,57,60,61,63,64
Blood-derived EPCs
Lin and colleagues had previously shown that circulating mature, differentiated ECs were present in peripheral blood and possessed proliferative potential in vitro, leading to the hypothesis of an endothelium origin as likely as a hematopoietic origin for the CD34+ EPCs.56,57,65 EPC isolation by Lin et al. differed from Asahara and Rafii by utilizing adherence enrichment, rather than depletion, of MNCs, which were expanded on type I collagen-coated substrates (Table 1). 66 In 2004, Ingram et al. expanded on the work of Lin and colleagues by seeking a more functional property to characterize EPCs than cell surface antigens: clonogenic and proliferative potential assays. 28 These assays, used to describe hematopoietic progenitor cells, identified a hierarchy of EPCs classified by proliferation potential, highly proliferative potential-endothelial colony-forming cells (HPP-ECFCs) found in umbilical cord blood (hCB-ECFCs) and low proliferative potential-endothelial colony-forming cells (LPP-ECFCs) found in adult peripheral blood (hPB-ECFCs) (Fig. 1E). HPP-ECFCs could give rise to at least 10 million progeny from a single cell without compromising telomerase activity, which protects against senescence. Other characteristics that distinguished cord blood EPCs from peripheral blood EPCs included 15-fold higher numbers of EC colonies per equivalent blood volume, faster appearance in culture and formation of capillary-like structures in vitro using a Matrigel™ assay, and expansion for 100 population doublings before demonstrating cellular senescence, in contrast to 20–30 population doublings for hPB-ECFCs.
Continued characterization of the EPC-derived ECFCs against EC colony forming unit ECs (CFU-ECs), which resemble the EPCs reported by Asahara, showed that ECFCs have significantly higher expression for EC-associated markers CD105, CD144, CD146, UEA-1, and lacked expression for the hematopoietic cell-associated markers CD14, CD45, and CD115. 47 Only ECFCs could form secondary EC colonies through a single-cell clonogenic assay, while CFU-ECs could form secondary hematopoietic colonies of myeloid lineage with methylcellulose assays. CFU-ECs and ECFCs were also shown to be clonally unrelated from experiments with polycythemia vera patients, who contain a mutated gene, Janus Kinase 2 V617F, in their hematopoietic stem cells. 47 All CFU-ECs contained this gene, in comparison to less than 4% of ECFCs. Only ECFCs could form capillaries within collagen and fibronectin gels, which do not recruit vessel ingrowth in murine species that anastomosed to host vasculature and were perfused.47,67 Hence, only ECFCs can be defined as true EPCs and recommended for use in the vascularization of engineered tissues.68,69 As well, based on the evidence that both LPP-ECFCs and HPP-ECFCs reside within mature vasculature, the most likely origin of CD34+ EPCs is the endothelium of all tissue vascular niches rather than a unique progenitor cell residing within the bone marrow.70,71
Human-induced pluripotent stem cell-derived EPC
Although the EPC subset of ECFCs has potential for autologous angiogenic therapy, the peripheral blood of patients afflicted with preexisting disease, such as coronary artery disease, can contain reduced numbers of circulating EPCs with impaired angiogenic function.72–74 iPSC technology offers a potential solution by developing mesodermal precursor cells.75,76 In particular, Lian et al. showed that human fibroblasts, treated with nonintegrating episomal vectors containing OCT4, SOX2, NANOG, LIN28, c-Myc, and KLF4 genes, could induce CD34+CD31+ EPCs through glycogen synthase kinase 3 (GSK3) inhibition and subsequent activation of Wnt/β-catenin signaling (Table 1). 77 The hiPSC-EPCs expressed VE-cadherin, formed capillary-like network structures in vitro, demonstrated clonogenic ability, and exhibited physiological EC response to stimulation with inflammatory and permeability mediators (Fig. 1C). 77 However, hiPSC-EPCs generated only 20 population doublings over 60 days of culture, which is more characteristic of mature ECs rather than EPCs. 66
At the same time of Lian's work, Yoder and colleagues found that previously reported methods for deriving hiPSC-EPCs involving OP9 mouse embryonic stromal cell coculture, three-dimensional (3D) embryoid body formation, or continuous transforming growth factor beta (TGF-β) inhibition, yielded heterogeneous cell populations that did not maintain a stable EC phenotype after several weeks of culture.78–81 Instead, the group developed a single-step, serum-free, two-dimensional (2D) adherent cell culture, utilizing growth factors involved in EPC specification from the mesoderm—activin-A, BMP4, FGF-2, and VEGF165—to generate a highly proliferative population of ECFCs from hiPSCs (Fig. 1D). 78 hiPSC-ECFCs were selected by fluorescence-activated cell sorting (FACS) for neuropilin-1 (NRP-1) and CD31 expression. Endothelial precursors express NRP-1 before CD34 and CD31. 82 The NRP-1+CD31+ hiPSC-ECFCs displayed similar phenotype and proliferative potential as HPP-ECFCs, expanding to over 1012 progeny by 90 days of culture, and maintaining homogeneous EC phenotype, as assessed through NRP-1, CD31, CD144 expression as well as lack of alpha-smooth muscle actin (α-SMA) expression. 78
Furthermore, the hiPSC-ECFCs possessed vasculogenic potential, evidenced by capillary network formation in vitro atop Matrigel, and functional microvessel formation in vivo within collagen gels that were subcutaneously implanted into severe combined immunodeficient (SCID) mice. 78 Addressing concerns of safety with hiPSCs, Prasain et al. found no teratoma formation of hiPSC-ECFCs after 6 months of implantation. The hiPSC-ECFCs showed therapeutic promise in oxygen-induced retinopathy within C57/BL6 wild-type mice, and in ischemic limbs within athymic mice. 78 Prasain et al. also showed that NRP-1 has a critical role in hiPSC-ECFC generation through VEGF165-mediated KDR signaling. Intriguingly, NRP-1 activation in ECs, derived from the blood and vasculature of patients with peripheral artery disease, could increase proliferation and reduce apoptosis and senescence. 78 Taken together, these findings offer promising technologies to obtain therapeutic numbers of vasculogenic EPCs, autologously derived from patients with cardiovascular disease.
