
Abstract
The cornea is an important barrier to consider when developing ophthalmic formulations, but proper modeling of this multilayered tissue remains a challenge. This is due to the varying properties associated with each layer in addition to the dynamics of the tear film. Hence, the most representative models to date rely on animals. Animal models, however, differ from humans in several aspects and are subject to ethical limitations. Consequently, in vitro approaches are being developed to address these issues. This review focuses on the barrier properties of the cornea and evaluates the most advanced three-dimensional cultures of human corneal equivalents in literature. Their application potential is subsequently assessed and discussed in the context of preclinical testing along with our perspective toward the future.
Impact statement
Most ocular drugs are applied topically, with the transcorneal pathway as the main administration route. Animal corneas are currently the only advanced models available, contributing to the drug attrition rate. Anatomical and physiological interspecies differences might account for a poor translatability of preclinical results to clinical trials, urging researchers to devise better corneal equivalents. This review elaborates on the emerging generation of three-dimensional in vitro models, which comprises spheroids, organoids, and organs-on-chips, which can serve as a stepping stone for advancements in this field.
Introduction
Modern drug development in the pharmaceutical industry is subject to high drug attrition rates, both before and during clinical trials. For example, 2019 saw the approval of 48 novel compounds for use in humans by the U.S. Food and Drug Administration (FDA). 1 At the same time, it is estimated that only 0.4% of drug candidates that enter preclinical testing receive FDA approval. 2 Combining both figures amounts to 48 out of 12,000 candidates that successfully completed the journey toward the market. This journey is notoriously expensive—the development of one therapeutic agent requires an average investment of 1.3 billion U.S. dollars, when accounting for failed trials and other indirect costs. 3 However, the investment required is not exclusively monetary as the drug development pipeline takes an average of 8.5 years to complete. 4
At the root of this tremendous cost is the low rate of success during clinical trials, due to the low predictive power of the models employed in preclinical studies. 5 Of the remaining candidates, estimations indicate that only one in five successfully pass clinical trials and obtain FDA approval after preclinical testing. 2 Better preclinical models are required that can identify and eliminate undesirable candidates early to expedite the drug development process, and to reduce costs, thereby saving pharmaceutical companies the time and effort of going through costly clinical trials with compounds that ultimately prove to be either toxic or ineffective.
In this context, the field of ophthalmology represents 2.5% of the global pharmaceutical market size6,7 and is associated with the second to highest average per-study cost with regard to clinical trials (excluding postmarketing surveillance costs), 8 which exhibit an overall probability of success that is greater than double the probability of success across all fields of research. 9 At first glance, the costlier clinical trials and increased likelihood of approval do not seem to be causally related; the latter might instead be attributable to the accessibility of the eye, facilitating localized therapies, in combination with the extremely high failure rates in the field of oncology, lowering the general probability of success.
The eye can be medicated in a variety of ways, including peroral administration, topical instillation, injection, and implantation. The majority of ophthalmic drugs are indicated for topical use, where the transcorneal route is widely regarded as the prime pathway for ocular drug delivery. 10 As such, testing the pharmacological properties of drug candidates by using a representative corneal model is extremely important during the preclinical stages of ocular drug development. Recent advances in the in vitro modeling of the human cornea have made it possible to simulate the in vivo characteristics of this tissue with unprecedented accuracy.
Hence, this review focuses on the emerging generation of three-dimensional (3D) in vitro corneal models. What follows is an introduction into the barrier properties of the human cornea and a concise overview of advanced in vitro models based on human corneal tissue, along with their strengths and limitations. Taking these aspects into account, the models are discussed further and compared with regard to their applicability in preclinical testing.
