
Abstract
To date, the therapeutic efficacy of human mesenchymal stem cells (hMSCs) has been investigated in various clinical trials with moderate or, in some cases, inconsistent results. The still elusive reproducibility relates, in part, with constitutive differences in the cell preparation, translated into variable “cell potencies.” Other factors include poor cell homing and survival, and age-/disease-associated host tissue impairment. It is well accepted that within in vivo niches, MSCs exist as heterogeneous cell populations with different stemness propensities and supportive functions. Phenotype-based MSC purification of homogeneous subsets can result in cell populations with distinct biological functions. In addition, preclinical studies have shown that MSC functionalization in vitro, through cell priming, can boost their immunomodulatory, trophic, and reparative capacities in vivo. Therefore, in this review, we discuss how phenotype-based MSC purification and MSC priming technologies can contribute to an improved MSC-based product for safer and more effective therapeutic applications.
Impact Statement
Culture expansion of MSCs has detrimental effects on various cell characteristics and attributes (e.g., phenotypic changes and senescence), which, in addition to inherent interdonor variability, negatively impact the standardization and reproducibility of their therapeutic potential. The identification of innate distinct functional MSC subpopulations, as well as the description of ex vivo protocols aimed at maintaining phenotypes and enhancing specific functions have the potential to overcome these limitations. The incorporation of those approaches into cell-based therapy would significantly impact the field, as more reproducible clinical outcomes may be achieved.
Introduction
S
Active research is being conducted to help minimize the reported variability, ultimately aiming at increasing clinical reproducibility. Efforts include thorough characterization of cell preparations, including the description of constitutive variations (i.e., cell heterogeneity), and methods to stimulate MSCs with chemical and physical conditions to induce and/or to modulate specific attributes of the cells (i.e., cell priming). In this review, we collect and discuss available evidence of such efforts, focusing on two main aspects: the identification of specific subsets/subpopulations of MSCs from different sources (heterogeneity) and the derivation of stimulatory protocols applied to the cells a priori to modulate cellular phenotypes and homing capacity (priming).
MSC Subsets/Subpopulations
The fact that standard MSC cultures consist of functionally heterogeneous cell subsets is well recognized in the literature; however, the reasons for such heterogeneity are poorly understood. In our opinion, such heterogeneity arises from two different factors: first, from an inherent heterogeneity of culture-initiating MSCs, and second, from culture-induced accumulation of senescent cells, which in later passages become predominant and therefore significantly impact cultures' potencies. As the latter phenomenon has been reviewed extensively,1,2 this section will focus on tissue-inherent MSC heterogeneity, which is closely linked to their local niches and tissue topography.
It is widely accepted that MSC populations within the different niches and tissues are highly heterogeneous; purification of homogeneous subsets with distinct biological functions is a challenge. However, isolated MSCs are generally analyzed for their clonogenic potential by the colony forming unit-fibroblast (CFU-F) assay and only limited information exists about markers that discriminate between developmentally, functionally, and morphologically distinct MSC subsets. 3 In this study, we summarize recent studies aimed to identify different MSC subsets in the bone marrow (BM) as well as adipose and synovial tissues.
Bone marrow MSCs
Location markers: CD146 and CD271
BM contains stem and progenitor cells for both hematopoietic and nonhematopoietic lineages. BM MSCs (referred also as multipotent stromal cells and skeletal stem cells) reside in the postnatal BM cavities and give rise to bone, cartilage, marrow fat, and hematopoiesis-supportive stroma following a specific sequence of events during postnatal development.4–6 A pioneering work by Tormin et al. has clearly demonstrated that human BM MSCs were present in two different regions within the BM cavities: in the perivascular niche, where they were characterized by CD146 marker expression, and in the bone-lining location, where MSCs lacked this marker. 7 Both, CD146+ and CD146Neg MSC subpopulations were CD271+ and had similar clonogenic capacities and gene expression profiles characterized by the simultaneous expression of multiple-lineage transcripts. Therefore, while CD271+CD146Neg cells are present in bone surfaces, CD271+CD146+ MSCs constitute the bona fide perivascular component of the BM (Fig. 1). Furthermore, adherent cultures derived from these two subpopulations had similar differentiation capacities in vitro and in vivo. In a more recent study, Espagnolle et al. have expanded BM MSCs at the clonal level and showed the presence of CD146high and CD146Neg/low clones. 8 In agreement with Tormin et al.‘s study, 7 they found no difference in the trilineage as well as hematopoietic support potentials between these two types of clones; however, clonal cultures derived from the CD146high subset had slightly slower growth rates and a more pronounced vascular smooth muscle phenotype, which was tested using functional cell contraction assay in a collagen gel. These findings supported Tormin et al.'s study, 7 in which BM CD146+ cells were shown to have a topography of pericytes. The significance of CD146 expression on these perivascular MSCs remains to be investigated. In regard to CD146 expression on BM MSCs, it is interesting to mention that Maijenburg et al. have also found CFU-Fs in the CD271NegCD146+ subset of fetal BM. 9 CD146 expression was shown to be dependent on calcium 10 and oxygen levels, 7 and can therefore reflect the niche environment in which this specific BM MSC subset resides. Less information is so far available with respect to CD146Neg BM MSCs. Because significant reduction of CD271+CD146+ population was found in elderly adults, compared with children, 9 it is possible that CD146Neg cells represent a subset of “aged” BM MSCs; however, this is not consistent with Tormin et al.'s data 7 that showed no difference in the clonogenicity between CD271+CD146+ and CD271+CD146Neg subpopulations.

MSC topography within BM. All BM MSCs are CD271+, whereas CD146 and CD56 help discriminate their differential presence within two distinct anatomical regions of the BM: (1) the perivascular niche (CD146+), where they interact with blood vessels (arterioles and sinusoids), and (2) the bone-lining niche (CD146Neg CD56+), where they interact with cells of the osteoblastic lineage (quiescent bone lining cells and active osteoblasts in places of active remodeling). BM, bone marrow; MSC, mesenchymal stem cell.