In summary, only ECFCs derived from adult peripheral blood and umbilical cord blood meet the criteria in Table 2 and are considered true EPCs.25,60,68 Ongoing EPC characterization efforts include advanced lineage tracing to assess whether ECFCs can generate the complete hierarchy of blood vessels, and gene expression analysis to determine whether ECFCs residing in the vascular niche of different organs possess similar angiogenic potential.25,68
EPC Vascular Morphogenesis and Differentiation
In the developing embryo, angioblasts, the embryonic EPCs, are derived from the mesoderm through the soluble effector, Indian hedgehog (IHH) and FGF-2, which act together to increase Flk-1/KDR expression.83,84 Flk-1+ EPCs respond to autocrine and endoderm-derived VEGF-A by differentiating toward a more mature EC phenotype, evidenced by expression of VE-cadherin, PECAM-1, Tie-1, Tie-2, and Flt-1.84–87 Extraembryonic vasculogenesis occurs first with migrating EPCs entering the yolk sac in response to chemotactic gradients formed by VEGF and semaphorins.87–89 Using αvβ3 integrins, EPCs coalesce with hematopoietic cells and form blood islands that fuse together on a fibronectin-rich extracellular matrix, generating the primary capillary plexi, shown in Figure 2.90–92 These primitive vascular networks remodel to form the vitelline circulation, the blood vessels responsible for transferring nutrients from the yolk sac to the embryo. 87
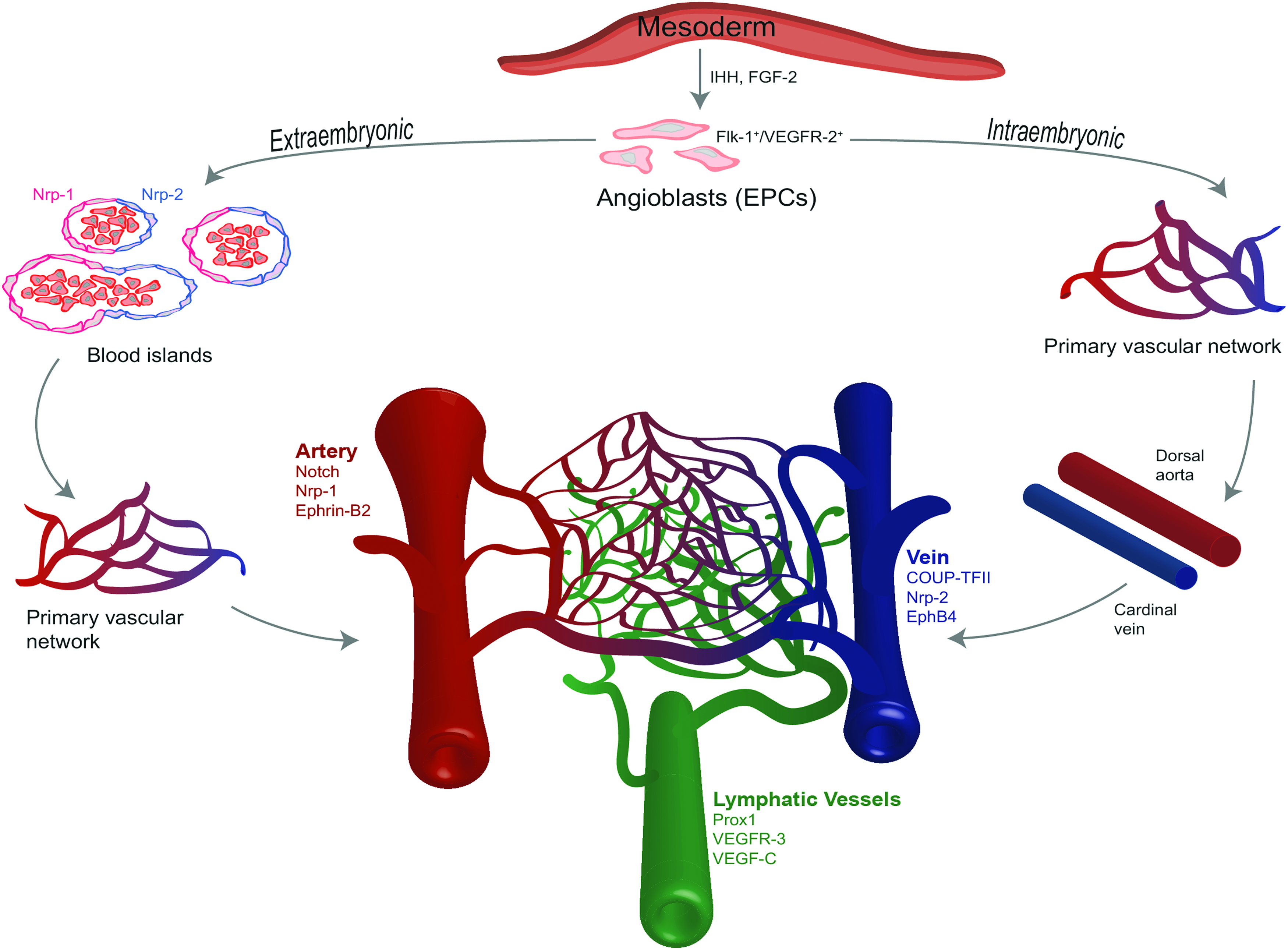
Overview of vasculogenesis within the vertebrae embryo. Mesodermal germ layer stimulation with FGF-2 and IHH proteins produces angioblasts/EPCs that express Flk-1/KDR/VEGFR-2. Locally produced VEGF-A in the extraembryonic yolk sac and intraembryonic embryo proper creates chemokine gradients that activate and recruit EPCs. VEGF-A stimulates EPC expression of VE-cadherin, PECAM-1, Tie-1, Tie-2, and Flt-1. In extraembryonic vasculogenesis, EPCs form an endothelial border enclosing primitive erythrocytes, generating blood islands. Expression of arterial Nrp-1 or venous Nrp-2 is present on blood island ECs before capillary formation and blood perfusion. Blood islands coalesce to create primary vascular networks that develop into the vitelline circulatory system. In contrast, during intraembryonic vasculogenesis, EPCs coalesce directly creating a primary vascular network that fuses to form the dorsal aorta and cardinal vein. Angiogenesis of the dorsal aorta and cardinal vein produces intersegmental blood vessels that further remodel to form the embryonic circulatory system, assisted by Notch signaling, Semaphorin, Netrin, and Ephrin proteins, as well as Slit ligands. Arterial specification is induced by VEGF-A, which binds to Nrp-1 and VEGFR-2/Flk-1 on ECs, inducing Notch signaling that results in Ephrin-B2 expression. Retinoic acid regulates venous specification by activating COUP-TFII to interact with Nrp-2 and inhibit Notch signaling, promoting EphB4 expression. A subset of venous endothelium expressing Sox18 interacts with COUP-TFII to upregulate VEGFR-3 expression, which acts together with VEGF-C to initiate lymphangiogenesis. Not shown are mural cells, pericytes, and vascular SMCs that closely associate with the vascular network, maintain endothelial quiescence, and regulate blood flow. COUP-TFII, chicken ovalbumin upstream promoter-transcription factor II; EC, endothelial cell; FGF-2, fibroblast growth factor; SMC, smooth muscle cells. Color images available online at www.liebertpub.com/teb