Components of the Corneal Barrier Function
The anterior ocular surface can be divided into two parts that are optically and functionally distinct. At the center lies the transparent dome-shaped cornea, which covers the iris and pupil (Fig. 1A). The area surrounding the cornea is occupied by the conjunctiva, which covers the white sclera and inner parts of the eyelids (Fig. 1A). Although the conjunctiva occupies 17 times greater a surface area than the cornea, the latter is considered to be the main passageway into the intraocular spaces for compounds that are applied topically. 11
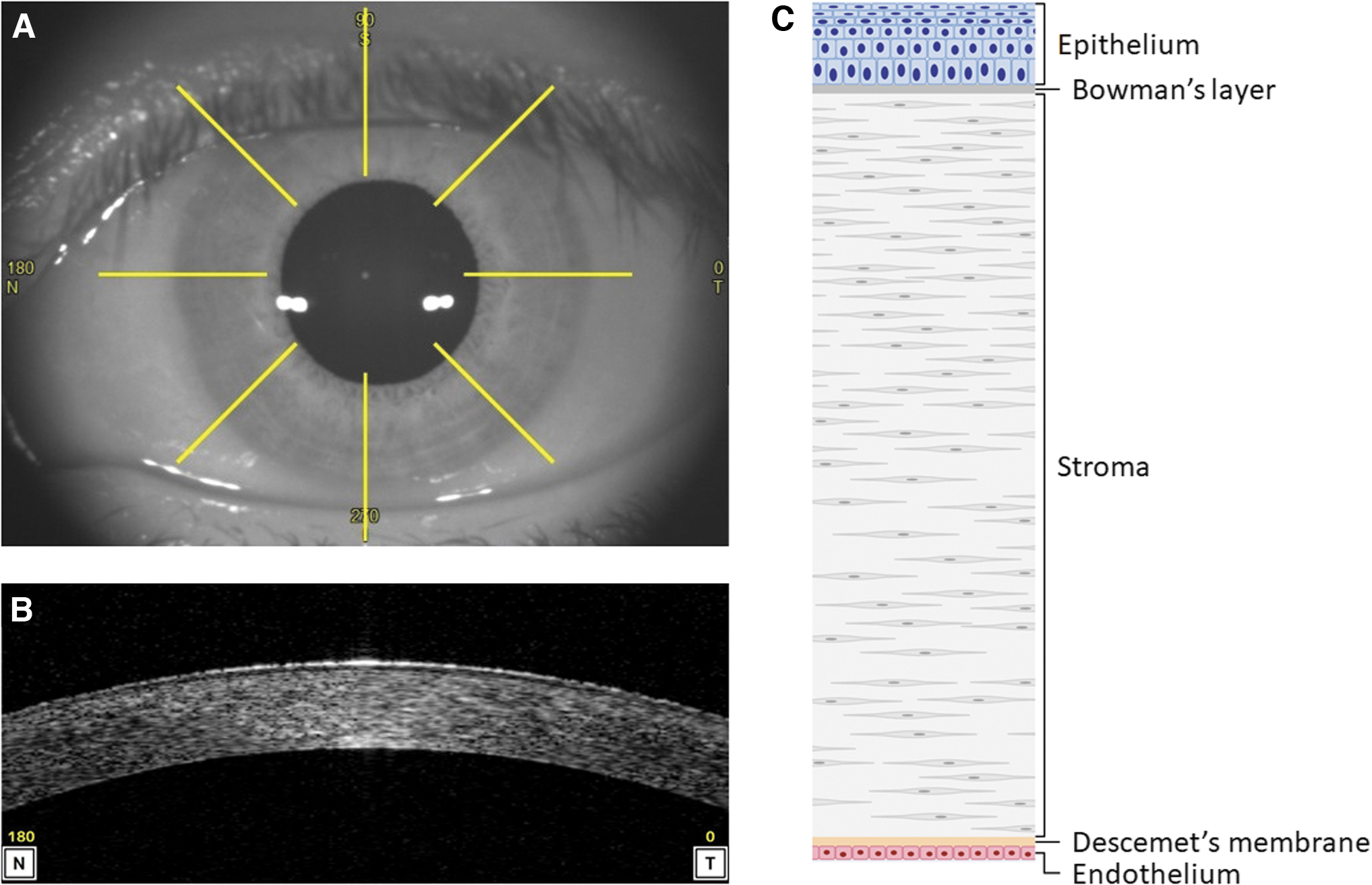
Anatomy of the human cornea.
Drugs attempting to enter the eye through the conjunctiva and sclera are instead diverted away by local blood vessels into systemic circulation, which are absent in corneal tissue. The cornea itself is ∼550 μm thick centrally and consists of, on the outermost surface, a stratified epithelium (corneal epithelium) supported by an acellular layer (Bowman's layer), a middle stromal layer (corneal stroma), and an inner basement membrane (Descemet's membrane) lined with a single sheet of simple cuboidal cells (corneal endothelium) (Fig. 1B, C).
Despite its accessibility, the transcorneal pathway for drug delivery proves a formidable barrier to drug absorption, resulting in limited intraocular bioavailability for topically applied compounds. Typically, at most 5% of the instilled dose is absorbed through the cornea, so the design of formulations that maximize bioavailability must take local and systemic toxicity into account.12,13 Table 1 provides a summary of the barrier properties pertaining to each corneal layer.