The perivascular localization concept of BM MSCs has been recently reinforced by Lin et al.'s pioneering study, showing that intra-arterially injected BM MSCs can serially engraft from the circulation into irradiated BM and proliferate in vivo, retaking the perivascular space around BM vessels and sinusoids. 11 Recent reports12,13 expanded the notion of MSC subpopulations beyond the CD146 and CD271 discrimination reported in human BM, incorporating additional markers thus far identified in mouse studies. For instance, the perivascular BM MSCs can be further divided into two main categories according to the blood vessel they associate with: the periarteriolar MSCs (Nestinbright, NG2+, αSMA+, CD271+, and CD146+) and the perisinusoidal MSCs (Nestindim, leptin receptor [LepR]+, CD271+, and CD146+). These subpopulations have distinct functions as they support hematopoiesis and probably are related with local homeostatic responses. Along those lines, Méndez-Ferrer et al. showed that Nestin+ MSCs contain all BM colony-forming unit fibroblastic activity, are spatially associated with hematopoietic stem cells (HSCs), and highly express HSC maintenance and retention genes such as those encoding the cytokines chemokine (C-X-C motif) ligand 12 (CXCL12) and stem cell factor (SCF).14,15 These perivascular cells known as CXCL12-abundant reticular (CAR) cells express Nestin, LepR, myxovirus resistance-1 (Mx-1), the transcription factor paired related homeobox-1 (Prx-1) that characterizes cells of limb bud mesoderm, and the transcription factor osterix (OSX or SP7) that regulates osteoblast maturation. 13 Importantly, Sacchetti et al. suggested that CD146+ hMSCs may be the in vitro counterpart of CAR cells as they acquire the same phenotype in in vivo transplantation models reconstituting the hematopoietic environment. 16 Therefore, except CD146+ MSCs' abilities to reconstitute bone and BM stroma after orthotopic and heterotopic transplantation models, they have an important role in HSC niche maintenance. To further support this notion, Corselli et al. showed that CD146+CD34NegCD45Neg perivascular cells support the HSCs' stemnesses, through cell-to-cell contact and Notch signaling activation, and HSCs' abilities to engraft to primary and secondary immunodeficient mice. In contrast, unfractionated and CD146Neg MSCs induce differentiation and inhibit ex vivo HSCs' maintenances. 17
In summary, the combined literature to date indicates that within human BM stroma, the CD146+ subpopulation has a preferential perivascular topography and hematopoietic support function, whereas CD271 is expressed on both perivascular and bone-lining MSCs; precise functions of these molecules on BM MSCs remain to be investigated.
Other markers
In recent years, a number of novel markers to identify distinct MSC subsets within the CD271+ fraction in human BM were proposed, including CD140b, 18 MSCA-1,19,20 CD90 and CD106, 21 CD140a, 22 and SUSD2 20 ; however, their expression in situ has not been performed; therefore, the precise tissue locations of these putative subsets remain unknown. Another surface marker of interest is CD56. Early studies have shown that CD56 molecule was highly specific for bone-lining cells, 23 in the same location of CD271+CD146Neg MSC subset in the Tormin et al.'s study 7 (Fig. 1); however, CD56 has not as yet been used to isolate BM MSCs as a single marker or in combination with CD271. An interesting study from Buhring's laboratory 19 showed a distinct morphology and potential chondrogenic bias of the CD271+MSCA-1+CD56+ BM MSC subset. These findings are awaiting an independent confirmation, but were confirmed, in part, by Cuthbert et al., who found MSCA-1 expression in the CD45NegCD271+ fraction of BM aspirates. 24 Busser et al. also found better chondrogenic potential of MSCA-1+-selected MSCs than unselected ones and higher chondrogenic capacity from BM MSCs grown from the CD271+ fraction. 20 On the contrary, Jezierska-Wozniak et al. found better chondrogenesis of cultures expanded from CD271Neg rather than CD271+ cells, after differentiation on gelated collagen microspheres. 25
CD271 marker is lost during culture expansion; therefore, other markers were studied to discover MSC subpopulations present both in fresh and early-passage expanded cells. When Qian et al. tested CD44 as one of those markers, they found that CD44 was absent in freshly isolated BM MSC population because the cells that co-expressed CD271 and CD146 and displayed the clonogenic and multipotent functions were CD44Neg. 26 However, CD44 marker was acquired by CD44Neg subset after in vitro expansion. This finding is unexpected because CD44 has always been considered a “well-known” MSC surface marker in expanded MSC studies (reviewed in Sousa et al. 27 ). In agreement with Qian et al.'s study, Busser et al. confirmed the higher clonogenicity of the CD44Neg fraction in the BM. 20 These authors also found that the CD44Neg fraction showed greater osteogenic potential than the total unselected population, but in contrast, no differences in chondrogenic potential were found between CD44Neg and CD44+ cells. 20 These authors also found greater osteogenic potential and clonogenic capacity in CD34+ than CD34Neg cells or unselected BM MSC population. Again, CD34Neg and CD34+ fractions did not show differences in the chondrogenic potential. As a result of different studies, CD34 had become another possible, but controversial marker for MSC subset selection from fresh BM aspirates (reviewed in Lin et al. 28 ).
Functionally selected MSC subpopulations
Other approaches use cell division as the functional parameter for identifying MSC subpopulations in early-passage MSCs, 29 or clonal expansion methods.8,30 After clonal expansion, Dickinson et al. performed a comparative analysis between greatest and poorest chondrogenic clones and found that the expression of ROR2 positively correlated with chondrogenesis. 30 In situ immunostaining corroborated the presence of ROR2+ cells co-located with CD146+ cells surrounding the blood vessels in the adult human BM. As mentioned, Espagnolle et al. 8 performed a selection of the expanded clones based on CD146 expression, for further analysis. Other markers were used to study heterogeneity in culture-expanded MSCs, such as ganglioside-based membrane microdomains. 31
Based on this information, it is fair to conclude that the existing literature supports the notion for at least two distinct topographies of MSCs in the BM (perivascular and bone lining); however, further work is needed to assess what effect these niches may have on MSC functionality assessed after their expansion and priming in vitro. It is possible that standard culture conditions lead to MSC convergence to a common phenotype 32 ; therefore, future investigations of BM MSC heterogeneity would benefit from a direct analysis of these cells immediately after purification.
Preclinical studies with MSC cultures derived from selected subpopulations
Some selected BM subpopulations, mainly grown from CD271+ cells, have been evaluated in preclinical studies. In one study, CD271+ subpopulation of cultured BM MSCs, when injected into infarcted murine heart, showed improved cardiac function. 33 Another study reported an improved repair of full-thickness cartilage defects in rats using MSCs sorted for CD271+ and expanded to form cell pellets that were placed in and covered with atelocollagen sponges. 34 Also, CD271-MSCs promoted significantly greater lymphoid engraftment than did plastic adherence MSCs when co-transplanted with HSCs in immunodeficient mice. 35 Other preclinical studies with BM MSC subpopulations have been recently reviewed in Mo et al. 36 and Lv et al. 37
Adipose tissue MSCs
Location markers: CD146 and CD34
Adipose tissue is a complex tissue of mesodermal origin that contains various cell types, including adipose tissue MSCs, preadipocytes and mature adipocytes, fibroblasts, vascular smooth muscle cells, endothelial cells (ECs), monocytes/macrophages, and lymphocytes. 38 Obtaining MSCs from adipose tissue, a waste product in several treatments, is popular due to the easy access to the tissue in large quantities. As for BM MSCs, two distinct MSC subsets have been discovered in human adipose tissue, but this time, they are CD146+CD34Neg pericytes and CD146NegCD34+ adventitial cells (both negative for CD45).39,40 In situ immunostaining of adipose tissue obtained from abdominoplasty showed CD146NegCD34+ cells localized in the outer vascular ring and CD146+/αSMA+ cells immediately adjacent to the vascular intima. 39 In one study, a third population of CD146+CD34+ was observed with a very low frequency by flow cytometry (∼0.5% of the nucleated stromal vascular fraction [SVF] cells), presumably a transitional population between the pericytic and supra-adventitial perivascular ring cells. Further studies are necessary to confirm these markers in situ. The frequency of these subsets differs significantly depending on donor demographics and the way of tissue processing, but is still unclear how these variables affect the different subsets.