In contrast, dispersed angioblasts/EPCs initiate vasculogenesis in the embryo proper, coalescing to directly form primary vascular networks without intermediary blood island formation.88,89 These networks fuse together and undergo bidirectional remodeling processes to generate the dorsal aorta and cardinal vein.87,93,94 ECs sprouting from these vessels are guided by positive and negative signals from VEGFR-2 and VEGFR-1, respectively, creating an intersegmental blood vessel that remodels to form the embryonic circulation system, with vascular patterns guided by Notch and VEGFR-2 signaling as well as Semaphorin, Netrin, Ephrin, and Slit ligands.95–97 Angiogenesis is initiated by proangiogenic factors VEGF-A and Ang-2 that activate ECs lining the interior of mature vessels to remove intercellular junctional contacts, secrete matrix metalloproteinases (MMPs), which break down basement membrane proteins, detach adjacent mural cells, and assume a migratory, proliferative phenotype with protruding filopodia.98,99 VEGF-induced vessel permeability during early angiogenesis releases plasma protein fibronectin, vitronectin, and fibrinogen that interact with interstitial collagens to create a provisional matrix, which assists in angiogenic growth by sequestering VEGF-A to form a haptotactic gradient that directs EC migration.100–102 In addition to VEGF-A, vascular patterns are regulated by interactions of EC ligands plexin D1 and roundabout receptor (Robo) with semaphorin and Slit proteins, respectively.100,103,104 The sprouting vessels migrate along the chemoattractant gradient using integrins αvβ3 and αvβ5. 105
Vascular ECs that are exposed to the highest, local VEGF-A concentration differentiate to tip cells that lead the sprouting vessels. 98 Trailing the tip cells are stalk cells, which maintain connectivity with the parent vessel and proliferate to elongate the vascular sprout, as depicted in Figure 3A.98,100 Lateral inhibition from tip cells prevent stalk cell differentiation to tip cells. 98 Tip cell VEGFR-2 interaction with VEGF-A induces expression of the transmembrane Notch ligand, Delta-like 4 (DLL4), which interacts with Notch receptors expressed on neighboring stalk cells to initiate Notch signaling. Stalk cell notch intracellular domain is activated to transcriptionally increase VEGFR-1 expression and decrease VEGFR-2 and Nrp-1 expression.100,106 VEGFR-1 traps VEGF, preventing tip cell differentiation of stalk cells by acting as an antagonist of VEGF-mediated signaling.98,100,107
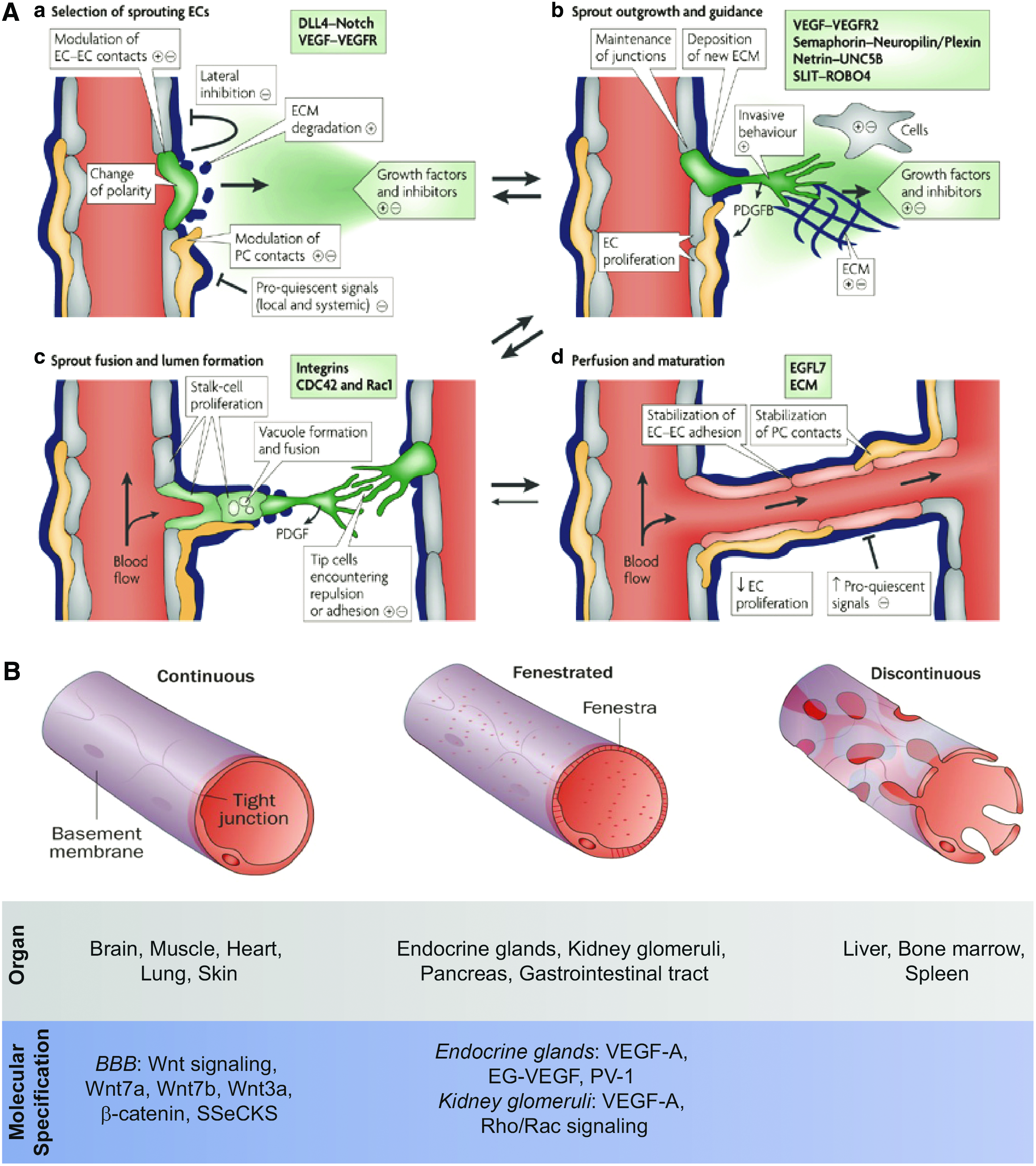
Review of angiogenesis and endothelium specification.