Overview of All Corneal Layers and Their Barrier Properties
TEER, transepithelial/endothelial electrical resistance.
Precorneal tear film
On administration of topical formulations to the cornea, they are first absorbed into a 5 μm thick tear film. 14 The tear film is a dynamic microenvironment that is designed to wash away foreign material (including drugs) and maintain a smooth surface, which protects the eye against external insults and allows for correct refraction of incident light. Its dynamics include tear displacement when blinking, tear evaporation between blinks, and a continuous turnover as a result of constant tear production followed by nasolacrimal drainage.
The film is composed of oily secretions from the Meibomian glands, aqueous secretions from the lacrimal glands, and mucous secretions by conjunctival goblet cells. 15 The differences in composition between secretions leads to the formation of separate layers with oily lipids on the surface, the aqueous forming a thicker middle layer and the mucous layer, adjacent to the cornea, maintaining “wettability” of the epithelial surface.
Under normal conditions, the entire tear film volume of 6 μL is fully refreshed every 5 min. 16 Remarkably however, between blinks, the aqueous layer remains relatively still, allowing the interaction of solutes with the corneal epithelium. 17 Lacrimation and blinking are stimulated during eye drop instillation, the intensity of which depends on both the manner of instillation and the properties of the formulation itself (e.g., tonicity, pH, and other factors that affect irritability). This tear reflex dilutes and promotes clearance of ophthalmic compounds, lowering the fraction that penetrates corneal epithelium, further reducing bioavailability. Fortunately, simply keeping the eyelids closed immediately after topical application and applying pressure to the nasolacrimal duct partly mitigates this instillation-induced reflex and improves drug residence time. 18
Corneal epithelium
The second barrier to transcorneal passage is the corneal epithelium. Its central region is composed of 5–7 layers of stratified non-keratinized cells with a total thickness of around 50 μm, whereas its peripheral region has more layers and increased thickness.14,19 These cellular layers form a tight lipophilic seal that prevents tears from entering the intercellular spaces, through cell adhesion proteins such as occludin and tight junction protein ZO-1. 20 The corneal epithelium further exhibits ridge-like microprojections that are coated with a negatively charged network of glycoproteins and glycolipids, called the glycocalyx.
The glycocalyx connects to the mucous layer of the human tear film and allows for a uniform distribution of tears across the otherwise hydrophobic epithelium. Whether or not the combined glycocalyx and mucous layer functions as an additional barrier to drug permeation has been investigated, but no conclusive evidence was found. 21
Regarding the permeability of corneal epithelium, transepithelial/endothelial electrical resistance (TEER), which is a measure for ion transport across a cellular barrier, is routinely used to quantify barrier integrity and permeability. However, the TEER values of fresh donor corneas are only available to a limited extent and vary substantially between publications. This variability might be attributable to the employed TEER measurement technique, but it could also arise from differences in tissue integrity as a result of insults before, during or after enucleation. For example, donor corneas have been reported to lose their superficial epithelial layers in storage. 22 * Hence, TEER values from artificially generated multilayered epithelium might prove to be more representative of native epithelial tissue. † TEER values ranging from 600 to 2700Ωcm2 have been reported,23,24 which is consistent with the poor permeability seen for hydrophilic compounds.
In addition to diffusion, active transport of drugs has been implicated in the corneal epithelium, so efflux proteins could prove an additional barrier to corneal absorption. 25 Several xenobiotic transporters, including multidrug resistance-associated protein 1 (MRP1), 26 MRP4, 27 and MRP5, 26 have been identified on the protein level in the human corneal epithelium. Moreover, the inhibition of such efflux transporters in rabbit cornea demonstrated an increased permeation of erythromycin into the aqueous humor in vivo.28,29 However, studies on active drug transport in the human cornea are sparse so their precise impact on ocular drug delivery remains unclear.
Corneal stroma
The corneal stroma comprises nearly 90% of the total human cornea, with a central thickness of 470 μm. 19 It is sandwiched between Bowman's layer and Descemet's membrane, both of which are considered negligible in terms of drug permeability. 30 Bowman's layer, previously considered a basement membrane similar to Descemet's membrane, is now defined as the acellular anterior 17 μm of the corneal stroma. 31 It is regarded as distinct from this stroma due to a different organization of collagen fibrils, which are mainly collagen type I, and embedded in a proteoglycan matrix. The collagen fibrils in Bowman's layer are arranged in a random fashion, whereas the collagen fibrils of the corneal stroma are organized in 200 to 250 interwoven lamellae that are responsible for maintaining the strength and shape of the cornea.
