
Abstract
Cell migration is an essential process in which cells move from one location to another with different modes, including mesenchymal, amoeboid, or collective movements. Migration occurs during development and in the maintenance of multicellular organisms for purposes of wound healing, tissue regeneration, and immune and pathophysiological responses. Cells in all three types of muscle: cardiac, smooth, and skeletal, are subject to and undergo migration, both general and adapted for tissue-specific needs. Cardiac cell migration is mediated by vascular endothelial growth factor (VEGF) through expression of VEGF receptors; it is not clear how cardiac cells migrate into a region of damage after infarction. In skeletal muscle, satellite cells, with dual roles as muscle precursors and self-renewing multipotent adult stem or stromal cells, are resident on muscle fibers and normally mitotically inactive. Their activation and subsequent migration critically mediate skeletal muscle repair. Nitric oxide and hepatocyte growth factor are important signaling factors and the only two chemical factors that activate satellite cells. Both induce satellite cell motility on fibers in culture and into a region of muscle damage in vivo. By comparison, vascular smooth muscle cells migrate in response to vascular injury, during the normal process of angiogenesis, and in the pathological process of atherogenesis and vascular thickening. Microfluidic devices are advantageous in their capability to control cellular microenvironments and thus offer a valuable approach for the quantitative study of cell migration in vitro: devices can be designed to incorporate conditions that mimic what is known of normal physiology and control of microenvironmental changes can model particular situations. In this direction, there is increasing interest in developing innovative microfluidic devices to enable investigation of the migration behavior of different muscle cells. The mechanisms of muscle cell migration and their physiological roles are discussed in context of the emerging development of microfluidics-based approaches to advance studies of muscle cell migration and highlight their potential applications.
Impact Statement
The essential interactions between and among cells in the three types of muscle tissue in development, wound healing, and regeneration of tissues, are underpinned by the ability of cardiac, smooth, and skeletal muscle cells to migrate in maintaining functional capacity after pathologies such as myocardial infarction, tissue grafting, and traumatic and postsurgical injury. Microfluidics-based devices now offer significant enhancement over conventional approaches to studying cell chemotaxis and haptotaxis that are inherent in migration. Advances in experimental approaches to muscle cell movement and tissue formation will contribute to innovations in tissue engineering for patching wound repair and muscle tissue replacement.
Introduction
Muscle cell migration
S
For single cells, migration begins with cell polarization in response to a promigratory agent. Then, cell protrusions extend along the direction of migration, driven by actin polymerization as protrusions are stabilized by attaching to extracellular matrix (ECM) proteins on the surrounding substrate. These adhesions act as tracking locators that let cells move forward over them, detaching when they disassemble at the rear of the cell. 4 This cycle can be a singular or ongoing response in a larger developmental or pathological process.
Migration has different modes: ameboid, mesenchymal, and collective, each specific to cell type, cell number, and the substrate environment (Fig. 1). Cells can move individually or collectively, depending on factors, including the type of substratum, adhesion strength, extrinsic signals, dimensionality, mechanical elasticity, and cytoskeletal organization. Interactions between cell-intrinsic factors and the environment will promote a particular migratory mode. For example, immune cells with a nimble, fast-moving and fast-turning mode, do not adhere strongly to the substrate as they lack a cytoskeleton organized for attachment; their movement is amoeboid. Interestingly, movement by extension of membrane blebs occurs in some tumor cells that lack a highly organized actin cytoskeleton.

The modes of cell migration. Cells demonstrate different modes of migration, based on the cell type and the substrate in or on which they are migrating; and also show different morphologies when they engage in different modes of migration. 5 Color images are available online.
Mesenchymal migration occurs with cell polarization as the cell follows its leading edge of lamellipodia. This mode varies in speed: epithelial cells and fibroblasts move more slowly than other types of cells, despite a complex cytoskeleton. It is fascinating to know that some cell types, depending on their environment, will switch between ameboid and mesenchymal modes of migration. Cells also move as collective groups in chains and layers, as in development. 5
Other processes also contribute to overall cell movement, including polarization, protrusions from and adhesion by the membrane, cell body translocation, and retraction of a following edge during cell movement. Such processes are organized by signaling networks, often with the same effectors, despite differences in cell type and migration mode. 5
Cells migrating in one direction show distinct ends. The leading or advancing edge extends flat, thin knobs called lamellipodia and filopodia during actin polymerization. Integrin receptors on their membranes allow cell attachment to integrin proteins in the ECM. At the following aspect of the cell, actin microfilaments, called stress fibers, depolymerize and shorten; this lets the cell detach from the back and move forward. Therefore, a retraction force is required to translocate the cell body during migration. Interestingly this force is mainly produced by myosin motors sliding on actin filaments inside the cell; the net difference between contraction and adhesion forces is the basis of cell movement.
Actomyosin contraction, cytoskeletal disassembly, and retraction force are essential for cell movement, including during cytokinesis in cell proliferation.6,7 Adhesive forces in attachment vary between cell types and near the edges of a cell used to form cell protrusions by actin polymerization, while the remaining force is applied in actin network flow. Actin filaments can be in a direction opposite to that of cell movement, as the leading edge moves forward. 6
Cell migration is fundamental in embryogenesis, often by collective movement of a homogeneous cell population. Migration is also a key feature that can underpin homeostasis and regulatory functions in adult organisms. For example, neural crest cells migrate into many tissues in developing brain, limb, and skin, and help regulate function of many other cell types in many organs. 5 Neural crest cells arise from the embryonic ectoderm, and give rise to several non-neuronal cells, including pigment cells in skin, cartilage, bone, and pericytes, and some have self-renewal capability.8,9
The role of actin in cell movement
The cytoskeleton provides a structural scaffold and is responsible for cell movement (Fig. 2) by using three protein filaments: actin, intermediate filaments, and microtubules, which differ in size and composition.

Cell movement. Actomyosin-mediated contraction pushes the cell body to move forward (indicated as locations on the substrate, numbered 1 to 6). The cell tail detaches from the substrate, first from location 1, then in consecutive images (from top to bottom of the panel) from locations 2, 3, and 4. Protease enzymes that are released by migratory cells into the ECM cut the proteins connecting the cell and its underlying substratum, and allow the cell to move forward. 94 In addition, nesprin proteins are thought to help position the nuclei, and coordinate and stabilize nuclear mobility inside the cell through interactions with the cell cytoskeleton as the cell moves forward.95 ECM, extracellular matrix. Color images are available online.
Microtubules, 25 nm in diameter, are composed of tubulins alpha and beta, two subunits that bind into long-strand protofilaments. Thirteen protofilaments form a microtubule. Microtubules extend from the cell centrosome and shape cytoplasmic structure. Intermediate filaments are 10 nm diameter; in contrast to homogeneous microtubules and actin filaments, they are composed of tissue-specific subunit proteins and are less dynamic. However, microtubule strength and supporting functions both depend on the intermediate filaments found in every cell type. For instance, desmin filaments are in muscle cells, neurofilaments are restricted to neurons, and keratins are localized in epithelial cells. In addition, intermediate filaments are not polar like tubulin in microtubules and actin filaments. 10
Actin monomers polymerize with a 166° rotation between nearest neighbors by head-to-tail interactions of the polarized molecule to form thin and flexible microfilaments 7 nm in diameter. Actin-binding proteins localize along the filaments and “manage” activities such as crosslinking into actin bundles and networks, and interactions with other filaments and organelles. Actin filaments are typically at highest density in the cell cortex, just beneath the plasma membrane, particularly in motile cells. In striated muscle, actin filaments are also organized internally into bundles within sarcomeres, the unit of striated muscle contractility.
Actin filaments have critical roles in mechanical support, changes in cell shape, movement of the cell surface across the cytoplasm, migration on a substrate, and proliferation; the configuration and role of actin filaments depend on the particular activities of a cell at a given time.
In vitro assembly of actin filaments is regulated by the ionic strength of actin in solution. At low concentration, actin filaments depolymerize to monomers; at higher ionic strength, monomers will polymerize. Typically, monomers are added more rapidly to the fast-growing end (called the plus or positive end) of an actin filament. Although ATP is not required for polymerization, it plays an important role in slowing monomer binding and filament dissociation at the end where ATP is bound as ATP-actin. The helical structure of actin filaments results in repeating cycles of polymerization and depolymerization, depending on internal ionic strength of the cell matrix.