Stalk cells develop lumen by first establishing apicobasal polarity through β1 integrin interactions with the underlying basal matrix, enabling protease activated receptor 3 (PAR3)-mediated redistribution of junctional EC proteins, zonula occludens (ZO-1), VE-cadherin, claudin-5, from the apical surface to the sprouting EC cord periphery.98,108 Signaling of VEGFR-2 and Rho-associating coiled kinase (ROCK) then recruits actomyosin proteins, such as F-actin, to build lumen.98,109 Epidermal growth factor-like domain-7 (EGFL7), secreted into the extracellular matrix by neovessel ECs, has also been shown to regulate tubulogenesis. 110 Fusion of intracellular vacuoles can also create lumen with the assistance of Rho family GTPases Cdc42 and Rac1.111,112 For example, EPC tubulogenesis in vitro was shown to occur through pinocytosis and fusion of intracellular vacuoles, regulated by MT1-MMP and Cdc42.17,113 Remodeling of the surrounding extracellular matrix occurs by release of additional MMPs and tissue inhibitor of metalloproteinases. 105 Angiogenesis ends when tip cells reach an adjacent vessel, initiating intercellular junctional contacts and anastomosis. 98 To further stabilize the nascent vasculature, tip cells secrete platelet-derived growth factor subunit B (PDGF-B), which binds to the surrounding extracellular matrix, creating a haptotactic gradient that recruits mural cells to PDGF-B via platelet-derived growth factor receptor beta (PDGFR-β).6,98,114,115 Once mural cells contact the vessel endothelium, ECs release sphingosine 1-phosphate, which recruits endogenous neural cadherin (N-cadherin) within ECs to create adherens junctions between ECs and mural cells. 116 TGF-β1 is then released by vascular ECs to induce mural cell differentiation toward a vascular smooth muscle cell (SMC) phenotype, which can regulate vascular blood flow.6,117 Basement membrane proteins also promote vessel stabilization through EC interactions with the laminin-α4 integrin, which promotes Notch signaling-induced EC quiescence.100,118
Differentiation of newly formed vasculature toward the arterial, venous, and lymphatic lineages is genetically predetermined, as evidenced by expression of arterial Nrp-1 and venous Nrp-2 on blood island EPCs prior capillary plexus formation and onset of blood flow.119,120 Yet, hemodynamic forces, such as the high pressure, high shear stress, pulsatile flow in arteries and the low pressure, low shear stress flow in veins, are essential to regulating and maintaining venous or arterial fate.120–123 Arteriovenous specification begins with notochord-secreted sonic hedgehog ligand, which promotes VEGF-A secretion from somites.120,124–126 A VEGF gradient is established, starting at the dorsal artery and continuing toward the cardinal vein.125,127,128 For arterial differentiation, VEGF-A binds to heterodimers of Flk-1/VEGFR-2 and Nrp-1 to activate Notch signaling, which increases Ephrin-B2 expression and suppresses EphB4 expression.88,120,124,129–131 Venous specification is initiated by retinoic acid, which activates a nuclear orphan receptor, chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII).120,125,132 COUP-TFII suppresses Notch signaling by interfering with Nrp-1 function, consequently increasing EphB4 expression. 132
Lymphatic vasculature differs from arterial and venous vasculature through lack of continuous basement membrane and loose intercellular junctions, and transport of interstitial tissue fluid instead of blood.122,133 Lymphatic ECs are produced by a subpopulation of cardinal vein cells that express lymphatic vessel hyaluronan-1 receptor-1 and induce Sox18 expression.125,134 Sox18 cooperates with COUP-TFII to stimulate prospero-related homeobox-1 (Prox1) transcription factor expression.120,135,136 Prox1 upregulates VEGFR-3 expression, which acts together with its coreceptor Nrp-2 to respond to VEGF-C.122,137–140 VEGF-C induces lymphatic ECs to create lymph sacs that have sprouting, migratory lymphatic ECs which form the lymphatic plexus that further remodels to form lymphatic capillaries and collection vessels and ducts.120,122,141,142 Still, mature vascular ECs are not permanently confined to arterial, venous, and lymphatic fate. Postnatal reprogramming ECs to an alternate macrovascular fate can occur when hemodynamic forces and levels of COUP-TFII and Prox1 expression are altered, consequently affecting Notch signaling and subsequent Ephrin-B2 and EphB4 expression.121,125,132,143–145
The entire macrovascular and microvascular hierarchy must undergo further differentiation to accommodate the unique function and metabolic demands of each organ. 125 Organ-specific specialization of capillary plexus EPCs is a consequence of local factors secreted by tissue cells.122,146 Currently, three known forms of endothelium structure exist: nonfenestrated continuous, fenestrated continuous, and fenestrated discontinuous, as illustrated in Figure 3B.125,147 Fenestrae are porous openings in the vascular wall that enable the swift exchange between tissue constituents and circulating blood.125,148 Fenestrae range in size from 60 to 200 nm and may contain diaphragms, spoke wheel-like subdivisions that filter blood based on solute size.125,148,149 Nonfenestrated, continuous endothelium is found in the brain, muscle, heart, lung, and skin. 125 These organs provide critical motor, respiratory, and protective functions that require endothelium with strong barrier properties. Accordingly, nonfenestrated continuous capillaries are characterized by tight junctions and adherens junctions as well as caveolae and vesiculo–vacuolar organelles that regulate permeability.125,150–152
At present, the understanding of endothelium specification toward nonfenestrated continuous phenotype is limited to studies involving the blood–brain barrier (BBB). The BBB tightly controls transendothelial passage of biomolecules and cells by tight junctional occludin and claudin proteins, and selective membrane transporter systems that comprised glucose transporter type 1 (Glut-1) and ATP binding cassette transporter proteins. 153 Together with canonical Wnt signaling, Wnt7a and Wnt7b genes promote Glut-1 expression.146,154–156 Claudin-3 tight junction expression in the BBB is regulated by the Wnt3a gene as well as EC-derived β-catenin protein, which induces claudin-3 expression while simultaneously reducing the expression of fenestra-promoting type II membrane glycoprotein plasmalemmal vesicle-associated protein-1 (PV-1).146,156 Brain-specific pericytes, astrocytes, and neurons provide local cues that assist in EC differentiation by regulating junctional protein expression and transport vesicles.125,157 As an example, to maintain BBB integrity, astrocytes produce Src-suppressed C kinase (SSeCKS), stimulating Ang-1 production while also suppressing VEGF, a permeability enhancer. 158
Kidney glomeruli, pancreas, intestine, and endocrine glands all contain fenestrated endothelium. 146 Endothelium differentiation to diaphragm-containing fenestrae found on endocrine glands is regulated by PV-1 and induced by VEGF-A and endocrine gland-VEGF (EG-VEGF).122,159–161 Although the fenestra of the kidney glomerular endothelium is also induced by VEGF, diaphragm expression is lost after development.125,161,162 Podocytes provide the local source of VEGF to induce EC differentiation. 163 The podocyte VEGF activates the Rho/Rac signaling pathway to reorganize the EC actin cytoskeleton, and form fenestrae.164–166 The current understanding of endothelium specification to fenestrated discontinuous phenotype is limited to anatomical descriptions.167,168 This endothelium type is found in the liver and bone marrow and characterized by a lack of continuous basement membrane and the presence of clathrin-coated pits and vesicles that regulate endocytosis. 125 The fenestrae are large, ranging 100–200 nm in diameter, and lack diaphragms, facilitating the release of niche cells and metabolites. 149
Tissue Engineering Vascular Niches with EPCs
Establishing design parameters for generating EPC-vascularized, tissue niches in vitro is a daunting task because cells, scaffold, and culture media must be optimized to provide the distinct biochemical and biophysical signals of each niche. Fortunately, several in vitro model systems for investigating microvessel formation utilizing vascular-derived ECs have been established.16,169–171 These systems mimic in vivo vasculogenic and angiogenic processes by incorporating ECs with stromal cells, such as mural cells and fibroblasts, within scaffolds that mimic the provisional matrix, supplemented with media containing VEGF-A and bFGF. These initial design parameters have been tailored to tissue engineer microvessels from EPCs.34,172 The EPC-derived microvessels described in this section are restricted to EPC subtypes that possess true EPC character, currently only hCB-ECFCs or hPB-ECFCs.25,60,68,69 Classifying the EPC progeny as ECFCs is due to the loss of expression for the progenitor-associated markers CD34 and CD133 during expansion, and is often interpreted as EPC differentiation toward a mature endothelial phenotype. 172 Yet, given that neither CD34 nor CD133 has been shown to regulate EPC vasculogenic function or differentiation toward organotypic endothelium, to describe expanded EPCs and differentiated ECs is misleading. Therefore, the terms EPC and ECFCs have been used synonymously.