Other markers
Bajek et al. 41 studied the differences in early-passage adipose-derived stromal cell (ASC) markers from tissues obtained either from mechanical or ultrasound-assisted liposuction, and they only found statistical differences in the expression of CD166. Other authors 20 did not find differences in CD166 expression, which was low in both stromal cells from abdominoplasty and lipoaspirate, although they observed that CD271+ selected ASCs were highly proliferative and clonogenic only in lipoaspirate samples, being the contrary for abdominoplasty ones (CD271Neg fraction). Interestingly, CD271 marker was not mentioned in many characterization studies.42,43 Importantly, Baer et al. 42 performed the first comprehensive phenotypic characterization of cultured ASCs isolated from lipoaspirates. They found that ASCs expressed the characteristic MSC markers CD29, CD44, CD73, CD90, CD105, and CD166, but there was high donor variability for 49 of the 242 markers tested, including CD34 and CD200. These findings can be explained by the fact that CD271 as well as other molecules present on native adipose MSC subsets can be lost in culture.
The difficulty in analyzing adipose tissue MSC literature is that adipose tissue is highly vascular and it is widely accepted that following digestion and removal of red cells, the remaining cells consist of leukocytes, stromal cells, and ECs. Subsequent dissection of stromal cells and ECs is complicated by the fact that many “MSC”-specific molecules—CD73, CD105, and CD146, and some authors even consider CD90—are also expressed on ECs (reviewed in Lin et al. 44 ). Conversely, a typical endothelial progenitor molecule CD34 can be expressed on adipose MSCs (reviewed in Sidney et al. 45 ), as stated above. So, these markers are not very useful alone for segregating MSCs from ECs; however, CD31NegCD45Neg combination can discriminate leukocytes and ECs from adipose MSCs as published by the ISCT together with the International Federation for Adipose Therapeutics and Science (IFATS). 43 These authors published a guidance for minimal criteria for stromal cells from adipose tissue, as previously performed for general MSCs. This guide differentiates between freshly isolated stromal cells as “uncultured stromal vascular fraction,” identified mainly as CD45NegCD235aNegCD31NegCD34+ subset, and cultured stromal cells (ASCs), characterized as CD45NegCD31NegCD36+CD106Neg cells. Overall, the literature agrees that CD34+ may be present on a subset of native adipose MSCs, but it is lost in prolonged culture expansion 46 and is not useful as a sole marker as CD34 is also expressed on ECs and other cell types.44,45 However, CD34 was considered a good marker for MSC enrichment from SVF cells by Busser et al. 20 as CD34+ ASCs were significantly more clonogenic and proliferative than parental cells. Furthermore, Bourin et al. 43 observed that both SVF cells and ASCs express CD73, CD90, and CD105, although CD44 expression is only present in the cultured ASCs, which seems to be similar to CD44 pattern of expression on BM MSCs. 26 Also, Bourin et al. 43 suggested additional positive markers with variable expression, as CD146. Despite this, one study did not find expression of CD106 and CD146 markers in culture-expanded ASCs, but passage number at which cells were analyzed was not specified. 47
Functionally selected MSC subpopulations
Different from BM, Busser et al. 20 found that adipose MSCs did not express SUSD2, MSCA-1/TNAP, and CD44 in situ, but these data need to be confirmed. Hardy et al., 48 performed enrichment of CD146+CD34Neg and CD146NegCD34+ subsets from adipose MSC tissue collected by lipoaspiration, focusing on the aldehyde dehydrogenase (ALDH) staining intensity. After culturing single-cell sorted cells, transcriptional profile analysis was done and ALDHbright cells were considered the most primitive cell population within both subpopulations. Considering that this study was only performed using one sample, further confirmation is necessary.
In general, different from BM MSCs, most of the studies using adipose MSCs do not study chondrogenesis. There also exists controversy about MSC variability due to age, body mass index, niche location, and the way of tissue processing that complicate data interpretation, but may lead, in the future, to discovery of many more subtypes of adipose MSCs, for example, the possibility of finding different MSC subpopulations that can support different types of blood vessels. So probably, different vascularity seen in lipoaspirates and solid fat tissues can be the reason for different types of MSCs found, but more work is needed to study the relationship between these blood vessels and MSCs. Despite all of these unanswered questions, surprisingly, SVF cells are being used for clinical treatment of human cartilage damage (reviewed in Pak et al. 49 ).
Preclinical studies with MSC cultures derived from selected subpopulations
In preclinical studies using AT MSCs, mainly CD34+, CD105+, and CD90+ subpopulations were tested. One study compared CD90+ and CD105+ subpopulations of cultured MSCs seeded on hydroxyapatite (HA)-coated polylactic-co-glycolic acid (PLGA) scaffolds in calvarial defects in mice and showed better bone formation from CD90+ subpopulation. 50 In calvarial defects in mice, cultures grown from CD105low-sorted cells and seeded on HA-PLGA scaffolds formed more robust bone than cultures grown from CD105high cells. 51 Other preclinical studies with selected AT MSCs have been reviewed in Johal et al. 52
Synovial MSCs
Location markers
The synovial membrane is a specialized tissue of mesodermal origin, lining the spaces of diarthrodial joints, bursae, and tendon sheaths. It contains two compartments, the intima inner continuous cell compartment composed of fibroblast-like synoviocytes, and the subintima compartment composed of few macrophages and lymphocytes, fat cells, and blood vessels. 53 Interestingly, the origin of synovium-derived MSCs in the synovial lining is still under investigation and can be related to infiltrated MSCs through vasculature or BM-originated MSCs that connect to intra-articular space. Recently, two studies showed that single or double positive Prg4-lineage and Gdf5-lineage cells, both present in the synovium, contribute to repair of articular cartilage injuries in mice.54,55 Despite the hypothesis of the involvement of synovium-derived MSCs in cartilage repair beginning to gain strength, 56 in vivo and in vitro studies about synovium MSCs are scarce compared with MSCs from other tissues such as adipose or BM. A comprehensive review of clinical studies utilizing intra-articular MSC therapy showed that only 7% of the studies reviewed used MSCs from synovial membrane. 57 The same applies to in vitro studies, in which much fewer investigations were focused on synovial MSCs, mainly culture-expanded cells, compared to BM MSCs.
In a pioneering study, Karystinou et al. 58 reported for the first time the presence of cell subsets with distinct characteristics within the synovial membrane. They performed cell cloning by limiting dilution and found variations in MSC proliferation and potency: all clonal populations were chondro-osteogenic and only 33% of them were also adipogenic, unlike the parental MSC population that was tripotential. Phenotype evaluation of tripotential extensively expanded clones by flow cytometry showed positivity for markers such as CD13, CD73, CD81, CD90, CD105, CD166, and SSEA-4. However, these authors did perform neither enrichment of these tripotential MSCs nor phenotypic analysis of bipotential clones. Besides, in this study, not all of the clones isolated were culture expandable. Taking this into account and also the fact that markers as CD271 are undetectable after extensive expansion,59–61 recent studies are being focused in early-passage expanded (passages 0–1) or freshly isolated MSCs.