The above process, called treadmilling, is associated with the dynamic behavior contributed by different monomer concentrations at minus and plus ends of actin filaments. Treadmilling requires ATP and influences filament assembly and disassembly during changes in movement and cell. 11 However, the role of treadmilling is still not clear. 12
Muscle Cell Migration and Regeneration
Migration by force-generating cells in cardiac, skeletal, and vascular smooth muscle is modulated by ECM components, peptide growth factors, cytokines, and drugs. Promigratory stimuli activate signal transduction cascades, which induce cytoskeletal remodeling and changes in cell adhesion to the ECM, and activate (phosphorylate) motor proteins that contribute to movement. Myosin is a common molecular motor, converting ATP to ADP by myosin ATPases during force production. This conversion of chemical into mechanical energy acts on actin filaments.
Contributions of actin polymerization and work by molecular motors lead to actin flow, the continuous movement of peripheral actin networks, in cell motility; remodeling of actin filaments, molecular motors, and focal cell contacts with the ECM are organized to mediate cell migration toward or away from chemical-gradient cues, by changes in matrix adhesiveness, composition, or stiffness.6,13,14
Skeletal muscle precursor cell migration and tissue regeneration
Muscle satellite cells, normally inactive in residence on muscle fibers, function as self-renewing stem cells and muscle precursors for growth, regeneration, and adaptation. Activation and subsequent migration of satellite cells critically mediate muscle repair since nuclei inside muscle fibers are postmitotic; only the fusion of satellite-cell progeny (myoblasts) to fibers can add nuclei. Muscle adaptation by forming new fibers in injury-repair depends on satellite cell mobility after activation. 15 Anderson and collaborators showed that nitric oxide (NO) and hepatocyte growth factor (HGF) are important chemical signaling factors that activate satellite cells; they are released from skeletal muscle during contraction or stretching. Inhibiting the enzyme that makes NO (NO synthase) blocks the immediate injury-induced activation of satellite cells and delays and impairs regeneration.16–18
The protein HGF is also called scatter factor; HGF promotes cell motility as seen in metastasis of cancer cells, and the migration of muscle cells in culture, on muscle fibers, and in vivo. 19 M2 macrophages in an area of muscle inflammation produce HGF which induces myoblasts to secrete semaphorin 3A (Sema3A), 20 a protein that is implicated in restoring the nerve and blood supplies to skeletal muscle during repair from injury.20–22
Myoblasts demonstrate a dose-dependent response to HGF, 23 and HGF increases the speed of their migration on cultured fibers and induces a more unidirectional movement. 22 In vivo, HGF originates from two sources: the cylinder of ECM where it is stored around fibers and single myoblasts (more of a point source). Thus, the structure of skeletal muscle tissue means that cells are responding to complex HGF gradients within the mixture of adhesive, stabilizing matrix proteins, including fibronectin and collagen. In trials of myoblast transplantation to promote muscle repair, cells essentially migrate only 200 μm before differentiating 24 ; this explains the need for multivariable approaches to myoblast transplantation and wound repair by tissue engineering that is more complex than initially envisioned in the late 1980s.
In addition, after fiber injury, tissue-resident macrophages and neutrophils are activated and release high concentration of growth factors, including HGF, chemokines, and cytokines, which attract in-migration of immune cells that interact with myoblasts and further promote repair. Interestingly, growth factors released by T cells, interleukin-2, insulin-like growth factor I, interferon gamma, and amphiregulin also impact migration and proliferation by myoblasts. 16 Satellite cell populations and their responsiveness to activating stimuli both decrease with age, 17 and immune factors released from muscle also change with age.
Also interesting is that macrophages express and release HGF after injury in skeletal muscle. 18 The microenvironment of old muscle could be modified by regulating such immune factors to improve myoblast migration, increase muscle growth, and improve function. 16 Matrix metalloproteases (MMPs) also help regulate myoblast migration, differentiation, and tissue repair as MMPs digest ECM components. The out-migration of activated satellite cells from fibers and their movement toward other myoblasts to fuse and form new fibers require their migration across the basement membrane that is remodeled during inflammation and regeneration. MMP degradation of the ECM thus has a critical role in satellite cell migration and differentiation. 25
Myoblasts can move significant distances, suggesting they are in a state of constant, variable-speed movement within the basement membrane on fibers.22,26 Even a very focal injury induces satellite cell migration toward the damage, although the migration between separate muscles is limited by connective tissues.22,27
However, activated satellite cells and myoblasts can migrate relative large distances as shown by Allen and colleagues, using a high-resolution SPECT (single-photon emission computed tomography) to follow movement by myoblasts injected into the systemic circulation as they “home” to regenerating muscle. 26 A repeat of that study in a sheep tested long-distance migration after bupivacaine injury to semitendinosus muscle. Myoblasts labeled with a lentiviral vector tagged with a green fluorescent protein (GFP) were injected 5 cm distal to the injury and 8 days later, GFP+ cells were observed in sections of the injured muscle (R.E. Allen, pers. comm.).
Cardiac muscle cell migration and regeneration
Diseases of the coronary arteries and myocardium result in loss of cardiomyocytes and an expansion of fibroblast populations in fibrotic scars; as heart muscle generally cannot regenerate its own cells, 28 after a cardiac infarction, the damage is chronic and function deteriorates. The small peptide thymosin-β4 is expressed in developing heart and has important effects on actin cytoskeletal organization. The peptide also has effects on cell migration that can promote skin and corneal wound healing, angiogenesis, and cell survival. 29
Vascular endothelial growth factor (VEGF) signaling facilitates cardiac progenitor cell (CPC) migration. The possibility that CPCs might migrate into a region of damaged heart tissue is still unclear. Promigratory stimuli activate signal-transduction cascades that induce cytoskeletal remodeling, signal changes in cell adhesiveness, and phosphorylate motor proteins during CPC migration to an ischemic area induced by cytokines, VEGF, and/or fibroblast growth factor-2.30,31
Vascular smooth muscle cell migration and regeneration
Smooth muscle cells (SMCs) migrate during development and branching of the vascular tree, after vascular injury, and during atherogenesis (pathological thickening of the vessel wall). Platelet-derived growth factor (PDGF) stimulates migration of smooth muscle precursor cells, and PDGF signaling is essential to smooth muscle migration and angiogenesis. Pericytes, small contractile cells present on almost all capillaries, also show phagocytic activity and help mediate angiogenesis, endothelial cell regulation, vessel stability, and blood flow regulation. 32
Pericytes are smooth muscle progenitor cells; after they migrate and proliferate, they can form an endothelial cell tube that becomes a normal vessel. SMC migration in vivo or in vitro starts with stimulation of cell-surface receptors and subsequent signals alter their cytoskeletal structure.13,14 A 165 kDa variant of VEGF can also induce migration of human aortic SMCs and mesenchymal stem cells through PDGF receptors. 30 Also, Sema3A regulates neurites, smooth muscle, bone, and vascular endothelial cells, and interacts with VEGF receptors to upregulate blood vessel formation. By inducing actin depolymerization, Sema3A helps cells lose their focal contact with a substrate, which in turn enables their motility. 20
Engineering Muscle Tissues
Skeletal muscle tissue
Tissue engineering of skeletal muscle has great promise for treating diseases of muscle wasting and clinical situations such as major trauma and warfare injuries that require significant muscle replacement to reconstruct a body segment. Tissue transplantation itself, causes functional loss and volume deficiency33–35 ; therefore, new methods to improve the success and efficiency of tissue engineering to produce large volumes of skeletal muscle composed of bundles of well-aligned myofibers are critical in a modern approach to therapeutics.