The highly ordered connective tissue of the corneal stroma is a product of its main resident cell type, the keratocytes, which are scattered throughout. The exact density of keratocytes in the stroma remains a subject of debate but has been found to be highest in the anterior stroma relative to the posterior stroma (Fig. 1C) and to decline with age.32–34 In healthy adult eyes, keratocytes are quiescent, although trauma to the stroma has been found to induce dedifferentiation into a proliferating fibroblastic phenotype that is associated with stromal tissue remodeling and scar formation.
The bulk of the stroma consists of the hyperglycosylated matrix that surrounds the cells, in contrast to the corneal epithelium and endothelium, which are almost exclusively composed of cellular layers. The carbohydrate moieties that coat the proteins of this matrix contain numerous polar groups, making the stroma hydrophilic, so much so that it generates a swelling pressure of around 80 mmHg. 35 The aqueous humor of the anterior chamber forms the drive of stromal hydration, given that the tear film has a higher osmolarity than the aqueous humor, and the closely packed epithelium limits water influx from the tear film.
Corneal endothelium
Descemet's membrane lines the innermost side of the corneal stroma and is a basement membrane of 10 μm that comprises other types of collagen than those found in the stroma. 36 This acellular membrane consists mostly of collagen type IV and is secreted by the corneal endothelium. The corneal endothelium is a hydrophobic monolayer of hexagonally shaped cells; it is 5 μm thick, and it regulates stromal swelling by establishing an osmotic gradient. 37
The hydrophilic glycosylated matrix of the stroma causes fluid to continuously leak from the aqueous humor into the stroma, which is compensated by active pumping of the corneal endothelium in the opposite direction. Failure of this pumping mechanism (e.g., due to trauma or genetic defects) results in swelling of the stroma, which is accompanied by a gradual loss of corneal transparency and, eventually, blindness. It is generally accepted that the collagen fibrils of the stroma have to be precisely arranged to correctly scatter incoming light, and it is the displacement of these fibrils due to stromal swelling that induces loss of vision. 38
Despite its barrier function, the corneal endothelium is regularly described as “leaky” due to its permeability to macromolecules larger than 150 kDa, which is reflected in a small TEER value of ∼20Ωcm2.37,39 An adult cornea has, on average, a cell density of 2500 endothelial cells/mm2, which gradually diminishes over time at a rate of about 0.6% per year. 37 Should the endothelial cell density fall below a density that can maintain the barrier, decompensation occurs and its integrity is compromised, leading to corneal-related loss of vision. 37
Precursor-Derived Corneal Models
The main cell types of the cornea are all derived from the cranial ectoderm during the development of the human eye. 40 After neural tube closure, protrusions at the level of the diencephalon extend to interact with the surface ectoderm, inducing a change in cellular morphology to create the lens placode. These protrusions invaginate and the lens placode detaches from the surface ectoderm, leaving behind a continuous epithelial layer that gives rise to the corneal epithelium. Later, neural crest cells migrate to the space between the presumptive epithelium and presumptive lens to establish the corneal stroma and endothelium. Such developmental events are guided by a series of molecular cues, which can be leveraged in vitro to generate 3D corneal structures from precursor cells. These structures can be divided into two types: spheroids and organoids.
Spheroids are cellular aggregates of a spherical shape that improve on conventional two-dimensional (2D) culture by introducing a 3D environment with increased cell–cell interaction (Fig. 2A). As a result, cell behavior in response to drug treatments is more similar to native tissue.41,42 Larger spheres resemble solid tumors and comprise a mixture of cells at different stages, with an outer proliferative layer, necrotic core, and quiescent zone in between. 43 This is the result of limited nutrient diffusion, hypoxia, and the accumulation of waste in the inner regions of large spheroids, which, in turn, might influence cell behavior. 44 Spheres can be generated in two fundamental ways: Culture conditions can facilitate spheroid formation by reaggregation, for example, when seeding in U-bottom low attachment plates, or by proliferation to obtain clonotypic spheroids, for example, with the addition of methylcellulose gel (Table 2).

Structural composition of advanced in vitro corneal models.