Hermida-Gómez et al. 62 performed the first MSC topographic analysis of the synovial tissue, showing CD271+ cells present not only in its intima lining in healthy donors but also along vascular subintima in osteoarthritic patients, a likely route for the mobilization of the MSCs to reach the cartilage damage. However, other authors associated the increase of CD271+ MSCs in synovial membrane of arthritic patients with proinflammatory function. 61 Hermida-Gómez et al. 62 found that CD271+ cells present in synovium had a high co-expression of CD44 and CD90, but low levels of CD105. The low positivity of CD105, in addition to CD166, in freshly isolated synovial MSCs, was confirmed by Jones et al. 63 using flow cytometry. Despite the low percentage of CD105+ MSCs present in synovial membrane, early-passage CD105-selected cultures had good chondrogenic capacity after spheroid formation, 64 but this study did not compare results with CD105Neg or total MSCs because these cells were not capable to form spheroids.
More recently, Mizuno et al. 65 proposed different phenotypes of MSCs in the osteoarthritic synovium based on their topography: CD55+ MSCs in the surface region, CD271−CD55− MSCs in the stromal region, and CD271+ MSCs in the perivascular region. However, these results need to be confirmed in healthy synovium.
Other markers
Another marker used for subset analysis and enrichment from early-passage culture-expanded synovial MSCs was CD73, which enriched cells with higher chondrogenic potency than CD106 marker. 66 Also, CD44 expression was correlated with chondrogenic capacity 63 ; however, no CD44 enrichment was performed in this study. Combinations of markers, such as CD73 and CD39, were later used for enrichment of synovial MSCs from early- and late-passage synovial cultures, and CD73+CD39+ subset was found to be more chondrogenic and osteogenic than CD73+CD39Neg subset. 67 However, when these authors compared culture-expanded and freshly isolated synovial cells, they found no difference in clonogenicity and chondro-osteogenic potential between CD73+CD39+ and CD73+CD39Neg subsets, confirming the necessity of performing this type of analysis using freshly isolated synovial cells. Furthermore, as commented above, markers such as CD271 are known to be downregulated following MSC culture expansion; similarly CD34 marker was detected in directly isolated synovial cells, but declined following MSC passaging. 60
To summarize, more work in this area is required to provide a convincing argument for the use of specific markers, for the isolation of any functionally relevant subset of synovial MSCs. To the best of our knowledge, no preselection of synovial MSC populations was performed in preclinical studies.
Altogether, while MSCs are being largely used in clinical trials as bulk-expanded heterogeneous preparations (reviewed in Squillaro et al. 68 and Kouroupis et al. 69 ), to our knowledge, there are no clinical studies reported on a comparison between different selected subpopulations. In this area, an unanswered question still remains: do phenotypically comparable subpopulations of MSC obtained from different tissues behave similarly upon implantation, or is it the tissue of origin rather than phenotype itself, that determines their effect?
MSC Priming
Efficacy and reproducibility of MSC therapies are not only affected by the composition of the cell preparation (above) but also by the capacity of the transplanted cells to consistently reach and interact with dysregulated tissues (i.e., homing and engraftment), and subsequently to predictably induce and/or modify specific host responses (i.e., therapeutic effect). In other words, after administration (e.g., injection and infusion), MSCs have to migrate, home, engraft, survive, sense the local environment, and reactively mount a paracrine reparative response. Previous studies have shown that systemically infused MSCs (e.g., intravenous or intra-arterial administration) are capable of migrating and homing to distant sites of active injury, including BM, intestine, liver, and lung.11,70–72 However, only a small percentage of infused cells (i.e., 0.1–2.7%) actually reaches the target tissues.73–78 Once at the injured tissue, it is well accepted that MSCs exert immunomodulatory (for both innate and adaptive immunity) and trophic activities,79,80 recently suggested to be collectively called “medicinal” signaling activities.81,82 These local activities are performed through both direct cell–cell communication (e.g., Notch receptor/Jagged-1 and PD-1/PD-L1/PD-L283,84) and locally secreted paracrine mediators. The latter involves a list of chemokines released by MSCs, including CXCL12, 85 SCF, 86 platelet-derived growth factor, 87 transforming growth factor β (TGF-β), 88 vascular endothelial growth factor (VEGF), 88 tumor necrosis factor-α (TNF-α) stimulated gene/protein 6 (TSG-6), 89 erythropoietin (EPO), 88 interleukin-6 (IL-6), 88 interleukin-10 (IL-10), 88 indoleamine 2,3-dioxygenase (IDO), 88 and prostaglandin E2 (PGE2). 90 Some of these factors (e.g., TSG-6) have been shown to also exert a remote paracrine therapeutic effect, demonstrated in myocardial infarction, 91 lung injury, 92 and corneal injury, 93 suggesting that the induction of its secretion even without interaction with the target tissue might be sufficient for a therapeutic effect. On the other hand, host tissue conditions (e.g., age and disease) also have an effect on the paracrine activity of engrafting MSCs.94–96 For instance, elevated levels of proteases such as elastase, cathepsin, and dipeptidylpeptidase (DPP) present in the aged tissue destabilize trophic factors induced by MSCs. Interestingly, the administration of a pharmacologic inhibitor of DPP before the therapy enhanced the stability of CXCL12 and increased the engraftment and function of CXCR4+ progenitor cells in an acute myocardial infarction mouse model. 97 Collectively, this evidence suggests that both the “pharmacokinetic” properties and therapeutic activity of MSCs can be modulated and/or boosted a priori, through multiple ex vivo priming protocols. We now discuss a few techniques proposed to effectively prime MSCs before their transplantation in vivo (Fig. 2).