To have a significantly large mass of engineered muscle tissue, a large volume of myoblasts must be produced by amplification in culture from an original sample without limiting the migration or differentiation capabilities of those cells. Induced pluripotent stem cells (iPSCs) could be used to form skeletal myoblasts that, similar to cells of the mouse-derived C2C12 cell line, would have an acceptable rate of differentiation and a very low chance of tumorigenesis.36,37
To improve prospects for engineering skeletal muscle tissue for therapeutic use, molecular mechanisms underlying muscle development; myoblast migration, fusion, and differentiation; and cell-cycling processes must be clearly understood, from transcription factors that “tune” the balance between growth by proliferation and myoblast differentiation to muscle that could become functional through innervation. 38
Other factors, including mechanical and electrical stimulation, and the role of the ECM are all critical to myoblast activities, including migration during muscle formation. Directed mechanical tension can mediate myoblasts to form new fibers that align exquisitely to the long axis of a muscle during contraction or passive movement; tension also influences cell number, fiber diameter, and muscle biochemistry. Electrical stimulation underlies voluntary control of skeletal muscle function; it promotes nerve regeneration into repairing muscle as well as myogenesis and angiogenesis in skeletal muscle tissue itself. 39
Myocardial tissue
The aim of cardiac tissue engineering is to reform an area damaged by infarction and reduce fibrotic scarring that limits pumping efficiency. While mechanical ventricular-assist devices or heart transplantation improves function, they are costly and many patients die before treatment. Therefore, engineering of heart tissue and stem-cell therapy are potential therapeutic options to “patch” an infarcted or scarred myocardium. 40
Skeletal muscle cells, embryonic stem cells (ESCs), and primary cardiomyocytes derived from neonatal rat hearts have been used to help repair cardiac tissue injury. A cell suspension can be injected into the myocardium or systemically, although this approach is not very effective as many cells are lost during delivery. The potential to engineer a patch of cardiac tissue or make a biomaterial scaffold as a vehicle to improve effective cell delivery to an injured area seems more promising. 40
Use of polymers that gel, such as collagen and alginate, to deliver cardiomyocytes into a scarred region aims to directly integrate donor cells into the host tissue by forming a new, hybrid myocardium through formation of intercalated discs and gap junctions that enable coordinated, effective cell contraction and electrical pacing.
Since adult cardiac myocytes have low proliferative capacity, cardiac stem cells have been identified (or induced from iPSCs) as a source of cells with greater potential for successful grafting and integration into the host tissue. Whether such cardiac stem cells will migrate to an infarcted region or remain in the location where they are injected, and whether they will differentiate and integrate into the surviving cardiac tissue after migration are not known. Therefore, a good understanding of cardiac stem cell migration and differentiation into cardiomyocytes is critical to future advances in cardiac tissue engineering for therapeutic use. However, we do know that transplanted cardiac stem cells respond to signals in an injured area and will migrate to it before they differentiate; this promising result was related to the extent of angiogenesis in the tissue. 40
Several trials have attempted to regenerate cardiac tissue in an area of infarction by injecting bone marrow cells, ESCs, skeletal myoblasts, or cardiomyocytes differentiated from primary CPCs. Any successful myocardial tissue engineering by injection or cell implantation requires minimally invasive approaches that do not lose cells during implantation approaches. An intravenous delivery would be more effective than direct injection into the heart, as direct injection cannot repair the important epicardial layer around the contractile tissue. Another approach, injecting cells within an injectable biomaterial, would provide a supportive matrix, but thus far, involves open-chest surgery. Injection of a biomaterial alone may be a useful approach as the artificial matrix could house autologous CPCs from the host. However, finding nonimmunogenic polymers is a challenge.
Use of left ventricular “restraints” of biopolymer “sheets” that wrap the ventricle, avoids cell injection, but interestingly, prevented cardiac tissue remodeling and repair. Ultimately, the ideal method will ensure that cells are delivered to the desired region with minimal injury or cell loss; tissue engineering could advance such approaches and help determine the optimal type of cell for implantation in a biopolymer or direct injection into the heart muscle. 41
Vascular tissue
Generating functional implants to replace damaged coronary arteries is a key goal of current biomedical engineering approaches to coronary vascular tissue. One of the drawbacks of current vascular prosthetic vessels engineered in vitro is the intimal hyperplasia that develops after grafting. The hyperplasia is caused by overproliferation of vascular SMCs (VSMCs) that are noncontractile. When SMCs lose their contractile proteins, they exit cellular quiescence and their proliferation, migration, and production of ECM proteins increase. As a result, grafted vessels narrow and the fatty deposit of atherosclerosis appears on the inside of the vessel wall. Figure 3 shows the processes involved in a shift from the typical contractile SMC phenotype toward a synthetic or proliferative phenotype.
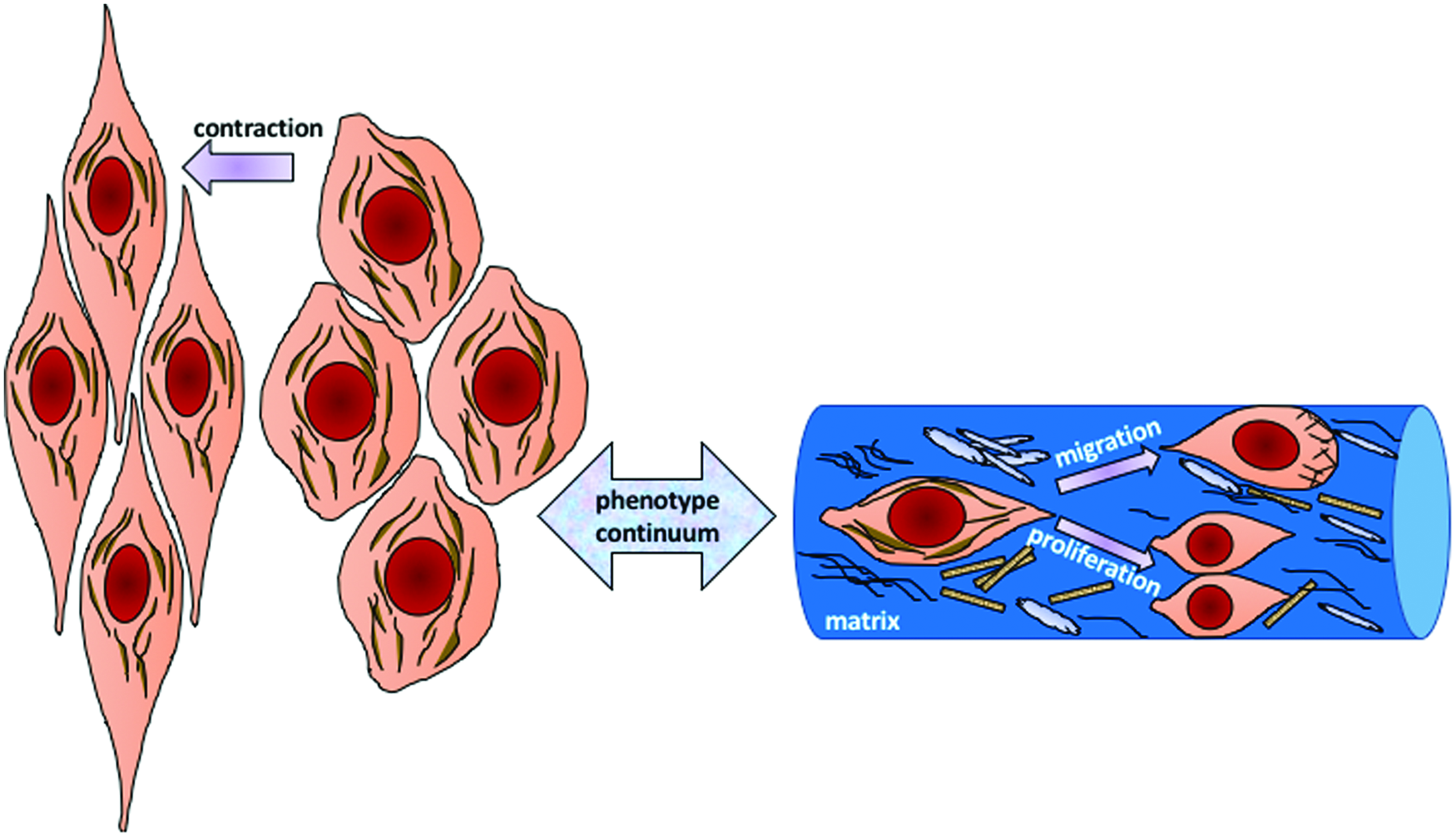
SMC phenotypes. A shift from a contractile phenotype to a synthetic or proliferative phenotype occurs in SMCs when they lose their contractility. 38 SMCs, smooth muscle cells. Color images are available online.