Beginning in 2004, Bischoff's group demonstrated safe expansion of hCB-ECFCs to therapeutically relevant numbers by 3 weeks in vitro, assessed through continued expression of CD31, VE-cadherin, vWF, E-selectin, and KDR. 172 The group also showed that mural cells were required for in situ hCB-ECFC microvessel formation within a polyglycolic acid-poly-L-lactic acid (PGA-PLLA) bioresorbable scaffold. 172 Without direct mural cell coculture, hCB-ECFCs and hPB-ECFCs fail to generate microvascular networks in vitro and in vivo.33–35,44,172
In 2008, Jain's group examined whether the de novo vasculature generated from hCB-ECFCs or hPB-ECFCs, in coculture with 10T1/2 murine mesenchymal progenitor cells (MPCs) within fibronectin/collagen gels, remained stable after several weeks of implantation into SCID mice. 173 hCB-ECFCs were capable of generating functional blood vessels that were durable for at least 4 months, as demonstrated by intravital microscopy of green fluorescent protein (GFP)-labeled hCB-ECFC microvessels perfused with rhodamine-conjugated dextran. 173 Intravital microscopy permeability measurements against 67 kDa Texas Red-labeled bovine serum albumin showed that hCB-ECFCs possessed permeability values similar to normal brain capillaries, 0.73 ± 0.21 × 10−7 cm/s.173,174 In addition, there were no significant differences between hCB-ECFC and host microvessels in red blood cell velocity or the ability to recruit leukocytes on treatment with interleukin-1β (IL-1β). 173 In contrast, hPB-ECFC and 10T1//2 cocultures produced unstable microvessels that regressed after 3 weeks of implantation in vivo. 173
Melero-Martin et al. expanded on the work of Jain's group by replacing the 10T1/2 cells with clinically viable mesenchymal stem/progenitor cells, derived from human adult bone marrow (bm-MPCs) or umbilical cord blood (cb-MPCs). 35 Both cb-MPCs and bm-MPCs could support hCB-ECFC and hPB-ECFC vasculogenesis in Matrigel constructs implanted within athymic mice for at least 1 week in vivo, differentiating toward α-SMA+, pericyte-like cells that covered EPC neovessels. 35 As well, hCB-ECFC microvessels resulting from bm-MPC cocultures were functional for at least 4 weeks in vivo based on erythrocyte perfusion. 35 Yet, for a tissue-engineered system to be considered as a therapeutic device, all design parameters must be clinically acceptable, prohibiting systems utilizing Matrigel, a xenogeneic-derived scaffold.
To date, a small number of studies have successfully tissue engineered vascular niches utilizing bona fide EPCs, hCB-ECFCs, or hPB-ECFCs, with translatable coculture cells and scaffolds. These include the adipogenic, vasa vasorum, dermal, osteogenic, and pulmonary tissue niches, summarized in Table 3. As well, a few groups have attempted to mimic the embryonic niche by incorporating both endothelial and MPCs within a synthetic scaffold.17,175 The advantage of this approach is the potential to generate the vascular niche of any organ, provided the appropriate differentiation cues are present within the scaffold and culture media. Few studies have attempted to understand the singular effects of coculture cell type, scaffold mechanical properties, or media composition on in vitro EPC network formation. Still, there are common themes across vascular niche design that are critical to generating stable, functional EPC microvessels in vitro.
All cells are derived from human sources.
3D, three-dimensional; AoSMC, aortic smooth muscle cells; BM-MSCs, human bone marrow-derived mesenchymal stem cells; CB-MNCs, cord blood MNCs; ECGS, endothelial cell growth supplement; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; FCS, fetal calf serum; FGM-2, fibroblast growth medium; GelMA, gelatin methacrylate; GPS, glutamine–penicillin–streptomycin; hASC, human adipose stromal cells; hDFs, human dermal fibroblasts; MMP, matrix metalloproteinases; MSCGM, mesenchymal stem cell growth media; NHLDFs, normal lung-derived fibroblasts; PDGFR-β, platelet-derived growth factor receptor beta; PEG, polyethylene glycol; PGA-PLLA, polyglycolic acid-poly-L-lactic acid; PNIPAM, poly(N-isopropylacrylamide); pOBs, primary osteoblasts; PSA, penicillin–streptomycin–amphotericin; vSMCs, vascular smooth muscle cells.
Coculture choice
In addition to mural cells, coculture with fibroblasts, stromal, parenchymal, and mesenchymal stem cells (MSCs) or MPCs, is critical to developing EPC microvessels in vitro and in vivo.17,34,176–178 These coculture cells serve as a source of angiocrine factors that function beyond stabilizing neovessel formation to initiating and supporting vasculogenic and angiogenic processes. For example, in vitro capillary formation from EC elongation and intercellular adherens junctions requires mural cell VEGF and EC-associated VEGFR-2 signaling, and cannot be simulated with exogenous VEGF or mural cell-conditioned media. 16 As well, in vitro capillary lumen formation from EC and fibroblast cocultures depends on the combined expression of fibroblast-associated extracellular matrix proteins collagen I, insulin growth factor-binding protein 7, procollagen C endopeptidase enhancer 1, transforming growth factor-β-induced protein ig-h3, and secreted protein acidic and rich in cysteine (SPARC). 179 Coculture cells also provide immunogenic shielding properties, as recently reported by Souidi et al., which showed that MSCs downregulated human leukocyte antigen-antigen D related (HLA-DR) expression on hCB-ECFCs in vitro and in vivo, reducing EPC-mediated allogenic T cell proliferation. 180