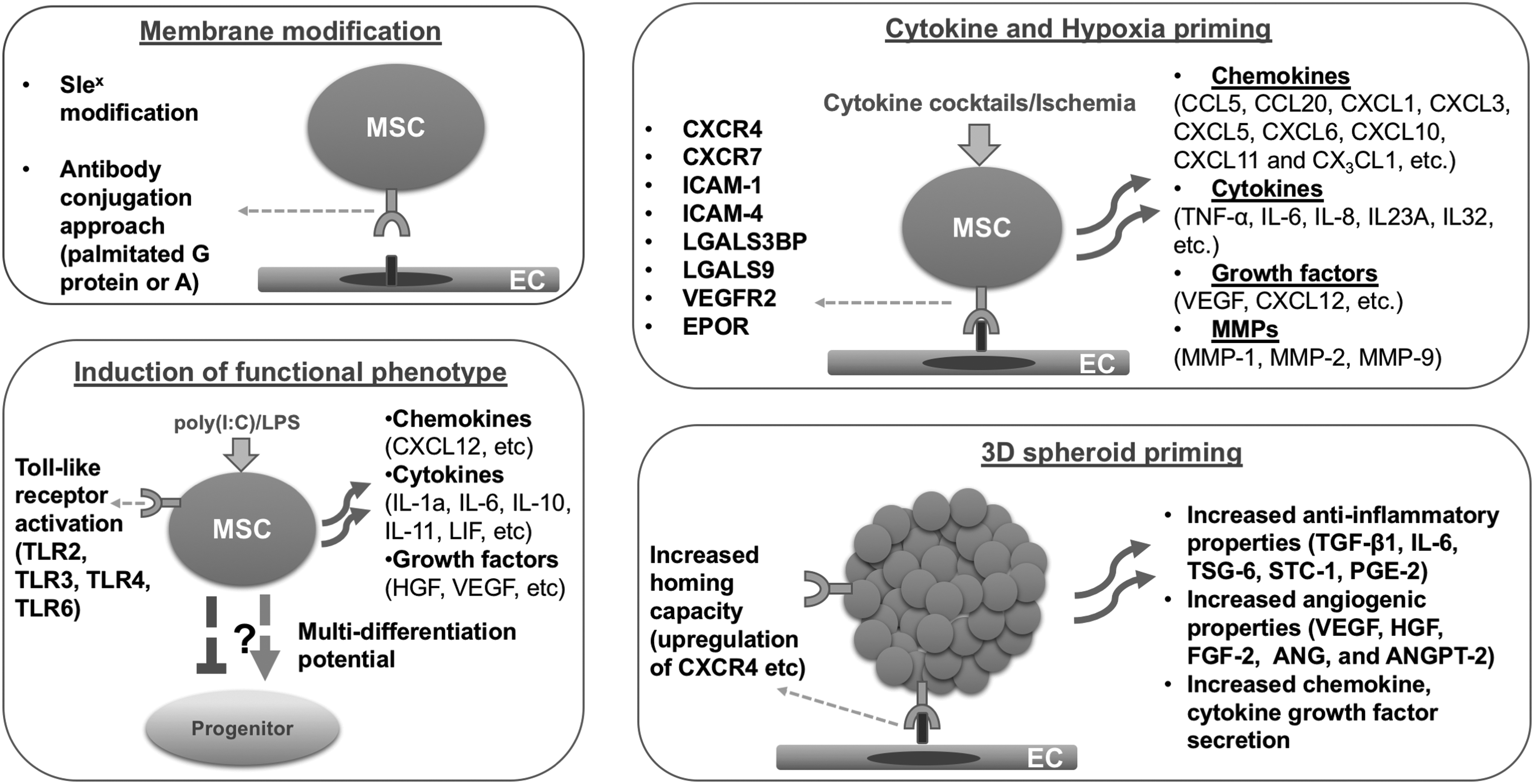
MSC priming strategies include membrane modification, cytokine and hypoxia priming, Toll-like receptor priming, and 3D spheroid priming. 3D, three dimensional.
Membrane and cytoplasmic MSC priming
MSCs express a variety of membrane adhesion molecules, including intercellular adhesion molecules-1 and -2 (ICAM-1 and ICAM-2), vascular cell adhesion molecule-1 (VCAM-1), L-selectin, CD18, CD24, CD29, CD44, and CD49a-f.98,99 Previous studies have shown that the adhesion molecule expression profile presents an intrapopulation heterogeneity, 71 which is not only determined by the tissue of origin 100 but also by isolation and culture procedures. For instance, Aldridge et al. showed that early- and late-passaged MSCs have different adhesion molecule profiles, with CD49d exhibiting variable expression within the same cell population. 71 Despite the potential deleterious effect this variability may have on cell adhesion processes, MSC homing to target sites can be enhanced through diverse in vitro membrane modification techniques that incorporate natural and induced processes such as (1) the HSC/leukocyte transendothelial migration process (i.e., diapedesis); (2) chemical or noncovalent interactions; and (3) biospecific recognition (Table 1). Early evidence has shown that engineering transmembrane glycoproteins on MSCs using characterized selectin ligands affects MSC migration through vasculature after systemic infusion in vivo, similar to leukocyte extravasation to inflamed tissues. 101 Rüster et al. identified that MSCs, like leukocytes, roll and adhere to postcapillary venules through cellular interactions with ECs engaging P-selectin as well as VCAM-1/(CD106)-VLA-4(CD49d/CD29) pathways.102,103 As MSCs do not express P-selectin ligands such as glycoprotein ligand-1 (PSGL-1/CD162) or CD24, alternative ligands have been proposed (e.g., CD44 glycoprotein, which binds hyaluronic acid). 104
BM, bone marrow; HCELL, hematopoietic cell E-/L-selectin ligand; MSC, mesenchymal stem cell; NOD/SCID, nonobese diabetic/severe combined immune deficiency; SLex, Sialyl-Lewis X; SPIONs, superparamagnetic iron oxide nanoparticles.
Hematopoietic cell E-/L-selectin ligand (HCELL), a specific isoform of CD44 that binds strongly E-selectin, 105 has been associated with MSC homing to BM in vivo 106 and thus, its modification constitutes a promising approach to enhance that process. Using enzymatic conditions, Sackstein et al. 104 converted the MSC membrane native CD44 glycoprotein into HCELL, allowing their efficient binding to E-selectin. The resulting sialofucosylated glycan moiety on CD44 is known as Sialyl-Lewis X (SLex), the active selectin-binding carbohydrate motif site for HCELL. Interestingly, intravenously infused HCELL+ MSCs infiltrated mouse BM within hours of infusion. 104 Enzymatic membrane modification targets only glycoproteins that already exist on the surface of MSCs, whereas further chemical alterations enable the cells to present several epitopes by the covalent bioconjugation method, potentially enhancing the effect. Importantly, Sarkar et al. used several approaches to covalently attach SLex on MSC membrane through biotin-streptavidin binding, conferring leukocyte-like rolling characteristics to MSCs without affecting their phenotype and multilineage differentiation capacity.107,108 These approaches resulted in increased MSC rolling on P-selectin at a physiological shear of 0.5 dyn/cm2 in vitro. In an ear inflammation mouse model, SLex-engineered MSCs exhibited a robust rolling response on inflamed endothelium. 109
Another method to improve MSC homing is through antibody conjugation approach. To localize MSCs at a target tissue, researchers have used palmitated protein G or A precoating of the cell membrane as a step to bind antibodies on the MSC surface. Interestingly, Dennis et al. 110 have precoated the membrane of MSC chondrogenic progenitors with palmitated protein G and bound to the protein G antibodies specific for cartilage matrix antigens. Effective homing of membrane-modified chondrogenic progenitors was observed to cartilage injury site in rabbit osteochondral explants. 110 In another study, homing rate and repairing efficacy of MSCs improved using anti-CD29 and anti-myosin light chain bispecific antibodies in a mouse myocardial fibrosis model. 111 Importantly, in this study, Deng et al. 111 also used highly focused ultrasound mediated stimulation of microbubbles, which significantly increases homing of MSCs to the mouse injured myocardium. 112 Finally, another nonconventional method to enhance MSC homing is the internalization of superparamagnetic iron oxide nanoparticles with exogenously guided magnetic targeting. This approach has been used by two separate studies showing enhanced MSC homing to mouse retina 113 and tail, 114 and rabbit and swine osteochondral defects. 115
Cytokine and hormonal priming of cultured MSCs
An alternative approach to enhance homing and therapeutic capacity of MSCs is to prime the cells with specific cytokines and growth factors (Table 2). Shi et al. showed that MSC priming with a cytokine cocktail containing SCF, hepatocyte growth factor (HGF), IL-3, and IL-6 increases their CXCR4 expression. 117 As a result, CXCR4 upregulation enhanced MSC chemotaxis to CXCL12 in vitro and homing efficiency to BM in vivo. Similarly, IL-1β-primed MSCs exhibited an enhanced homing capacity to inflammatory sites in an ulcerative colitis mouse model, also by upregulation of CXCR4. 118 Mechanistically, IL-1β-primed MSCs express multiple cytokines, including TNF-α, IL-6, IL-8, IL-23A, and IL-32, and chemokines CCL5, CCL20, CXCL1, CXCL3, CXCL5, CXCL6, CXCL10, CXCL11, and CX3CL1, as well as matrix metalloproteases (MMPs) and adhesion molecules ICAM-1 and ICAM-4. 119 Interferon-γ (IFNγ) is another molecule widely used to prime MSCs.120–122 It is a major proinflammatory mediator that has been shown to increase MSC homing capacity to inflamed intestine in an ulcerative colitis mouse model, by upregulating CXCR7 and lectins LGALS3BP and LGALS9 in infused cells. 123 A major effect of IFNγ relies on the induction of the expression and activity in MSCs of the enzyme IDO, involved in the direct suppression of T cell proliferation and activation.124,125 IFNγ priming also upregulates the production of IL-6 in MSCs, 126 a cytokine with known anti-inflammatory and reparative effects in vitro and in vivo (e.g., liver fibrosis mouse model 127 ). Therefore, due to its high potency in inducing immunomodulatory factors, IFNγ has been proposed by many studies as a priming cytokine to enhance MSC efficacy in vivo. Other priming methods with positive effects include exposure to complement component 1 subcomponent q (C1q), which enhances MSC homing by inducing chemotactic responses to SDF-1 gradient. GSK-3β (glycogen synthase kinase 3β) inhibitors have also been used for MSC priming, resulting in increased MSC migration through the upregulation of β-catenin, phospho-c-Raf, ERK, and phospho-β-PAK-interacting exchange factor, 128 enhancing CXCR4, MMP-1, and MMP-2 expression.