Overview of Precursor-Derived Constructs Based on Human Corneal Tissue
hESC, human embryonic stem cell; hpSC, human parthenogenetic stem cell; iPSCs, induced pluripotent stem cells; ULA, ultra-low attachment.
Corneal spheroids are usually generated through reaggregation, are derived from primary tissue, and are limited to a single cell type (Table 2). Sphere-forming cells have been reported to exist in all layers of the cornea45–49 and in the limbus,45,50 which is the transitional zone in which the cornea, conjunctiva, and sclera meet. It can take up to a month to generate mature corneal spheres, but there is substantial variation in differentiation times between reports, which might, in part, be attributable to the type of cell used for spheroid generation (Table 2).
Spheroids are predominantly employed in the field of oncology, as they represent miniature solid tumors. They can be used, among other things, to investigate drug absorption, drug response, and cell–cell interactions. However, the usage of corneal spheroids in the field of ophthalmology has so far been limited to the isolation of highly proliferative cells with sphere forming assays. The cancer-like properties of spheroids make them less suitable for modeling the organized structure of the cornea, which is required for proper pharmacological testing. In addition, the 3D environment created through spheroid formation has been observed to promote the “stemness” of cells in vitro. 51 Hence, the main potential application for corneal spheroids may be in (stem) cell generation for use in tissue engineering or regenerative therapies (Table 2).
Although spheroids are a step up from 2D cultures, they are relatively simple when compared with organoids. * Organoids can be seen as the spiritual successor to the spheroids but they differ in a few key areas. They are derived from stem cells, whereas spheroids are mostly generated from differentiated tissue or cell lines. Embryonic stem cells have been used in the past, 52 but the field has moved toward the use of induced pluripotent stem cells (iPSCs), given that they are more readily available and pose fewer ethical concerns.
The differentiation process of organoids is vastly more complex when compared with spheroids, as the resulting 3D constructs self-organize into complex organ-like functional units and include multiple cell types, in both the early and late stages of cellular differentiation (Fig. 2B). However, the improved similarity of organoids to in vivo tissue comes at the cost of a longer differentiation period, which generally takes place over the course of multiple months (Table 2). Another drawback, which is especially present in iPSC-derived organoids due to the additional reprogramming step, is the increased variability between individual organoids established with the same protocol. 53 This is evidenced by the most recent publications on corneal organoids, which describe the generation of corneal organoids alongside retinal organoids when using culture conditions that favor retinal differentiation.22,54 In addition, corneal organoids have been shown to include non-corneal cell types. These include ocular cell types from anatomically proximal tissues such as the eyelid, limbus, and conjunctiva, 54 but also from more anatomically distant tissues such as the retina. 22
Similar to spheroids, organoids could provide a source of cells for transplantation, but the longer differentiation time and increased variability make this less evident. Similarly, although corneal organoids recapitulate the organized structure of the cornea, their use in preclinical testing is less appealing due to the associated variability. *Instead, the sequential nature of organoid differentiation might make organoids a useful tool to study corneal development and holds the potential to elucidate disease mechanisms that previously could not be modeled otherwise.
Microphysiological Models of the Cornea
The concept of miniaturization is most commonly associated with electronics, in particular with the evolution of personal computers. Although early computers filled entire rooms, they have since become much smaller and can, for example, be worn as a wristwatch. An additional benefit of striving to downscale the base components of any system is the resulting increase in efficiency with which they can be produced. Likewise, miniaturized systems have found their way into the fields of chemistry and life sciences, where extensive usage of specialized reagents can be financially draining. These miniaturized systems are called “lab-on-chips” because they recreate a (bio)molecular process, usually performed in a fully equipped laboratory, on a small plastic or paper-based “chip.”
The chips contain microfluidic channels that are designed to guide and mix reagents in a controlled manner. When infused with biological material, these characteristics can be leveraged to construct in vitro models that mimic the composition and physiology of native tissue, known as organs-on-chips (Fig. 2C). Organs-on-chips are microphysiological systems that further build on the concept of organotypic constructs, which are static 3D cultures commonly cultured by using transwell supports. Although such static organotypic models are interesting in their own right, they are outside the scope of this review because they are increasingly being superseded by this novel generation of dynamic chips.