Parenchymal cells are often the initial coculture cell choice for engineering vascularized tissues, grounded on the assumption that mature niche cells can provide differentiation cues to EPCs, resulting in the desired endothelium phenotype. Wu et al. mimicked the vasa vasorum niche by coculture of hCB-ECFCs with human saphenous vein-derived SMCs within PGA-PLLA constructs, yielding lumenized microvessels after 6 days of ex vivo culture. 172 In addition, hCB-ECFC and human aortic SMC coculture, with minimal supplemental growth factors, gave rise to neovessels that contained properties of physiological microvessels, such as quiescence and lumen formation. 32 When encapsulated in a polyethylene glycol (PEG) hydrogel system, the hCB-ECFC microvessels expressed intercellular EC junctional proteins VE-cadherin and connexin 32, endothelial nitric oxide synthase (eNOS), perivascular localization of α-SMA+/PDGFR-β+ cells, and basement membrane formation with collagen IV and laminin, shown in Figure 4A–C. 33 Fuchs et al. determined that human osteoblasts could support microvessel formation of hPB-ECFCs, generating lumenized microvessels with intercellular tight junctions after 1 week of in vitro culture. 15
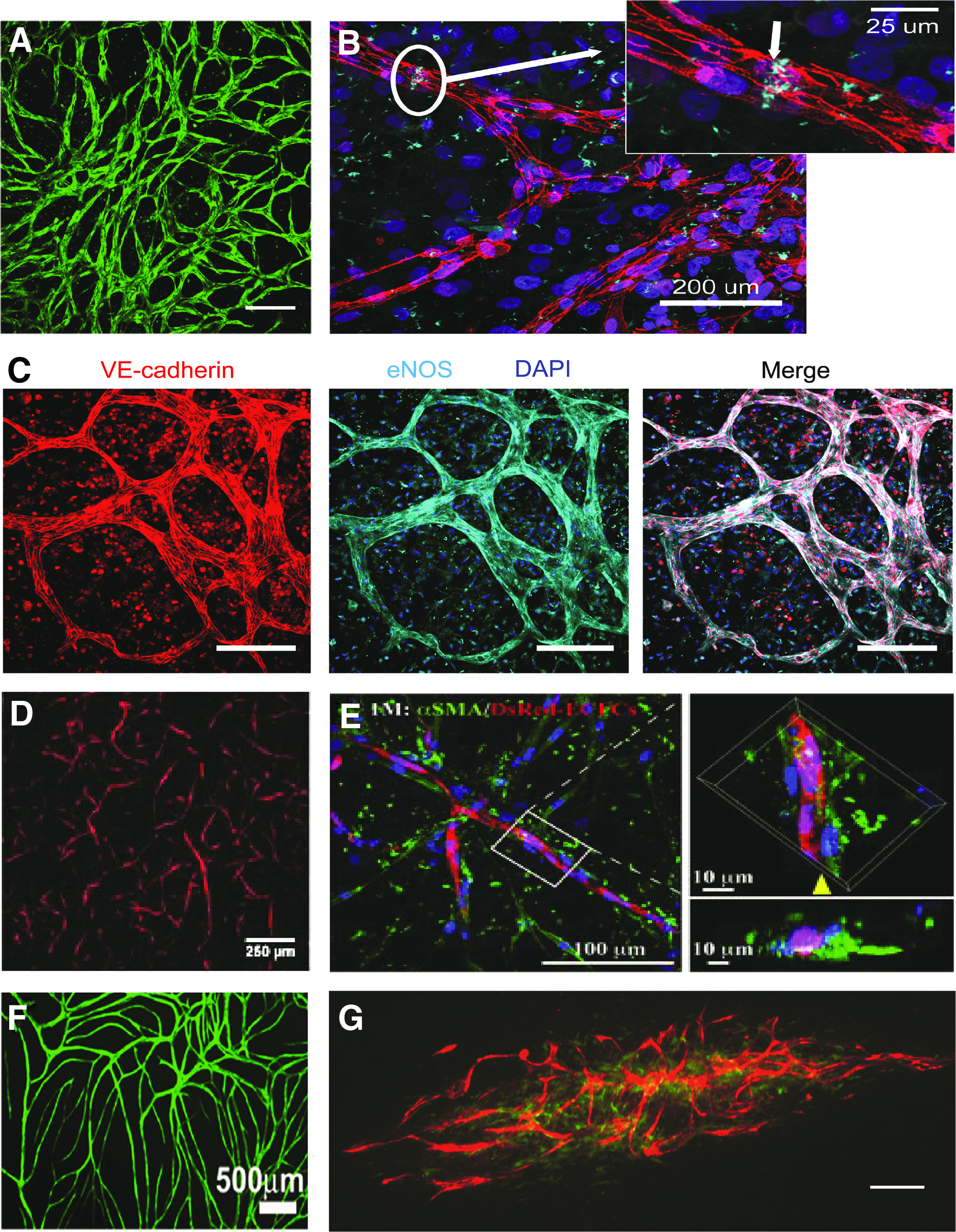
Representative images of microvessels that were tissue engineered from hCB-ECFC cocultures in vitro.
Even so, mural and parenchymal cells have major drawbacks limiting their application in regenerative medicine, including invasive isolation procedures and limited expansion potential. Tissue stromal cells, such as dermal fibroblasts and adipose stromal cells, require less invasive isolation, have expansion potential, and can support EPC microvessel formation.176–178,181 For instance, Athanassopoulos et al. demonstrated hCB-ECFC and dermal fibroblast cocultures could vascularize clinical-grade, dermal-substitute scaffolds. 181 George's group also found that fibroblasts, derived from human lung tissue, supported hCB-ECFC microvessel formation within fibrin gels in situ and differentiate toward an α-SMA+ myofibroblast, pericyte-like phenotype that encircled the EPC vessels. 176 Furthermore, the prevascularized fibrin constructs anastomosed with host vasculature and were perfused within 1 day of implantation. 176 Offering promise for reconstructive surgery application of EPCs, Traktuev et al. revealed that coculture with liposuction-derived adipocytes and stromal cells within collagen/fibronectin gels could support hCB-ECFC microvessel formation, express α-SMA+, exhibit perivascular localization, and protect against hCB-ECFC apoptosis within SCID mice. 178
All the same, stromal cells possess limited differentiation potential toward niche parenchymal cells and, in the case of lung fibroblasts, are not easily accessible. Using multipotent MSCs from adipose, bone marrow, umbilical cord blood, and amniotic fluid can overcome this barrier. During vasculogenesis and angiogenesis, MSCs/MPCs undergo mural cell differentiation on recruitment to neovasculature by means of EC-secreted TGF-β1 and gap junctions.122,182,183 Chen et al. demonstrated that bone marrow-derived MSCs (BM-MSCs), cocultured with hCB-ECFCs within gelatin methacrylate hydrogels, could support vasculogenesis and differentiate toward mural lineage cells expressing α-SMA and smooth muscle-myosin heavy chain (SM-MHC) within 1 week of culture in vitro (Fig. 4D, E). 17 Athanassopoulos et al. also demonstrated BM-MSC support of hCB-ECFC microvessel formation in vitro, with no significant differences in total tubule length, number of tubules, or number of junctions in comparison to dermal fibroblast cocultures after 2 weeks of culture. 181 Encouraging pediatric applications with EPCs, MSCs, and MPCs derived from amniotic fluid and umbilical cord blood have been shown to support hCB-ECFC network formation in vitro (Fig. 4F, G).43,184
Adipose-derived stem cells, another promising autologous coculture cell choice, contain multilineage differentiation potential as well as support EC network formation.185,186 Adipose-derived stem cells can be produced following clinical-grade procedures and used safely in patients. 187 Mural cells can also be directly transdifferentiated from blood-derived ECFCs through myocardin (MYOCD) expression, exhibiting SMC phenotype and function through smooth muscle aortic alpha-actin (ACTA2) and myosin heavy chain 11 (MYH11) expression, contractility, and calcium signaling activity. 188 This finding offers promise for utilizing blood as a single tissue source to isolate both mural and ECs.