To minimize intimal hyperplasia, current research aims to engineer vessels containing cells that progress to a well-differentiated, contractile SMC phenotype. 42 A promising area of research in engineering vascular tissue for use in grafting regional and intraorgan vessels after damage is applying biodegradable scaffolds that incorporate smooth muscle stem cells and can maintain a layer of healthy endothelial cells, and produce an effective, nonhypertrophic connective tissue matrix. 43
Notably, while engineering skeletal, cardiac, and vascular muscle tissues has many common requirements, it is essential that host and engineered vascular tissues become integral to those tissues and are able to expand into any transplantable, implantable, or pathological (i.e., regenerating) patches of cardiac or skeletal muscle for effective, long-term function.
Conventional Cell Migration Assays
Studies of cell migration utilize a variety of in vivo and in vitro approaches, often microscopy methods to record migration, that is, changes in cell position and shape. Different aspects of cell behavior during migration and cell responses to stimuli are often imaged with fluorescence or phase-contrast microscopy before analysis with computer software, often NIH ImageJ.
Approaches to the study of directed and spontaneous cell migration are exemplified by studies of leukocyte migration in which cells migrate from wells punched into a layer of agarose gel, into the surrounding agarose layer; this is a better approach for studying directed and random migration than the Boyden chamber, since cell migration is measured as the distance cells move from the edge of the well toward an applied chemotactic factor. A chemotactic index of migration is calculated at a microscopic scale with higher resolution, and the measurement of migration distance is standardized against migration toward a control medium. 44 An Optimax image analyzer can be used to automate measures of cell migration. 45 A capillary chamber migration assay is similar to the Boyden chamber assay. 46
A high-throughput study of migration using the now-classic in vitro “scratch assay” was established to reveal potential toxicities that affect development of human neural crest tissue. The analysis was at the level of individual cells, typically tracked by time-lapse microscopy over time, using automated Introtox software for image processing. 47 The scratch or in vitro wound-healing assay is inexpensive and relatively easy to conduct; it is often used to study cell migration in 2D. The scratch assay is conducted by scraping a line or area into a monolayer of cultured cells; the scratch simulates a tissue wound and a simulated healing response can be visualized in real time by tracking cells microscopically as they move from the intact area toward and into the scratch-injured part of the culture.
An alternative approach applies electrical stimuli to injure cultured cells; this approach is recommended as it is considered to be a more physiological injury. “Tscratch” automated software can be applied to analyze the pattern and speed of cell migration in such an approach. In vivo migration assays of mesenchymal stromal cells (often called stem cells in the literature) were conducted in mice subjected to a chronic wound that was not allowed to heal.46,48
An option that avoids these disruptive methods, creates a “cell-exclusion zone” in a culture or tissue that prevents cell growth into that area; later, the barrier is removed and cells are allowed to migrate inward to fill the gap. An advanced version of this approach is called a fence assay, which allows the rate of movement to be measured using automated digital-image analyzers.
The microcarrier bead assay cultures cells onto beads and then beads are transferred to a 2D culture area. Cells that migrate from the beads onto the culture dish are then incubated and beads are removed by suction. Cell migration in these approaches is monitored by light microscopy.
The spheroid migration assay 49 uses a combination of 3D and 2D microfluidic technologies to study cancer cells. This assay is similar to that using cells grown on beads, with the movement of cells off the 3D spheroid onto a 2D substrate being monitored under a microscope. Both assays simulate the close cell–cell contact observed in cell migration studies.
Chemotaxis studies
Chemotaxis is the motion of cells directed by a soluble chemical gradient. Chemotaxis is typically described in contrast with more random-impact Brownian motion.
Chemotactic signals are important for growth, migration, and cell differentiation within a dynamic, 3D microenvironment since gradients provide critical influence on cell responses to processes such as inflammation, wound healing, and cancer. In traditional cell cultures containing medium, it is essentially impossible to generate spatial gradients with reliable control or stability, as isotropic and unbounded diffusion of molecules in solution limits gradient stability in the absence of a reservoir and outflow. Fortunately, microfluidic technology can manufacture a wide variety of remarkably complex devices that help route fluids accurately over micron-scale dimensions; such devices also deliver the capability to create gradients based on fluid flow at a scale appropriate for studying cell migration in real time. 50
Traditional chemotaxis assays
Traditional chemotaxis assays generate gradients in vitro; their application has instrumentally shaped our current understanding of gradient signaling, although they can barely produce stable, and well-defined gradients with reliable spatial and temporal profiles. 50 The Boyden chamber, developed in 1962, is used for Transwell migration assays and has greatly advanced our understanding of chemotaxis. Migration is quantified as cells respond to chemokines over a range of concentrations.
The Boyden chamber has two sections separated by a membrane or filter (pore size typically 2–12 μm). A putative chemoattractant or chemorepellent solution is placed in the lower chamber and cells are incubated in the upper chamber. The motion of cells squeezing through the porous membrane simulates the movement of cells through the interstitial space and the ECM, and between other cells in vivo. In some studies of leukocytes, endothelial cells are grown on the upper surface of the membrane and cells migrate in response to the differential gradient generated between two types of cells on either side of the membrane. For analysis, cells are fixed, stained, and counted. The Boyden chamber assay is easy to use and enables observations and analysis of chemokine effects on cells that are more accessible to study than in a typical culture dish. 51
Use of a Boyden chamber or Transwell assay is limited by the difficulty of sustaining a chemokine gradient in the chamber for more than a few hours due to diffusion, and challenges of visualizing cells in real time as they pass through the membrane. It is also impossible to study the integrated response to multiple gradient signals. Also, the effects of shear stress on the cells, for example, from fluid movements while the cells are migrating, cannot be distinguished from chemokinetic effects on the cells.50,52 Modern applications such as IncuCyte® for live cell imaging during chemotaxis address some of these limitations.
Biological hydrogels of collagen, fibrin, or agarose can be manipulated to establish in vitro gradients around cells. Cells or tissue explants can be cultured on a gel surface or mixed within a liquid hydrogel solution before it gels. A gradient of one or more biomolecules can be formed in two ways in this assay, either by co-culturing test cells or tissue with a second cellular source of a putative or known chemokine or by molding the gel or making holes in it that can be filled with a soluble form of one or more potentially active molecules. Hydrogel modifications thus enable real-time observation of cells responding to one or multiple chemokines.
Recent advances generated biological hydrogel gradients by spacing lines or drops of a biomolecule on the surface of a gel so it diffuses into the gel matrix. The droplets are placed by pump or inkjet printing and form a concentration gradient by diffusing in space and time, out from the source and into the gel where the cells are cultured. Advantages include the ease of making these gels that better resemble in vivo tissue than cells in conventional 2D culture dishes. Chemokine movement in a biological hydrogel is based on free diffusion, unaffected by bulk movement, and the position of a biomolecular source is quite feasibly controlled. However, several drawbacks interfere with use of this method, including endogenous secretion of biomolecules, (possibly the same as that under study), by the cells themselves.
As it is technically challenging to detect nanomolar concentrations, the concentration-dependent response to a given biologically active molecule is not easily detected by protein assays of the gel. The porosity and polymer chemistry of the gel network also reduce the ability to control the gradient patterns, and once a biomolecule is loaded into a source cavity, concentration, shape, and diffusion of the gradient cannot be modified. This adds between-experiment variability as cells and biomolecule source compartments are established manually. Finally, single-cell responses are not easily visualized due to the 3D architecture of the culture system and gel itself, due to the optical properties of some gels (e.g., containing refractile collagen fibrils) that obscure cells in phase-contrast microscopy.
Therefore, cells in hydrogels have to be tracked using Z-series image stacks by confocal imaging or 3D-tracking algorithms. Overall, biological hydrogels have advantages over Boyden chamber assays: they are easy to use without complex equipment, and produce reasonably controllable gradients of more than one biomolecule. 50
Micropipette-generated gradients use glass micropipettes to generate biomolecular gradients. A fine glass tip with an internal diameter 1 μm or less is pulled from a heated glass capillary tube. A biomolecule solution is suctioned into the tip which is then placed at a known distance from cells of interest using a manipulator under a microscope. The solution is released in proximity to the cells using a pump to control the volume and frequency of ejections that generates a gradient. 53
A variant of the micropipette method uses pneumatically ejected biomolecules that diffuse passively from a micropipette tip and induce cellular responses, for example, by primary neurons and neutrophils.54,55 In contrast to cells on or within biological hydrogels, single-cell responses to chemokine gradients at a known distance or direction can be measured since cells can be visually distinguished from the background. The method can create multifactor gradients by adjusting multiple micropipettes around a cell of interest.