Several microphysiological systems based on human corneal tissue have been reported, including chips based on primary tissue 55 and cell lines.23,56–58 These can be limited to only the epithelial layer or contain an epithelialized stroma construct, that is, epithelium and stroma (Table 3). Polydimethylsiloxane (PDMS) remains the standard chip material of choice for prototyping due to its favorable properties, including transparency, non-fluorescence, thermal stability, biocompatibility, gas permeability, capacity for surface modification, general versatility, and commercial availability.59,60 In addition to the microfluidic channels, organs-on-chips can be rationally designed to include other features such as TEER sensors 58 or even a biomimetic eyelid. 55
Overview of Microphysiological Constructs Based on Human Corneal Tissue
C-6, coumarin 6; Gel-MA, methacrylated gelatin; PC, polycarbonate; PDL, poly-D-Lysin; PDMS, polydimethylsiloxane; PET, polyester; PS, polystyrene; rCol1, rat tail collagen I.
After chip assembly and cell seeding, tissue generation is rapid and generally takes between a week and a month (Table 3). When compared with precursor-derived corneal models, these chips offer decreased variability and have a generation time similar to spheroids while encompassing increased complexity. Another benefit of combining cell culture with a chip-based miniaturization approach is that it favors mass production, facilitates standardization, and allows for high-throughput preclinical research. For example, multiple chips could be run in parallel and coupled to online measuring equipment. Therefore, microphysiological systems make more sense with regard to preclinical safety and efficacy testing than precursor-derived constructs.
However, a complete cornea-on-chip has not yet been realized and instead there is currently a major focus on accurately simulating tear dynamics and shear stresses. Flow conditions such as continuous,23,56,58 pulsatile, 23 bidirectional, 57 and mechanically stimulated 55 flow at various shear stresses have been reported. Although tear flow is important to account for, there is currently no consensus as to how it is best simulated.
The reasons for producing epithelialized stroma instead of a full-thickness cornea might be twofold. First, several corneal cell lines have been shown to vary substantially with native tissue61–63 and between laboratories, 24 which advocates the usage of primary cells. Culturing primary corneal endothelial cells is not without its challenges; namely, they possess limited proliferative capacity and tend to lose their phenotype through endothelial to mesenchymal transition when cultured. 64
Second, there is a notion that corneal stroma presents the most important barrier after corneal epithelium. This notion might be premature and ignores the possible impact of endothelial active transport on drug permeation. Moreover, a large-scale literature analysis revealed that the second most important barrier structure of the cornea varies, depending on the nature of the applied medicine. 39 Although the corneal epithelium is, indeed, the most formidable barrier of the cornea, resisting both large (radius >10Å) and hydrophilic molecules, small lipophilic molecules can readily pass through and penetration enhancers can be used to increase epithelial permeability. 65 In these cases, it becomes important to consider the barrier properties of the stroma and endothelium. The results from the literature analysis suggest that corneal stroma forms a greater barrier for larger compounds whereas the endothelium is slightly more impermeable for small lipophilic molecules than the stroma. 39 This highlights the importance of a full-thickness cornea-on-chip over an epithelialized-stroma-on-chip.
Chip material is another possible area of improvement. Due to its advantages, PDMS has been preferred for rapid prototyping. Uncured PDMS polymers, however, have been shown to leach into microchannel media and integrate into cell membranes. 66 In addition, PDMS demonstrates a selective absorption of hydrophobic molecules, which could interfere with studies on corneal permeation. Fortunately, chips are highly customizable and material can be chosen to best suit the experimental setup.
Clinical Perspective
Ocular pathologies have a substantial impact on a patient's quality of life. Vision is one of the most fundamental senses for most people and the prospect of losing it can be highly detrimental to a patient's well-being. 67 Globally, it is estimated that at least 2.2 billion people suffer from some form of vision impairment and this number is projected to rise due to the increasing elderly population, who carry the highest risk for developing eye conditions.68,69 Topically applied ocular drugs are primarily indicated for anterior segment diseases such as blepharitis, corneal epithelial and stromal keratitis, glaucoma, and conjunctivitis, and to a lesser extent for posterior segment diseases such as bacterial endophthalmitis, retinitis, and macular degeneration. 70 One of the benefits of topical instillation is its ability to bypass the liver, avoiding the first-pass metabolization.
However, as discussed, the cornea is quite resistant to drug permeation, leading to the majority being absorbed into systemic circulation through vasculature of the conjunctiva, sclera, and nasopharyngeal mucosa. This is especially relevant when considering the main target population, the elderly, who have an increased susceptibility to adverse drug reactions due to factors such as decreased renal function and polypharmacy. 13 Hence, new topical formulations require rigorous testing to ensure therapeutic efficacy, while keeping local and systemic toxicity to a minimum.