CCl4, carbon tetrachloride; DSS, dextran sulfate sodium; FGF-2, fibroblast growth factor-2; HGF, hepatocyte growth factor; IFNγ, interferon-γ; IL, interleukin; MLNs, mesenteric lymph nodes; SCF, stem cell factor; TNBS, trinitrobenzene sulfonate; TNF-α, tumor necrosis factor-α.
Hormonal priming of MSCs has been proposed as an additional method to improve MSC survival and to induce paracrine actions post-transplantation in vivo in various conditions such as myocardial infarction 131 and brain injury 132 (Table 3). Mias et al. also showed that MSCs primed with melatonin stimulated angiogenesis and proliferation of renal proximal tube cells in vitro through increased HGF and fibroblast growth factor-2 (FGF-2) expression, and accelerated renal recovery in vivo following similar mechanisms and increased cell survival after transplantation. 133 Melatonin MSC priming also showed beneficial effects in focal cerebral ischemia in rats, where it not only increased MSC survival post-transplantation in vivo but also reduced brain infarction and increased angiogenesis and neurogenesis. 132 On the other hand, oxytocin-primed MSCs reduced cardiac fibrosis and macrophage infiltration, while enhanced cardiac repair for at least 4 weeks post-MSC transplantation in vivo. 131 In a recent study, Liu et al. 134 showed that angiotensin-II hormone MSC priming results in reduced cardiac fibrosis and infarct size, improved cardiac function, and increased expression of VEGF and von Willebrand factor in the ischemic myocardium, but no MSC differentiation toward cardiomyocytes in vivo.
VEGF, vascular endothelial growth factor.
Hypoxia priming of cultured MSCs
Rapid loss of implanted MSCs in vivo has been associated with hypoxic stress at the ischemic target tissue, which can initiate cellular apoptosis. 135 It is worth mentioning that MSCs are typically cultured at a pO2 level of 142 mmHg (∼20%), while the oxygen tension in the BM niche ranges from 1% to 7%, and in ischemic tissues from 0.4% to 2.3%. 136 In a pioneering study, Annabi et al. identified that hypoxic environments (i.e., 1% oxygen, 5% CO2, and 94% nitrogen) induce in vitro MSC migration and three-dimensional (3D) capillary-like structure formation through the secretion of VEGF and MMP-1. 137 Moreover, this increased migratory behavior is explained by Akt signaling activation and induction of cMet expression, the main receptor of HGF, 138 whereas the promotion of the MSC-dependent angiogenic effect is directly associated with the activation of hypoxia-inducible factor-1α (HIF-1α)-78-kDa glucose-regulated protein (GRP78)-Akt signaling pathway. 139 In general, MSC priming under hypoxia enhances the upregulation of several signaling molecules, including CXCR4,140–142 CX3CR1, 140 CXCL-12,140,141 VEGF 141 and vascular endothelial growth factor receptor-2 (VEGFR-2), 141 MMP-2, 142 MMP-9, 142 brain-derived neurotrophic factor, 135 glial cell-derived neurotrophic factor, 141 EPO 141 and its receptor, 141 and focal adhesion kinase. 143 The in vivo administration of hypoxia-primed MSCs shows increased homing capacity, enhanced vascularization, and restored function in various ischemic conditions, including myocardial infarction, and cerebral and hind limb ischemia138,140–145 (Table 4). Mechanistically, two separate studies in mouse and rat myocardial infarction models showed that hypoxia-primed MSCs improve infarcted heart function through enhanced survival of implanted MSCs, increased angiogenesis, and prevention of cardiomyocyte apoptosis through cell survival factor secretion.146,147 These results were extended to diabetic cardiomyopathy, in which anoxia-primed MSCs improved cardiac function through not only antiapoptotic effects but also attenuation of cardiac remodeling. 148 In acute kidney injury, hypoxia-primed MSCs showed beneficial effects by improving renal function, increasing angiogenesis, and reducing the levels of proinflammatory cytokines.149,150
Even though hypoxia-derived MSC priming has a positive influence on migration and homing capacities in several conditions, there is evidence of potential unwanted effects that require further investigation and may indicate that hypoxia priming may be application specific. This includes a negative effect on MSC osteogenic differentiation capacity, 151 and the accumulation of reactive oxygen species that alter their transcriptional factor profile. 152 In contrast, Estrada et al. showed that low oxygen levels enhance cell proliferation and genetic stability by favoring a natural metabolic state of increased glycolysis and reduced oxidative phosphorylation. 153 Therefore, further studies should carefully consider the long-term effects of hypoxia-primed MSCs in models of disease.