However, these micropipette-generated gradients are not particularly controllable between experiments due to changes in the free solution that generates the biomolecule gradients, thermal variation and evaporation of the solution, and variations in the size and shape of the micropipette tips. Commercial tips are pulled under well-controlled conditions, but still show geometric differences that will generate variation among gradients under the same experimental conditions. These 3D gradients also require confocal microscopy and fluorometric dyes for quantification during an experiment, and careful positioning of the micropipette tip or tips relative to a cell, on a crowded microscope platform with equipment to control humidity, carbon dioxide, and temperature.
Therefore, complex on-stage environmental controls are typically most feasible in such experiments on nonmammalian cells that do not require 37°C or cells that respond fairly quickly to a gradient (within 1–2 h). Despite these limitations, the micropipette method is well suited to studying single-cell responses, although less suitable for measuring cell responses to multiple gradients. 50
The Zigmond chamber was developed in 1977 to study neutrophil chemotaxis, as it allows direct visualization of cells. The device has a glass slide with a ridge and an overlying coverslip; the slide and coverslip are separated by a gap 3–10 μm high and 1 mm wide where a gradient is formed. Cells near the ridge are observed under a microscope. The precise geometry of the Zigmond chamber can produce a uniform, linear uniaxial gradient, which can be studied in detail over time.
The important drawback is that the gradient has a short lifetime (stable for 1 h), so the Zigmond chamber device is used to study only fast-responding cells such as sperm and neutrophils. A further limitation is that the fluid under the coverslip evaporates; this can affect the fluid flow that generates the gradient and protects cell viability. However, the Zigmond device is very useful for studying cell migration under a reliable gradient for up to 1 h and the gradient can be monitored with fluorometric dyes. 50
The Dunn chamber is similar to a Zigmond chamber, but less sensitive to evaporation. Source and sink chambers in the Dunn device are made of concentric circles and cover slipped to avoid evaporation. Limitations and strengths of this device are generally similar to those of a Zigmond chamber, and a multifactor gradient can be generated only in one axis, similar to Boyden and Zigmond chambers. While the Dunn chamber is geometrically symmetrical, evaluation of cell responses in a radial direction is difficult. 50
Haptotaxis studies (cell-ECM interaction)
Cell adhesion, the interaction between cells or with an ECM, is a very broad topic of biological study due to its functional importance. Interactions inherent in cell adhesion sustain normal cell physiology and behavior and influence development of pathology, wound repair, and tissue engineering. Ultimately, advanced tissue engineering will only be achieved when cells can attach, grow, and be viable on biological substrates during synthesis of implantable tissues.
A microfluidic device was used to model the in vivo regulation of cell adhesion behavior with a small number of cells to investigate changes in cell adhesion under pathological conditions. The device used passive fluid pumping, where liquid is pumped indirectly through a microchannel between the inlet and outlet ports due to the surface tension of a small drop of liquid (cell medium or a potential cytokine solution); this reduces the wasted volume of cytokine. Also, applying an oscillatory flow through channels was able to mimic normal physiological conditions (e.g., in the cardiovascular system) by providing the stimulus of pulsatile mechanical shear stress to a cell population. A large population of cells was examined in a single experiment to save time and reagents; this revealed interesting interactions between cells that are not as accessible in studies of cells in culture dishes. 56
The directional motility of cells or parts of cells (e.g., axonal outgrowth from a neuron) is usually toward a gradient of cell adhesion sites or substrate-bound molecules that chemoattract the cell. Such gradients in the ECM also operate during angiogenesis; gradients can be artificially placed on a biomaterial substrate by controlling the concentration of a particular protein on the polymer surface. Cells may sense and respond to cell-adhesive and/or chemoattractant substrates. Responses recognized by changes in cell morphology, migration speed, cell–cell interactions, or even a “memory effect” by which some cells will migrate back to their starting point.
The ECM is also continuously remodeled since cells in any tissue produce ECM molecules over time. This dynamic feature of tissue formation rearranges cells and/or the matrix components in a tissue and is a critical aspect of embryonic development and tissue repair. The ECM provides stability and mechanical support to cells and contributes important biological signals that impact cell cycling, motility, and other behaviors. The majority of cardiac ECM are collagens type I and III that are remodeled by cardiomyocytes in response to physiological and pathological stimuli, including infarction, pressure and/or volume overload, and inflammation. 57
A prototypical ECM protein is tenascin (TN-C), and is expressed in the early stages of inflammation, wound healing, tissue remodeling, and embryogenesis. TN-C has a notable anti-adhesion property that can destroy cellular interaction with fibronectin in the ECM and result in loss of focal adhesion sites. 57
Microfluidic Devices for Studies of Muscle Cell Migration
Microfluidic studies of muscle cell behavior have recently investigated contractility and migration with applications in cancer therapy, drug delivery, and the development of “organ-on-a-chip” models. In general, a microfluidic chip is about the size of a universal serial bus drive and consists of micrometer-scale channels to study cell behavior under conditions that mimic one or more aspects of physiology. 58
Important in this review, many microfluidic devices are designed to generate controllable chemical gradients that comprise putative stimuli for cells of interest. Their flow-based mechanisms can generate stable gradients, the shape of which can be manipulated due to the device design. Typically, the gradients are uniaxial and cells in the device channels are subject to shear stress.
Using this type of microfluidic gradient device, our lab demonstrated the influence of a gradient of epidermal growth factor on the growth and migration of adult rat adipose-derived stem cells. 59 By contrast to a flow-based microfluidic gradient device, 3D gel-based microfluidic devices better mimic the physiological ECM and maintain a stable, somewhat less-controlled gradient. For example, a large source and sink in the device will create a stable linear concentration gradient in a small hydrogel-filled channel at the equilibrium state of mixing. This approach is useful for studying directed cell migration in a controlled 3D microenvironment.
A microfluidic device with a narrow channel was used to mimic the geometry of a lymphatic capillary and create a platform for studying differential single-cell migration patterns under a chemotaxis gradient.
A single-layer poly-dimethyl siloxane (PDMS)-based device with two inlets and two outlets was developed to have a central migration channel of single-cell capture sites, connected to left- and right-side flanking channels; the right channel contained a test agent. Cells were loaded from the left and flowed toward the right channel, influenced by chemotaxis to the gradient and the influence of gravity. Cells separated by their chemotactic response were collected from outlets for further analysis. Cells with higher elasticity showed a higher capture rate than those with lower elasticity. This type of device simulated how certain cancer cells in a heterogeneous tumor cell population might flow into capillaries during metastasis. 60
Microfluidic devices have also revealed the migratory behavior of endothelial cells under a VEGF gradient, while in co-culture with smooth muscle or cancer cells within a collagen-gel scaffold. Importantly, both the gradient of growth factor and the substrate condition (i.e., the stiffness of the collagen gel) influenced cell movement. Microfluidic devices can be used to generate gradients in the substrate as well, for use in studying other mechanisms of cellular movement, including actin filament movement on myosin heads on a substrate surface, without interference from bulk flow that would produce larger-scale disturbances and obscure very fine-scale movements of myosin–actin interactions. 61 Microfluidics-based studies of the behavior and migration by the three types of muscle cells and other relevant applications are highlighted below.
Cardiac cell isolation and in vitro studies using microfluidics
Cardiac muscle cells demonstrate electromechanical responses, growth, and migration; sorting cardiac myocytes from nonmuscle cells in cardiac tissue will be critical for developing applications such as transplantation of cardiomyocytes or isolating stem-like cells for use in implantation to regenerate infarcted heart muscle. The growth of cardiac muscle tissue and cardiomyocyte migration are closely associated. The effects antitumor and other drugs on the growth of spheroids formed from cardiomyocytes can be probed in detail using devices such as a commercial Ibidi μSlide® microfluidic device for long-term 3D culture of cardiac cell spheroids at high-throughput. This device is composed of six channels running in parallel between inlet and outlet ports. Cell spheroids were seeded in the channels and exposed to test drugs, while the device was rocked on a tilting platform to prevent spheroid attachment. The approach was used to quantify the number of cells that migrated over time, outward from the cluster of cells forming the spheroids.