Before a novel compound can begin first-in-human trials, preclinical evidence must be gathered and deemed satisfactory. The current impasse of drug development is that promising preclinical data are often not corroborated during human testing, meaning that the currently employed models have poor predictive power. 5 Cell culture is, by far, the most efficient way to acquire data due to its ease of use, scalability, and access to standardized protocols. However, traditional 2D cultures fail to recapitulate the complexity of tissues such as the cornea and, therefore, do not accurately predict human drug response. For example, many studies focus on the corneal epithelium given that it is considered the rate-limiting barrier for drug permeation. 39 If cultured as a monolayer, this model does not accurately represent in vivo epithelial permeability because the fully differentiated epithelium is a multilayered structure with cells that gradually transform from a basal columnar into an apical, flattened phenotype (Fig. 1C) and it is only the superficial epithelial cells that possess the tight junctions that limit drug permeability. 70
Consequently, preclinical evidence is required to contain data from experiments on animals (EU Directive 2001/83/EC; M2 5.2 b). 71 Non-human primates are phylogenetically closest to humans and would be ideal for investigating ocular diseases, but practical and ethical constraints make them inaccessible to the majority of researchers. Instead, rabbits are primarily used for in vivo ophthalmic research over other mammals as they are relatively easy to house and handle while sharing many anatomical features with humans. 72 Nevertheless, rabbit and human corneas are not completely alike. The rabbit corneal epithelium is strikingly similar with regard to thickness and the amount of layers, whereas the stroma is thinner and the endothelium shows increased proliferative capacity.73,74 Moreover, the lower blinking frequency and presence of nictitating membranes, which is not seen in humans, might contribute to disparities between rabbits and humans in drug trials.
Other commonly used animal models are porcine and bovine, which are rather used in ex vivo experiments as opposed to in vivo experiments to eliminate the need for large animal confinement areas. These exhibit increased epithelial layers and thickness when compared with human corneas, which are expected to correlate with a lower permeability that has to be accounted for during animal-to-human translation of results. 73 Taken together, conventional preclinical models for the human cornea are either too simplistic or exhibit substantial interspecies differences, thereby diminishing the probability of correctly predicting human drug response.
Conclusion
Advanced in vitro models hold the promise to bridge the gap between traditional cell culture and animal models. Each in vitro model discussed has unique properties that can be leveraged for different purposes (Fig. 3). Corneal spheroids are less suitable for pharmacological testing when compared with tumor spheroids, as this 3D model is unable to represent the complex-layered structure of the cornea. However, the relative simplicity of spheroids allows generation on a large scale and in a reproducible manner, which could provide a convenient source of corneal cells for grafting. 75