Still, differences exist among mural, stromal, and MSC/MPC ability to support EPC network formation, which may be masked from the presence of angiogenic growth factors commonly used in the culture medium. Supporting this hypothesis, we found that when culture media contained no exogenous angiocrine factors beyond those present in serum, only vascular SMCs and cb-MPCs, in contrast to brain pericytes and BM-MSCs, could support hCB-ECFC network formation.43,44 Moreover, without the addition of exogenous VEGF, FGF-2, EGF, hydrocortisone, insulin growth factor-1, ascorbic acid, and heparin, hCB-ECFC networks formed in coculture with BM-MSCs regressed and underwent cell loss by 2 weeks of culture, possibly due to trace amounts of CD45+ cells with phagocytic potential. 44 As well, ex vivo expanded BM-MSCs secrete less than half of the angiocrine factors produced by amniotic fluid-MSCs and dermal fibroblasts, which includes molecules that regulate tubulogenesis, FGF-2, MMP-9, and endothelium differentiation, EG-VEGF, VEGF-C. 184
In addition to coculture choice, parameters of coculture ratios, seeding densities, and coculture spatial arrangement can also influence EPC network formation.32,33,176,181,189 The optimal coculture design parameters depend on whether the cells are grown atop a 2D biomaterial or within a 3D scaffold. In the context of cell-sheet tissue engineering technologies, 2D systems with mixed coculture spatial arrangements yield more connected, homogeneous networks than lamellar systems. 32 Alternatively, a sandwiched coculture system containing a layer of hCB-ECFCs between fibroblast monolayers can also produce interconnected, lumenized microvessels. 177 Uniformly distributed cells enable greater sprouting and interconnected networks than spheroid cell aggregates in 3D systems.17,33,176,189 As well, the ideal coculture ratio to promote hCB-ECFC microvessel formation is 1:1 for 3D systems and near 1:4 for 2D systems.17,32,33,176,181,184 Increasing densities of supportive stromal cells can enhance the rate of microvessel formation in vitro and anastomosis rate in vivo in 3D systems, but this effect was not observed in 2D systems.176,181
Scaffold
Fibrin gels have been approved for patient use, possess excellent cytocompatibility, and have shown success as a tissue engineering scaffold. 190 Fibrin gelation occurs by recapitulating the blood coagulation cascade using thrombin, fibrin, and calcium chloride. Calcium activates thrombin to cleave fibrinogen, generating fibrin. 191 As well, fibrinolytic inhibitors aprotinin and tranexamic acid can adjust fibrin degradative properties. 191 The mechanical properties of fibrin gels can be tuned by varying thrombin concentrations. 192 Although collagen and fibrin gels can support EPC microvessel formation in vitro and in vivo, they possess batch variability and allow limited control over degradation rate and mechanical properties.17,193,194
On the contary, while having tunable mechanical properties, semicrystalline polymers, such as PGA-PLLA, have limited ability to incorporate bioactive cues to imitate tissue niches.172,195 A potential solution proposed by Okano's group is cell-sheet-based tissue engineering technology, which has the advantage over 3D scaffolds in mimicking microenvironments of niche cells by conserving their extracellular matrix and intercellular connections formed during ex vivo culture. 196 As well, cell-sheet tissue engineering offers a lower risk of toxicity to cells that can occur from polymer degradation by-products.195,196 Cell sheets are produced from tissue culture plastic covalently grafted with poly(N-isopropylacrylamide) (PNIPAM) polymer chains that swell at temperatures below its lower critical solution temperature of 32°C, due to increased aqueous solubility, repelling the cell monolayer from the culture dish as a continuous cell sheet. 196 Sasagawa et al. demonstrated that EPCs, sandwiched between human dermal fibroblast layers, formed network structures by 3 days in vitro that contained partially formed lumens. 177 These preformed networks connected with host vasculature and were perfused with blood by 1 week after subcutaneous transplantation. 177 Yet, the cell-sheet engineering approach is limited by poor control over microarchitecture and mechanical strength.
Chemically modifying extracellular matrix proteins to create hydrogels can improve the mechanical properties of naturally derived gels while maintaining their biofunctional properties. Gerecht's group has explored hyaluronic hydrogel design factors of matrix viscoelasticity and adhesion peptide density to support EPC capillary formation and uncover underlying mechanisms of vascular morphogenesis.113,197 Hyaluronic acid is a glycosaminoglycan present during embryonic development. hCB-ECFCs express hyaluronic acid receptors and can interact with hyaluronic acid hydrogels through surface membrane CD44. 197 Gerecht found that crosslinking thiol-modified hyaluronic acid-gelatin hydrogels with a chemical crosslinker, PEG diacrylate, allowed greater control over physical properties and microarchitecture.
Specifically, soft hydrogels with a Young's modulus of 10 Pa supported hCB-ECFC vascular formation, from monoculture, to a significantly greater extent than 73 Pa hydrogels, as assessed by tube length and tube area. 197 In contrast, hCB-ECFCs remained rounded without observable vascular morphogenesis in 640 Pa hydrogels. The extensive network formation in the 10 Pa hydrogel was correlated with 200-μm average diameter microchannels that were not observed in the 73 or 640 Pa hydrogels. 197 Similarly, Khademhosseini's group chemically functionalized gelatin with methacrylate groups to produce a photocrosslinkable hydrogel that could support hCB-ECFC microvessel formation in coculture with BM-MSCs in vitro and within athymic mice in vivo. 17 They found that the degree of methacrylation affected microvessel morphology and MSC mural differentiation, with softer, 1 M methacrylate gels (2.0 ± 0.18 kPa) stimulating BM-MSC expression of α-SMA and SM-MHC, increasing microvessel density and lumen diameter in vivo in contrast to stiffer, 10 M methacrylate gels (4.5 ± 0.33 kPa). 17
Despite these advantageous, chemically modified biological gels do not allow precise control over biochemical and biophysical cues to recapitulate each tissue niche. The extracellular matrix composition is unique to each tissue niche, with varying amounts of constituent proteins, such as collagen, elastin, and fibrin, which activate separate cell signaling pathways to create the distinct cell phenotypes of each tissue. 198 A promising candidate for tailored scaffold microenvironments in vascular niche tissue engineering is PEG hydrogel. PEG hydrogels are considered a “blank slate” that resists protein adsorption, can incorporate specific cell-adhesive and protease-sensitive peptides in addition to angiogenic factors.199,200 We found that PEG hydrogels, (18 ± 5 kPa) containing fibronectin-derived RGDS adhesive peptide and MMP-2/9 sensitive peptides, provided an adequate microenvironment to induce vasculogenesis of hCB-ECFCs within SMC coculture, with minimal supplemental growth factors. 33 Importantly, this system used a mild photocrosslinking process, suitable for encapsulation of progenitor and stem cells.33,201
Culture media
Currently, almost all tissue-engineered models of EPC microvessel formation utilize culture media enhanced with angiogenic growth factors and animal-derived serum (Table 3). This is problematic as continuous supply of angiogenic factors may not occur on implantation of the construct in vivo, leading to vessel regression. Although a few systems can support EPC microvessel formation without additional angiogenic factors, animal-derived serum is still required, which is immunogenic and clinically unacceptable.33,43 Fortunately, xenogeneic-free methods exist for EPC expansion that replaces animal-derived serum with human platelet lysates, which contain VEGF, PDGF-AB, and PDGF-BB, and preserves EPC vasculogenic potential in vivo.202,203 Palecek's group has also developed a translatable culture media to support hiPSC-EPCs, consisting of basal media and ascorbic acid. 204
In summary, when combined with appropriate coculture cells in natural or synthetic biomaterials, EPCs from hCB-ECFCs can form microvascular structures in vitro and in vivo that mimic vascular tissue niches. Conversely, hPB-ECFC-derived microvessels lack stability and durability in vitro and in vivo.173,181 Cell engineering strategies, notably iPSC technology, have the potential to improve the expansion and vasculogenic potential of hPB-ECFCs, significantly broadening EPC therapeutic applications.