In this application, the 3D culture of cardiomyocytes in dynamic conditions also resulted in proliferation, noted by the presence of more nuclei than in spheroids cultured under static conditions; results implicated combined effects of rocking and drug exposure in mediating cardiomyocyte cycling. 49
Cardiac physiology and development both depend on tissue responsiveness to rhythmical mechanical stress on cardiomyocytes. Since most cells and tissues are under constant mechanical influences, typical cell-culturing methods cannot accurately reproduce or even closely simulate the development of high and low pressures acting on the heart in vivo. A microfluidic approach can more closely simulate the influences of normal and pathological dynamic mechanical conditions on cardiomyocytes. The coronary blood supply perfuses cardiac muscle during diastole when the tissue is relaxed and blood is returned to the atria and the systemic circulation during systole, when the ventricles contract. 62 A microfluidic method simulated this pattern of blood flow: a multilayer device was assembled to create a cell culture chamber of PDMS with a flexible substrate; the chamber was sandwiched between two polycarbonate plates with micromachined channels through which flows could be directed through the chamber.
The above device was further integrated with valves and pressure sensors that allowed simulations of normal and disease states affecting cardiac hemodynamics, while cardiomyocyte responses were measured. The model also enabled experimental control of the balance between the physical loading of cardiac cells and their contractile behavior or cell–cell interactions by regulating stimuli such as pressure stress, stretch or strain, and “beat” frequency.
Cell migration studies typically note changes in cell shape and size as important features in characterizing a migratory phenotype; for this reason, it is useful to provide an example using microfluidics to sort and isolate functional cardiomyocytes from a heterogeneous cell mixture to improve studies of myocyte growth and proliferation. 63
In this case, the PDMS-based microfluidic device used a diffusive filter for cell isolation and was composed of a middle channel connected with microsieves to two side channels. Tygon tubing was connected to the inlets and outlets to pump the cell suspension and medium into the main channel. At the end of the experiment, the isolated cells were collected from the outlets and analyzed for viability, size, number, proliferation, and differentiation.
Another PDMS-based microfluidic device for cell sorting delivered cells in buffer from an inlet into the center of a wide sheath of medium; cells and medium were streamed through an array of posts, shifted in position from row to row, further into the device. The cells streamed around the posts, depending on their size. High-purity collections of homogeneously sized cardiomyocytes were sorted from debris and smaller cells by their lateral displacements through the array that hydrodynamically focused the fluid streamlines. The purity of cells collected at particular outlets was compared by flow cytometry to the inlet (source) of mixed cells.
Experiments with this device avoided intercellular binding by suspending cells in a medium of buffered saline containing albumin supplied from an input port separate from that used for the biological buffer containing cells. The flows of both the sheath fluid and the cells suspended in buffer in the channel were generated by an external syringe pump containing a steel ball coated with PDMS that created turbulence in the syringe itself. The sorting system was integral to this microfluidic device as the pattern of fluid flow toward the outlets helped prevent the larger cells clog the chamber; instead, larger cells collected to one side of the stream as they were directed toward the outlet. Sorting did not damage cell membranes or reduce cell viability, and collected cardiomyocytes showed reliable growth and elongation in both 2D and 3D cultures.
Therefore, while flow-related features of the microfluidic device were used to sort the cells, the precise characterization of cell architecture during the isolation was integral to phenotypic behavior of the cells, in that the myocytes formed contractile groups in 3D culture. The study illustrated that well-characterized cell behavior was a useful complement to the theoretical aim to develop a “patch” of engineered cardiac tissue. Another benefit of this particular cell-sorting microfluidic device was that it did not require any cell labeling or treatment, making the approach useful in clinical applications to patch damaged heart tissue. 64
A microfluidic method was also able to mimic the in vivo activities of ventricular muscle in the heart 65 by controlling the cell environment and mechanical stimuli. Cardiac myocytes were cultured on an elastic membrane together with fibroblasts, and mechanical stretch and cell alignment were controlled by using the properties of the elastic membrane inside the device.
A PDMS-based microfluidic device was used to pattern collagen deposition on the elastic membrane, and then the device was removed and cells were added for culture. This was reported as a reliable model for studying the reaction of myocardium to mechanical, biophysical, and/or biochemical stimuli in culture. Mechanical stretching was used to stimulate the production of proteins that cardiomyocytes use in calcium handling and regulating beat frequency and contractility. Close interaction between fibroblasts and cardiomyocytes co-cultured in the microfluidic device therefore simulated the in vivo conditions of atrial and ventricular tissue, since these cultures displayed myocytes that were aligned and surrounded by parallel fibroblasts. A more detailed experimental protocol for this method was also reported. 66
These studies show the variable application of microfluidic devices in studying cardiomyocytes: use of valves and pumps to create different hemodynamic conditions; use of laminar flow to separate different cell types based on their lateral displacement; use of PDMS devices to pattern collagen on a membrane that, in turn, patterned the growth of cardiomyocytes; and migration of cardiac cells out of a spheroid in a microfluidic device under continuous flow generated on a tilting platform. This wide range of research applications on cardiac muscle cells, including cell isolation methods and preparations for controlling culture conditions, enables studies of cardiomyocyte growth and responsiveness. The migration behavior of myocytes outward from a spheroid was made accessible using a microfluidic approach.
SMC culture and migration using microfluidics
SMC migration is an important determinant of successful wound healing as it underpins the new vessel formation (angiogenesis) that is essential to tissue repair. A microfluidic device to quantify VSMC migration used a PDMS-based device with a simple Y-shaped channel. 67 The design allowed a monolayer of VSMCs to be cultured in the channel and a wound edge was created using a gravity-based laminar- flow interface of trypsin. Using this device, differences in VSMC migration were demonstrated to depend on factors including microchannel height, coating substrate, and chemical stimulation. Specifically, cells were more migratory in a taller channel on a fibronectin substrate, and under stimulation by growth factors and cytokines.
In another study, a microfluidic device was used to examine migration by primary VSMCs. 68 This PDMS-based device had a loop design with multiple interspersed wells and reservoirs for medium with and without growth factor to generate a flow-free linear gradient that was stable up to 72 h at the equilibrium state of free diffusion. Using this device, dose-dependent chemotaxis and chemokinesis were stimulated by a growth factor, and the effects depended on treatment to inhibit cell proliferation. These microfluidic studies were able to show a higher-sensitivity cell-migration response to the very low concentration of growth factor in a gradient than studies using a Transwell assay, and were concluded to better characterize gradient-dependent cell migration by VSMCs.
The potential of microfluidic approaches to generate functional muscle tissue from SMCs is also under investigation. The ability to control the structure and contractility of vascular muscle tissues could ensure the tissue was functional for efficient blood transport after grafting. A microfluidic device that integrated an elastic PDMS film and a fibronectin substrate patterned by microcontact printing was used to simulate contraction by smooth and cardiac muscle cells. 69 Several designs based on a similar device-fabrication method were developed to compare responses by one or more muscle types in parallel, using an electrode setup to stimulate contraction and study effects of an inhibitor of muscle contractility.
A circular microfluidic device was used to characterize and control the structure of an engineered vascular tissue, specifically to investigate the alignment of SMCs around and along a channel as a model of a vessel. The device improved the effectiveness of tissue engineering in manufacturing vascular tissue of various calibers, a major focus in the field that aims to understand the 3D vascular architecture so vessels can be engineered for grafting.