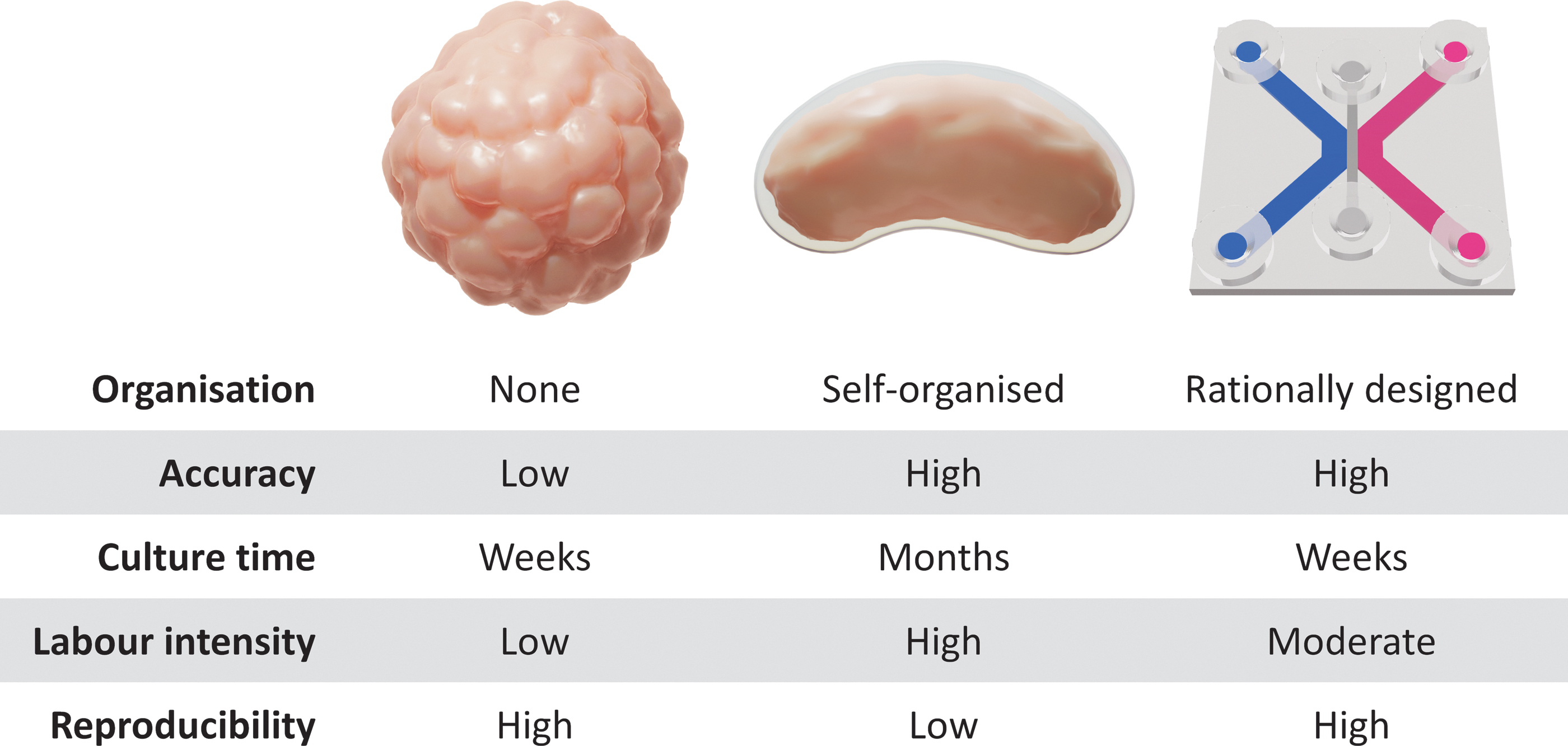
Comparison of advanced in vitro corneal models. Color images are available online.
Corneal organoids progress through the stages of corneal development and establish structures that include every layer of the cornea, including extracellular matrix components. Similar to in vivo eye development, corneal organoid generation takes several months to complete and involves complex differentiation steps. The resulting variability between individual corneal organoids along with their lengthy protocols might result in less reproducible data if employed for preclinical testing. Instead, corneal organoids are a valuable tool for studying corneal development and developmental disorders.
Another approach to modeling corneal tissue is to artificially combine each layer of the cornea in a cornea-on-chip. Such microphysiological systems are highly customizable and can be tailored to the design of the experiment. Although it could be argued that diffusion chambers such as Franz cells 76 and Ussing chambers 77 containing human corneas make these systems redundant, miniaturized chips are much more scalable and have the potential to eliminate interindividual variability by incorporating cell lines.
At present, the concept of tissue-engineered chips is gaining traction and has already proven its merits in other fields of research. Pulmonary edema, for example, has successfully been modeled in a lung-on-chip and has been used to validate the efficacy of a novel compound. 78 Corneas-on-chips, however, are still in their infancy and a long road lies ahead before they can be used for robust pharmacological testing.
Apart from preclinical testing, corneas-on-chips provide a highly controllable environment that can be used to develop better in silico models. As organs-on-chips further mature and mathematical models become more robust, we envision a synergistic effect that both reduces costs and accelerates drug development while downscaling the number of animals used in preclinical research.
Footnotes
Authors' Contributions
J.V.M.: The author contributed to the drafting of the article. S.N.D.: The author contributed to general supervision and editing. C.K.: The author contributed to general supervision and editing. B.V.d.B.: The author contributed to general supervision and editing.
Disclosure Statement
J.V.M.: The author declares that the research was conducted in the absence of any commercial or financial relationships that could result in a conflict of interest. S.N.D.: The author declares that the research was conducted in the absence of any commercial or financial relationships that could result in a conflict of interest. C.K.: The author declares that the research was conducted in the absence of any commercial or financial relationships that could result in a conflict of interest. B.V.d.B.: The author declares that the research was conducted in the absence of any commercial or financial relationships that could result in a conflict of interest.
Funding Information
This work is supported by an internal BOF-DOCPRO4 grant from the University of Antwerp.
*,†
*