Induction of specific functional MSC phenotypes
One of the main therapeutic mechanisms of MSCs at the target site is to immunomodulate local responses.80,154 However, depending on the molecular composition of the instructive environment at the injury site, interacting MSCs exhibit a therapeutic responsive polarization into either anti-inflammatory (MSC-2) or proinflammatory (MSC-1) phenotypes, tightly coupled to M2/M1 macrophage skewing.155,157 Toll-like receptors (TLRs) are the most studied pattern recognition receptor family in MSCs, which sense pathogen-associated molecules involved in the regulation of innate immunity. Previous studies indicate that hMSCs consistently express TLR1, TLR2, TLR3, TLR4, TLR5, and TLR6, whereas TLR7, TLR8, TLR9, and TLR10 expression are dependent on the MSC origin.158–162 As elaborated below, several studies indicate that activation of specific TLRs in MSCs in vitro before transplantation has a profound effect on the immunomodulatory capacities of MSCs (Table 5). TLR3 and TLR4 are particularly important for MSCs' downstream effects, which can be in vitro activated by dsRNA mimetic polyinosinic-polycytidylic acid [i.e., poly(I:C)] and lipopolysaccharide (LPS), respectively. 155 Waterman et al. showed that TLR3 stimulation of MSCs supports their immunosuppressive effects preferentially through enhanced fibronectin deposition, whereas TLR4 stimulation of MSCs provides a more proinflammatory signature, in part, through release of TGF-β and collagen deposition. 155 In that respect, low dose of poly(I:C) for 24 h results in TLR3 activation and induction of VEGF, CXCL12, IL-6, IL-10, IL-11, leukemia inhibitory factor, and HGF without upregulation of inflammatory cytokines. 163 Therefore, it is not surprising to observe that injection of poly(I:C)-treated MSCs in a hamster heart failure model resulted in cardiac functional improvement with a 50% reduction in myocardial fibrosis, 40% reduction in apoptosis, and 55% increase in angiogenesis. 163 On the other hand, a recent study showed that both TLR3 and TLR4 activation comparably enhance MSC-mediated T regulatory cell (i.e., Treg) induction through Notch signaling and upregulation of DL1 (Delta-like 1), critical cellular mechanisms for the immunomodulatory properties of MSCs. 164 In a rat acute myocardial infarction model, Yao et al. showed that LPS-dependent priming results in increased MSC survival post-transplantation in vivo, coupled with reduced fibrosis of the infarcted myocardium, increased neovascularization and earlier recovery of the cardiac function. 156 Transcriptional profiling of LPS-primed MSCs showed that several chemokines, cytokines, and adhesion molecules were highly upregulated, including CXCL10, CCL20, IL-8, CXCL1, IL-6, CCL2, IL-1B, CXCL2, IL-1A, CXCL6, ICAM-1, VCAM-1, and SELE. 166
LPS, lipopolysaccharide; poly(I:C), polyinosinic-polycytidylic acid; Th1/17, T-helper type 1/17; TLR, toll-like receptor.
Different studies have indicated contrasting effects of TLR activation on MSC multidifferentiation potential. In a comparative study performed by Raicevic et al., TLR activation of MSCs with postnatal and perinatal origin resulted in differential osteogenic potential depending on MSC tissue of origin. 169 TLR2 and TLR4 activation in umbilical cord blood-derived MSCs promote chondrogenesis and osteogenesis with different intensities, whereas adipogenic differentiation is not altered by such TLR activation. 170 MSCs harvested from the umbilical cord showed differential responses in terms of osteogenic potency, with TLR4 activation increasing and TLR3 activation decreasing it, respectively. 171 On the contrary, both TLR3 and TLR4 downstream signaling promote BM MSCs' osteogenic potencies, 172 through activation of Wnt3α and Wnt5a signaling (TLR4). 173 Several groups have reported comparable increased osteogenic potency with activation of TLR2-, TLR3-, TLR4-primed adipose-derived stromal cells with unaffected adipogenesis.158,174 A novel pattern recognition receptor, the triggering receptor expressed on myeloid cells (TREM), identified to regulate myeloid cell function in vitro, 175 has also been associated with MSCs. Zhang et al. indicated that one of TREM family members, TREM-2, is expressed in MSCs and its knockdown reduces TLR2, TLR4, and TLR6 expression, impairing MSCs' multidifferentiation potentials. 176 Thus, we could speculate that TREM-2 ligands would induce TLR-specific responses in favor of cell differentiation.
Priming MSCs in 3D spheroid cultures
Adult MSCs possess a remarkable ability to coalesce and assemble in 3D structures, reminiscent of their innate aggregation as limb cell precursors in the mesenchymal condensation during early skeletogenesis. In that context, 3D organoid formation in vitro closely recapitulates the in vivo MSC niche by providing spatial cell organization with increased cell–cell interactions. As a matter of fact, MSCs cultured in 3D spheroid cultures show stable immunophenotypic profile, with a significant enhancement in survival, 177 homing, 178 stemness features, and differentiation potential,179,180 angiogenic effect, 177 and anti-inflammatory properties. 181 For example, in a mouse model of hind limb ischemia, MSC transplantation as 3D spheroids improved their survival compared with two-dimensional (2D) expanded MSCs, by suppressing a key apoptotic signaling molecule (Bax), while activating antiapoptotic signaling (BCL-2). 177 These positive effects can also be attributed to improved resistance to oxidative stress-induced apoptosis exerted by hypoxia-induced genes (e.g., VEGF-A, HIF-1α, and MnSOD), elevated by the hypoxic conditions at the spheroid core.182,183
Additional benefits of 3D cultures account for the established reduction in size of individual MSCs (about 0.25–0.5 the volume of an average 2D cultured cell), which reduce cell entrapment in the lungs when systemically infused. 181 MSCs' stemness features are also improved in 3D MSC spheroid cultures, evidenced by the following: (1) higher expansion and colony-forming activities 180 ; (2) enhanced differentiation capacities179,184,185; and (3) changes in the epigenetic status of genes indicative of a more pluripotent nature (NANOG, SOX2, and OCT4). 179 As expected, given the presence of a stimulating variable oxygen tension within the spheroids, angiogenic properties of MSCs are positively affected by 3D spheroid priming. This trophic enhancement is produced by upregulation of key angiogenic factors, including VEGF, HGF, FGF-2, angiogenin (ANG), and angiopoietin 2 (ANGPT-2).178,183,186 Murphy et al. showed that MSC spheroids embedded in fibrin gel secrete up to 100-fold more VEGF compared to dissociated MSCs in fibrin gel. 187 In addition, other groups have reported an increased homing capacity of MSC spheroids through a significant upregulation in the expression of the CXCL12 chemokine receptor CXCR4.178,180 Enhanced anti-inflammatory effects of 3D cultures have been reported by previous studies, indicating that MSC spheroids highly express TGF-β1, IL-6, TSG-6, STC-1 (stanniocalcin 1), and PGE2 anti-inflammatory molecules.181,188,189 As mentioned earlier, MSCs acquire effective anti-inflammatory properties after being primed with proinflammatory cytokines. Interestingly, Bartosh et al. showed that MSC aggregation into 3D spheroids activates the expression of IL-1 in an autocrine secretion manner, thus initiating an “auto-priming” effect. 181 Contrarily, Redondo-Castro et al. reported that the combination of IL-1 stimulation with spheroid priming resulted in significantly increased expression of IL-1Ra, VEGF, and G-CSF molecules without anti-inflammatory effects on LPS-treated microglial cells in co-cultures. 190 The discrepancies of the data underline the necessity for optimization of the priming methods and culture conditions. As a recent effort, Ylostalo et al. proposed specific protocols to efficiently prime MSCs in 3D cultures under chemically defined xeno-free conditions and how to administer the primed MSCs in vivo. 191
Finally, MSC-based 3D spheroids have been applied in various preclinical models, including wound healing,182,193 bone and osteochondral defects,194–196 and cardiovascular diseases (Table 6).192,199 Two separate groups applied MSC spheroids for wound healing in chemotherapy-induced oral mucositis 182 and in a model of diabetic healing impaired (leptin receptor deficient) mice. 193 In both cases, the MSC spheroid group provides better therapeutic efficacy compared with the traditional MSC suspension group. Using a rat calvarial defect model, MSC spheroid implantation resulted in full-thickness bone formation that efficiently filled the generated bone defects. 196 Intramyocardial transplantation of MSC spheroids in rat 192 and porcine 199 myocardial infarction models resulted in greater heart function improvement compared with MSC suspensions.