In this device, two halves of a microchannel were made with PDMS and bound together by exposure to ultraviolet light. Highly viable SMCs were aligned around a tube orthogonal to co-cultured endothelial cells that lined the inner “lumen” of the vascular construct. Such approaches could lead to the development of therapeutic applications to treat vascular disease. 70
SMCs are also located around other hollow organs and tubes, such as respiratory airways, where they interact with other cells. Interaction between endothelial and SMCs was examined using microfluidics to model lung airway-on-a-chip that eventually may be useful to treat chronic lung disease. The approach was based on suspending a biocompatible gel in a microfluidic device to generate interaction between cells in separate upper and lower chambers. The lung-airway microdevice was fabricated from polymethyl methacrylate polymer as three layers: the top layer was exposed to air, a middle channel contained the suspended hydrogel, and the bottom channel was the media source. The two cell types in the device could be co-cultured in long-term experiments. 71
These studies illustrate various applications of microfluidic devices to study SMCs: use of geometric patterns and laminar flow to create a wound assay; use of thin-film and microcontact printing to simulate smooth muscle tissue on a chip; use of a circular microfluidic device to simulate blood vessels and study SMC alignment; simulation of lung airway by endothelial and SMC co-culture; and SMC migration under influence of a chemotaxis gradient. Microfluidic devices were highly advantageous in advancing this research on SMC function and migration related to vascular tissue engineering.
Skeletal muscle cell culture and migration using microfluidics
C2C12 cells of a murine skeletal muscle cell line derived from satellite cells, are a common reagent for studying myogenic differentiation. C2C12 myoblasts express unambiguous markers of skeletal myocyte differentiation, and have been used in various 2D and 3D culture configurations. 72 C2C12 myoblasts in culture fuse to form multinucleate syncytia, called myotubes, an in vitro form that displays many of the morpho-functional features of typical multinucleated muscle fibers found in vivo, including contractile sarcomeres, and adaptation to regulatory influences such as neural and hormonal signals. 73
Endothelial cells and skeletal myoblasts respond to mechanotaxis by distinctive changes in cell alignment and migration, depending on dynamic and static cues that modify mobility, proliferation, differentiation, and cell spreading on the substrate. 74 Similarly, cell-strain experiments without a microfluidic device showed differential responses in cell dimensions by primary fetal rabbit myoblasts, lung type-II alveolar cells, and bone cells subjected to cyclical stretch and release. 75
A microdevice of a biocompatible polymer, poly(lactic-co-glycolic acid) melt-casted from PDMS mold, was used to form microgrooves of various depths to culture C2C12 cells and study their migration and proliferation. 76 Cell alignment responded to the dimensions of the grooves by increasing expression of filamentous actin, and migration by pseudopodia responded to groove depth. 76 The 3D microgrooves also induced cell proliferation, consistent with observations that a smooth substrate reduced cycling, possibly by reducing adhesion.
The 3D configuration of the ECM, cell alignment, and tissue architecture are all important influences on skeletal myoblast proliferation and migration, and are modified by mechanical stretching. 77 Microfluidic devices have been used to study combinations of mechanical stretching, shear forces of fluid movement, and cell alignment on geometrically patterned substrates, including alignment of C2C12 cells. 78 While reports relevant to muscle cell behavior and migration are not always consistent due to the variety of substrate configurations and properties, 78 the control of such features can be facilitated by the use of microfluidic devices.
The responses of C2C12 cells among others were studied using a microfluidic device after cells were plated on different ECM substrates; cell growth and motility responses to mechanical stimuli were tracked. 79 Isotropically modified substrates patterned by microgrooves, lanes, and aligned electrospun nanofibers in the device facilitated cell alignment after plating; subsequent protocols stretched cells or subjected them to shear forces from flow within the device. The evolution of intracellular calcium concentrations, expression of phosphorylated (activated) focal adhesion kinase, and visualization of cell orientation were used as direct measures of cellular responses to shear and stretch. C2C12 cells oriented themselves perpendicular to the stretch direction on a nonpatterned surface, but showed irregular orientation or realigned in parallel to a micropatterned fibronectin substrate. 79
The contraction of muscle cells during culture is also available for study using microfluidic devices. A PDMS-based microfluidic device was constructed by soft lithography and C2C12 myoblasts were seeded in collagen inside microchannels and cultured for a week to study the contraction of the differentiated myotubes that model fibers. Myotubes demonstrated contraction by internal structural displacements upon electrical stimulation, and by their expression of α-actinin in sarcomeres of stimulated cells. 80
Another microfluidic culture system allowed study of skeletal muscle myoblast contractility after electrical stimulation; muscle cells were co-cultured with nonexcitable cells such as monocytes. In such cultures, both cell types are influenced by myokines, proteins released from contracting muscle that promote muscle cell movement. C2C12 cells were cultured around a conductive polymer wire composed of poly(3,4-ethylenedioxythiophene) and polyurethane; the wire was attached to the PDMS chamber containing cell medium. This approach was able to regulate muscle cell contraction during culture using controlled ionic currents generated by depolarization. 81
In our laboratory, microfluidic experiments are investigating how an HGF gradient regulates myoblast migration and differentiation under the influence of various substrates, alone or in combination; the substrates are also established in a gradient pattern across the migration channels of triple-docking devices 82 and other novel 2D or 3D devices, as represented by examples in Figure 4. These studies are aimed to help improve methods of engineering skeletal muscle tissue to grow “bulk muscle grafts” that would be able to replace large volumes of muscle lost through trauma or other injuries.

Triple-docking microfluidic device. This microfluidic device (left) is designed to enable investigation of the effects of a gradient of HGF (depicted as a triangle in the upper middle of the panel) in combination with 2D gradients of the substrate (middle). Cells are introduced into the device at cell-loading ports (shown in green on the left, with one loading port per channel [in black]) inside the device. Channels are prepared with or without an HGF gradient after they are coated with a substrate of fibronectin, collagen, or fibronectin plus collagen (shown in the middle part of the figure, representing the three channels coated under various conditions). Migration by cells in the experimental channels (measured in conditions with a particular substrate coating and/or an HGF gradient) is compared to cell migration in control channels without a substrate coating and/or HGF. 83 HGF, hepatocyte growth factor. Color images are available online.
Another interesting study used a microfluidic device to estimate cell responses to different modes of delivering culture medium in long-term culture experiments. The PDMS-based microfluidic device was composed of five parallel channels connected to tubing to allow repeated refreshment of medium inside channels. Such lengthy cultures will be the basis of developing future tissue-on-a-chip technologies that can reproduce the biology and physiology of normal tissue, and could be used to promote functional repair of muscle.
Since continuous flow provides more cell heterogeneity in cells cultured in a device, over the longer term, such cultures result in abnormal cell morphologies and vesicle formation in the cytoplasm. These findings suggested fluid delivery should be pulsatile in such devices, a feature that was achieved by pumping medium at a specified rate to maintain normal cell morphology and viability. 83
A different microfluidic device for long-term culture of C2C12 myoblasts was used to investigate cell proliferation without medium exchange.84,85 A PDMS-based device was fabricated to form multiple channels in elastic silicone rubber; channels varied in thickness and had extended cell cultivation channels and two cell-seeding ports.
This microfluidic device was combined with a so-called “Braille display,” a level surface with a grid of over 300 vertically moving pins; the pins were used to power an integrated system of pumps and valves and induce local deformations of the channel networks. A transparent heater was used to maintain temperature outside the incubator and avoid changing the culture medium, so cell responses could be studied with finely controlled fluid movement patterns.
These two reports revealed an exciting new possibility for portable cell-culture systems that could be visualized in long-term time-lapse studies, while maintaining healthy cells with minimal medium.
A later study on spatial differentiation of C2C12 skeletal myoblasts investigated effects of culturing in medium with a linear gradient of two bone morphogenetic proteins, BMP-2 and BMP-7. A microfluidic device fabricated as layer-by-layer films of poly(
A micro-bioreactor array was developed to cultivate two different cell lines together, including C2C12 myoblasts and nondifferentiated human embryonic stem cells (hESCs), in different configurations of flow. In this study, a PDMS-based microfluidic device was fabricated with 12 culture wells connected to the main channels. After binding to a glass slide, coating with fibronectin, and seeding with cells, the device was covered with a thin layer of PDMS. Inlets and outlets were connected to a syringe and filled with cold medium, and the whole device was placed in an incubator.