In Vitro Differentiation of EPCs to Organotypic Endothelium
The intimate associations between microvascular ECs and parenchymal cells enable exchange of soluble and membrane-bound angiocrine signals that provide essential induction signals for organogenesis. 205 For example, during liver development, hepatocyte invasion is significantly impaired in Flk-1/VEGFR-2-negative embryos.205,206 In addition, without the presence of microvascular ECs during pancreatic tissue development, insulin production is stopped and pancreatic cell differentiation is inhibited.205,207 In the adult, vascular ECs serve as professional niche cell regulators of tissue function, producing angiocrine factors that orchestrate stem cell self-renewal and fibrosis-free organ regeneration.205,208,209 Vascular ECs also convey cues between the intravascular to extravascular space that control tissue metabolic activity and homeostasis. 205
Expanded hCB-ECFCs possess an intermediate arteriovenous phenotype based on similar levels of gene expression for EphB4, Nrp-2, Notch4, and Ephrin-B2. 210 Intriguingly, hPB-ECFCs contain populations of both blood vascular and lymphatic ECs, based on expression of VEGFR-1 and VEGFR-3/Prox1, respectively, suggesting potential for use of blood-derived ECFCs in blood vascular and lymphatic tissue engineering. 211 Yet, the molecular basis for EPC specialization to arteriovenous and organ-specific vasculature is not well known. Methods to induce tissue-specific endothelium differentiation in vitro include coculture with parenchymal cells, directed differentiation, and direct reprogramming. 212
For example, Boyer-Di Ponio et al. found that early passage hCB-ECFCs, indirectly cocultured with rat-derived astrocytes, differentiate toward cerebral microvascular cells that possess characteristics of the BBB based on decreased permeability and increased expression of tight and adherens junction proteins. 210 Specifically, after 2 weeks of coculture, hCB-ECFCs, plated atop polyester cell culture inserts, demonstrated a significant decrease in permeability against 457 Da fluorescent small molecules, at 2.05 × 10−5 cm/s, in comparison to hCB-ECFC monocultures. Furthermore, hCB-ECFCs increased gene and protein expression of Glut-1 and occludin, and more homogeneous expression of VE-cadherin, ZO-1, claudin-3, and claudin-5 than hCB-ECFC monocultures. 210 Moreover, Boyer-Di Ponio et al. used directed differentiation of hCB-ECFCs to an arterial phenotype by treatment with 50 ng/mL of VEGF, which promoted Notch signaling and increased gene expression of arterial associated markers Ephrin-B2, DLL4, Notch 3, HES1, HEY1, and HEY2. 210 Palecek's group utilized directed codifferentiation of hiPSCs through activation of Wnt/β-catenin signaling to produce neural progenitor cells and cerebral microvascular ECs 213 The cerebral ECs displayed permeability of 0.37 × 10−5 cm/s against 180 Da glucose molecules and continuous cell–cell contact expression of ZO-1, occludin, and claudin-5. 213
Examining gene transcriptional profiles for organotypic endothelium may identify molecular candidates to induce EC specification. Providing data for this approach, Nolan et al. performed a systematic investigation to determine the unique molecular signatures of vascular endothelium from murine liver, bone marrow, heart, brain, lung, spleen, kidney, muscle, and testis. 26 They showed that each tissue expresses distinct clusters of angiocrine factors, with the ETS transcription factor family being likely responsible for regulating organotypic endothelium differentiation.26,214 Specifically, SFPI1 was correlated with differentiation toward fenestrated, discontinuous endothelium due to its high expression in the bone marrow and liver vasculature. 26
Future Perspectives
Although significant progress has been made toward utilizing EPCs for vascularizing engineered tissues, there is no evidence of EPC integration with tissue niche parenchymal and stromal cells to produce organotypic endothelium in vitro. Contributing to this lack of progress is the common approach to generate vascularized tissues in vitro, which is mixing EPCs or ECs alongside tissue stromal and progenitor cells, utilizing previously optimized growth conditions for the tissue of interest. The fallacy in this concept is the underlying assumption that the growth conditions for avascular tissue development will also promote vasculogenesis and that the angiogenic conditions will not interfere with parenchymal differentiation of tissue progenitor cells.
Organ development is an elaborate feedback control system in which temporally and spatially secreted factors by tissue niche cells provide positive and negative regulation. 205 The use of angiogenic initiators, such as VEGF, can redirect parenchymal progenitor differentiation toward vascular lineages. 215 As a consequence, novel techniques are required to guide orthogonal differentiation of vascular and parenchymal progenitor cells in situ. The use of reductionist, in vitro models utilizing synthetic hydrogels and chemically defined media can provide a translatable platform to probe the intricate relationship among endothelial, stromal, and parenchymal cells. For instance, Lutolf and colleagues demonstrated VEGF could be covalently bound with protease-sensitive peptides within synthetic PEG hydrogels and released by cell-secreted proteases, enabling studies of vascular morphogenesis within a chemically defined microenvironment. 216
In addition to angiocrine factors, angiogenic transcription factors, such as the transcription factor hypoxia-inducible factor 1, could also be identified in reductionist, synthetic hydrogel models of vascular morphogenesis. Activators for the master angiogenic regulators of each tissue niche could then be incorporated within the scaffold to indirectly stimulate the production of angiocrine factors in mural and parenchymal cells surrounding the vascular cells.37,205 This approach would enable complex angiocrine profile production at physiological concentrations, confined to localized regions, thus eliminating the complexity of releasing multiple angiocrine factors within the scaffold in a temporally and spatially controlled manner.217–220 As well, clustered regularly interspaced short palindromic repeat-mediated gene activation may be utilized to regulate the angiogenic transcription factors. 221
Also, deriving EPCs and tissue-specific progenitors from mesodermal precursors would closely mimic the process in organ development and eliminate the need for multiple cell isolations. Gerecht, Palecek, and colleagues have provided proof-of-principle for this approach, generating EPCs or organotypic ECs and parenchymal progenitors from hiPSCs.175,213,222 In addition, inducing EPCs to form macrovasculature within the prevascularized construct could create a vascular axis within the construct, enabling microsurgical connection with the host vasculature and instantly perfusing the scaffold with blood.46,223 Assisting efforts to advance EPC vascularization research is omnidirectional, direct-contact bioprinting that can recapitulate the distinct 3D hierarchical vascular networks for each tissue, as well as mimic the local matrix microenvironment by finely controlled deposition of biomolecules.224–226 Thus, combining reductionist in vitro models with recent advances in polymer science, genetic engineering, and 3D bioprinting can provide the sophisticated systems needed to develop the next generation of tissue-engineered models that can further EPC vascularization research.
Footnotes
Acknowledgments
E.B.P. was supported by the NIH T32 National Institutional Research Service Award T32 HL07670, and gratefully acknowledges Dr. George Truskey for review of this article. This review is dedicated to the living memory of Gregory Duke Brown.
Disclosure Statement
No competing financial interests exist.