2D, two dimensional; 3D, three dimensional.
Collectively, MSC priming strategies aim to yield cellular products of high quality and potency by enhancing homing, survival, stemness, differentiation, anti-inflammatory, and other MSC properties (Figs. 2 and 3). As mentioned before, various methods exist to manipulate MSC properties that possess advantages and disadvantages mainly related to the quality of the cellular product (Table 7).
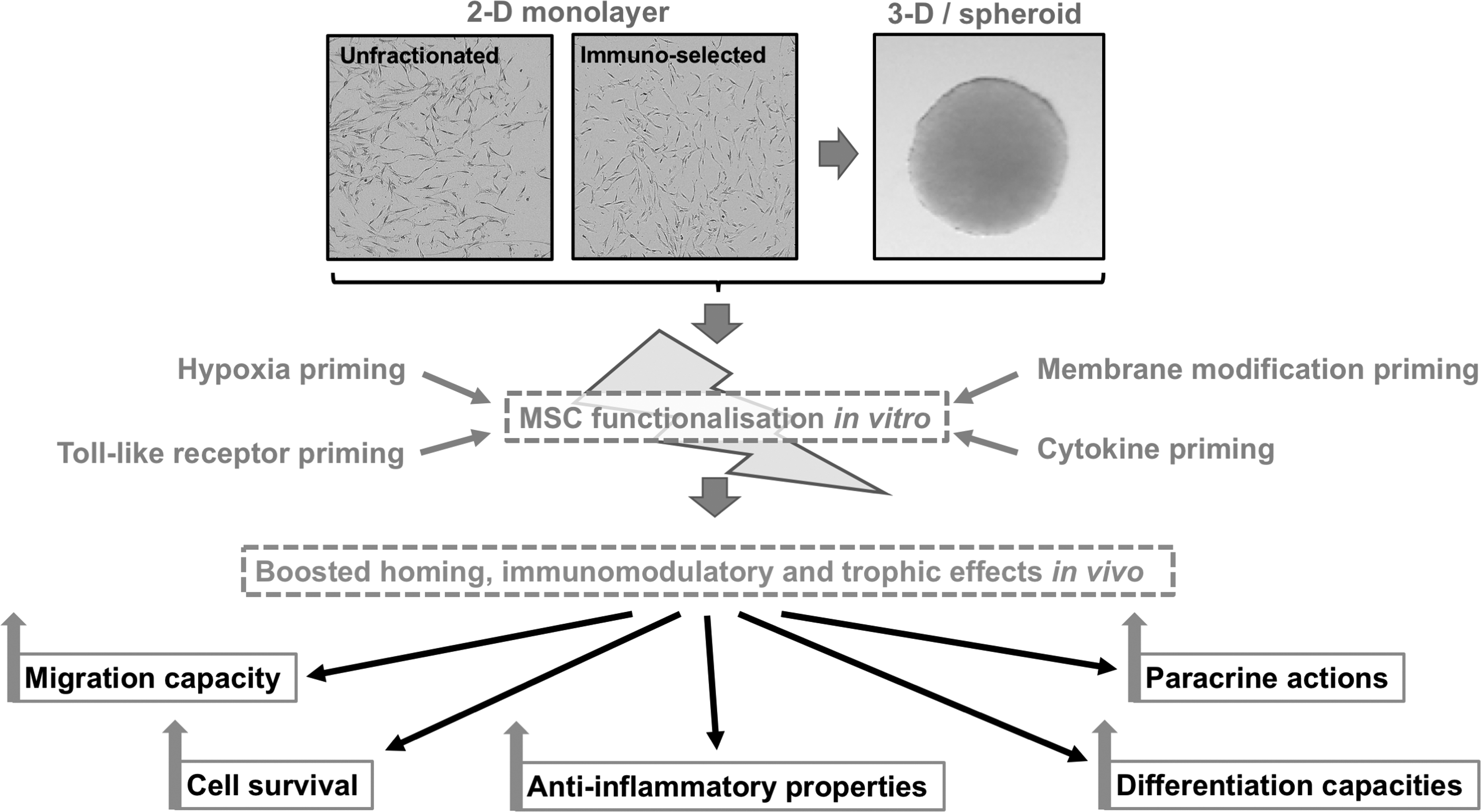
MSC functionalization by priming in 2D and 3D cultures results in boosted homing, immunomodulatory and trophic effects in vivo. 2D, two-dimensional.
Clinical Studies Using Functionalized MSCs
Up-to-date, no clinical trials have been executed to evaluate the efficacy of MSC subpopulations. On the other hand, three clinical trials are currently registered in www.clinicaltrials.gov using primed MSCs. Despite the fact that various priming methods have been developed to enhance MSC properties in vitro, some with preclinical validation, these clinical studies center on hypoxia/ischemia as the priming method to enhance MSC trophic properties. The phase 3 “STARTING-2” study (NCT01716481) evaluates the efficacy of intravenously infused autologous BM-derived MSCs preconditioned with autologous “ischemic serum” to treat stroke patients. A phase 1/2 study (NCT01849159) investigates the effectiveness of intravenously infused allogenic BM-derived MSCs primed in vitro under 1% hypoxia to regenerate the lungs of patients suffering from pulmonary emphysema. Finally, the “TPAABPIHD” phase 1/2 study (NCT02504437) investigates hypoxia-primed autologous BM-derived MSCs for their effectiveness to treat patients with ischemic heart diseases. Consequently, additional clinical research is needed to assess the safety and efficacy of “functional enhancing” strategies to MSCs before administration, including cell selection and priming protocols.
Conclusions
It has become increasingly clear that current MSC culture-expansion methods, although proven to be clinically safe, do not guarantee the preservation of specific native MSC characteristics and attributes, thus yielding cell-based products of variable quality, and more importantly, potency. These manufacturing limitations come in addition to inherent differences in the product performance, secondary to various factors including origin of the cells (i.e., autologous vs. allogeneic), donor age, underlying pathological conditions, and the recipient's implantation site microenvironment. The resulting compromised standardization may account for the observed inconsistent therapeutic outcomes. Therefore, obtaining more homogeneous MSC preparations (e.g., through selection and/or induction by priming), in which the critical features are preserved, may help circumvent the lack of reproducibility, while enhancing their therapeutic effects (Fig. 3). Thus, based on preclinical data, MSC subpopulation(s) selection and priming protocols may offer therapeutic advantages compared with the use of bulk/heterogeneous preparations in a number of clinical indications.
Footnotes
Acknowledgments
We are in gratitude with the Soffer Family Foundation and the Diabetes Research Institute Foundation (DRIF) for their generous support. C. Sanjurjo-Rodriguez is beneficiary of a postdoctoral fellowship from Xunta de Galicia (Consellería de Cultura, Educación e Ordenación Universitaria).
Disclosure Statement
No competing financial interests exist.