Cell type-specific comparisons spurred interest in understanding the more complex regulatory pathways of hESCs as they grow and differentiate in culture. For comparison, C2C12 myoblasts and rat cardiac myocytes were also cultivated and examined by automated image analysis in situ in the microfluidic device, to investigate the expression of genes that are used to mark the progression of cell differentiation. 72
Microfluidic devices have thus proven to be valuable tools in research on cardiac, smooth, and skeletal muscle. Their use has fostered major advances in our understanding of the migration behavior of fast-responding cells, including immune cells, and cells that seem to respond more slowly. In turn, ongoing refinement of microfluidic devices designed specifically for testing individual hypotheses encourages further exploration of mechanisms underlying the many types of differential response patterns even among the three types of muscle cells. Additional enhancements to make multigradient devices have more recently enabled even more in-depth studies of proliferation, migration, and differentiation of several cell types under a wide range of culture conditions.50,82
The features of skeletal myoblast differentiation, clustering, alignment, and fusion into myotubes in vivo in development and regeneration,87,88 and in culture, 89 can therefore be modeled in the channels and on the patterned substrates of a variety of PDMS-based microfluidic devices. 90 Findings that myoblasts migrate to regions of high myoblast density in culture and toward fused myotubes 91 are consistent with observations that small muscle wounds heal very quickly in comparison to large-volume injuries.
An important gap in our understanding is identifying how a differentiated myotube by itself can slow or sufficiently overwhelm the migration of a myoblast to trigger the return of the myoblast to the quiescent state as a stem cell on a newly repaired fiber in a muscle in vivo, after injury. Our laboratory is currently addressing this gap using a newly designed microfluidic device to load C2C12 cells in two waves; the first will be induced to differentiate into myotubes before the device channels are reseeded with myoblasts that will be observed for their movement and differentiation behavior. 92 Figure 5 provides a schematic of potential experiments. 82
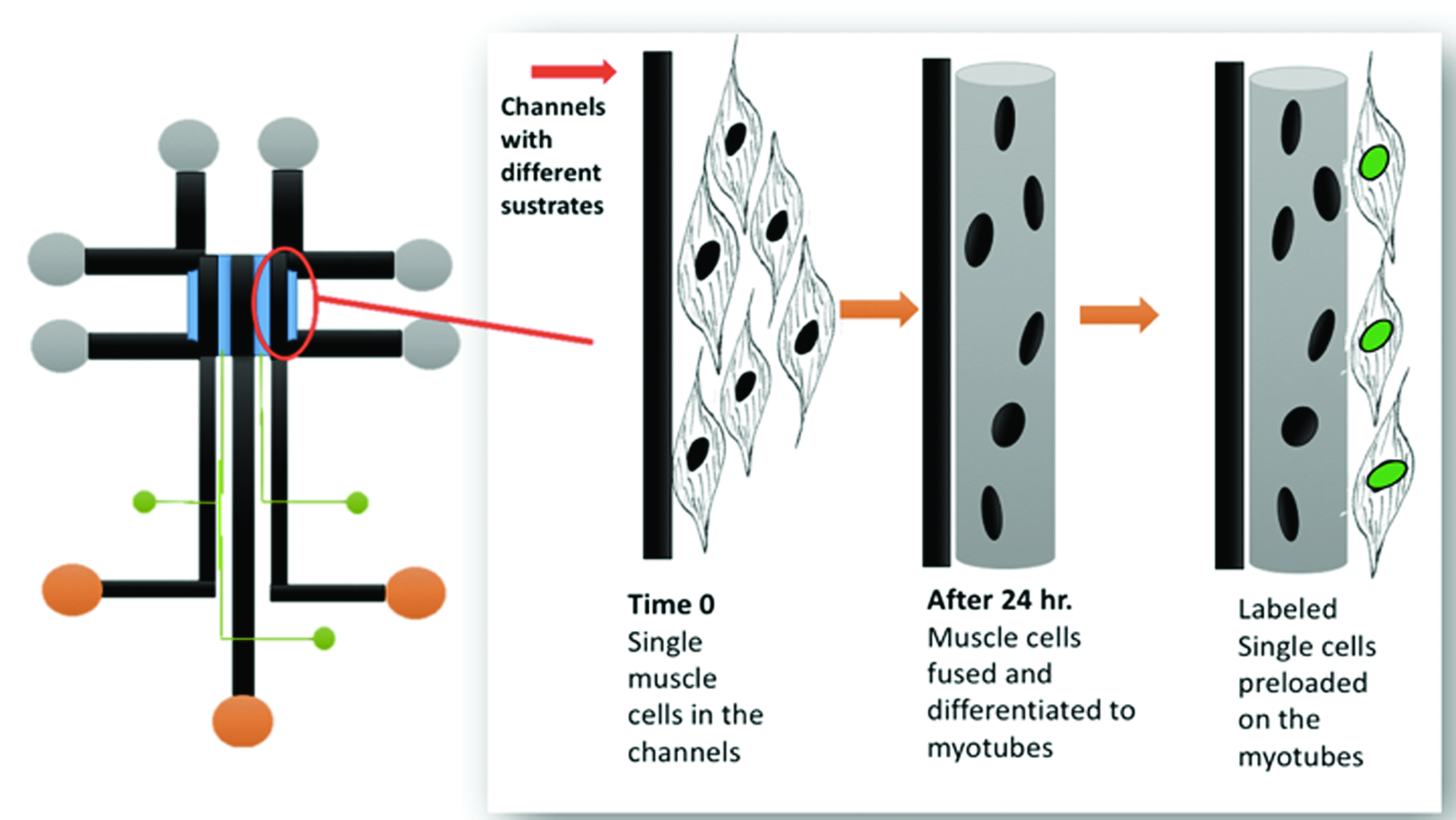
Future application of microfluidic technology to study myoblast migration on myotubes. A novel design for a triple-docking device will enable time-lapse observations of myoblasts under controlled conditions for investigation of the initial cell differentiation into myotubes, and the behavior of a second wave of myoblasts that is introduced into channels containing the myotubes. Preliminary results suggest cell–cell communication by myoblasts during migration is highly attuned to the substrate composition and the state differentiation in neighboring myoblasts. 83 Color images are available online.
Conclusions and Perspectives
This review aimed to integrate current information on the importance of understanding muscle cell migration under controlled environmental conditions in the assessment of cellular activities such as proliferation and differentiation. These activities ultimately lead to and distinguish the functional capabilities of cells in morphologically and phenotypically distinctive lineages of smooth, cardiac, and skeletal muscle.
The synthesis of fields of microfluidics and muscle biology highlights the strong scientific potential of collaborative research. That will ultimately help us take the major step from understanding the biology of cell migration in biomedicine to reaching the full potential for effective therapy of injury and disease. Achieving this step will specifically help us understand how to employ tissue-engineering approaches to replace pathological or absent muscle tissue.
The potent strengths of using microfluidic technologies are promising, given the need to overcome many current deficits in our knowledge of muscle tissue biology. Reaching that goal requires a new integration of information on the biological and biomechanical features of muscle cells, together with ongoing refinement of technologies, including microfluidic devices, to advance our capability to engineer tissues and organs.
The many clear and important distinctions among smooth, cardiac, and skeletal muscle cells during tissue development in vivo can be at least partially replicated by modeling their growth and migration behavior using microfluidic devices (represented by examples in Fig. 6).

Representative microfluidic studies of migration by different types of muscle cells. Skeletal muscle cells (left, MSCs), cardiac muscle cells (middle panel, CMCs), and vascular SMCs co-cultured with HUVECs (right panel). CMCs, cardiac muscle cells; HUVECs, human umbilical vascular endothelial cells; MSCs, skeletal muscle cells. Color images are available online.
Ongoing avenues avenues of cell biology research using microfluidics now require intensive investigation and then careful validation in vivo before their potential for application, for instance as tissue patches, can be realized in humans. These are still “early days” in the field employing microfluidic approaches to study migration of muscle cells and muscle stem cells, and yet exciting new experiments with microfluidic devices promise to provide solid documentation of the many tantalizing features of cell migration.
Footnotes
Acknowledgments
Authors are grateful for research support from the Natural Sciences and Engineering Research Council of Canada (03833-2015 to J.E.A.; 04789-2014 to F.L.), the Faculty of Science Collaborative Research Grant program (to J.E.A. and F.L.), and the Graduate Enhancement of Tri-Council Stipends program from the Faculty of Graduate Studies at the University of Manitoba (to F.L. and J.E.A., toward support of graduate research by Z.R.).
Disclosure Statement
No competing financial interests exist.