
Abstract
Soon after bone fracture, the initiation of the coagulation cascade results in the formation of a blood clot, which acts as a natural material to facilitate cell migration and osteogenic differentiation at the fracture site. The existence of hematoma is important in early stage of bone healing, but the persistence of hematoma is considered harmful for bone regeneration. Fibrinolysis is recently regarded as a period of critical transition in angiogenic–osteogenic coupling, it thereby is vital for the complete healing of the bone. Moreover, the enhanced fibrinolysis is proposed to boost bone regeneration through promoting the formation of blood vessels, and fibrinolysis system as well as the products of fibrinolysis also play crucial roles in the bone healing process. Therefore, the purpose of this review is to elucidate the fibrinolysis-derived effects on osteogenesis and summarize the potential approaches—improving bone healing by regulating fibrinolysis, with the purpose to further understand the integral roles of fibrinolysis in bone regeneration and to provide theoretical knowledge for potential fibrinolysis-related osteogenesis strategies.
Impact statement
Fibrinolysis emerging as a new and viable therapeutic intervention to be contained within osteogenesis strategies, however to now, there have been no review articles which collates the information between fibrinolysis and osteogenesis. This review, therefore, focusses on the effects that fibrinolysis exerts on bone healing, with a purpose to provide theoretical reference to develop new strategies to modulate fibrinolysis to accelerate fibrinolysis thus enhancing bone healing.
Introduction
Large bone defects caused by trauma, tumor, or bone-related diseases result in significant social issues and remain as major treatment challenges in reconstructive surgery and orthopedic surgery. 1 Bone has an excellent intrinsic capability of self-healing within a small defect, whereas large segmental bone defects fail to undergo spontaneous healing.2–4 Delayed or incomplete healing of common fractures leads to serious compromise of overall health status, calling the need for advanced therapeutic approaches.
Bone injury is often accompanied by the damage of blood vessels, thus resulting in the formation of hematoma followed by a coagulation cascade situated between the broken fragments. The hematoma (fibrin network) formation is the first stage of bone regeneration, which has long been considered to stimulate the local inflammation and facilitate the recruitment of mesenchymal progenitor cells in the bone defect area. 5 The fibrin network will then be degraded by plasmin. The timely fibrinolysis is crucial for bone regeneration, as the persistent fibrin in the cartilage callus (during bone fracture repair) results in the failure of new blood vessel invasion into the tissue of the callus, thus causing delayed bone healing; conversely, the removal of fibrin in the fracture sites can lead to normal bone regeneration.6,7 Similarly, in a rat model, the inhibition of the dissolution of fibrin (fibrinolysis) by applying aprotinin and ɛ-amino-n-caproic acid (ɛ-ACA) is detrimental to normal wound healing. 8 Those findings indicated that fibrinolysis may be a potential regulatory target to facilitate bone regeneration.
Additionally, amounts of evidence has shown that the intrinsic conformation of the blood clots, which facilitates fibrinolysis, results in enhanced bone healing, compared with those blood clots resistant to fibrinolysis.9–14 Moreover, the components of fibrinolysis system, such as plasminogen (PLG) and plasmin, have crucial influence on bone metabolism and osteogenesis-associated cells.15,16 Furthermore, the products of fibrinolysis, for example, fragment D can affect the recruitment of inflammatory cell and the release of inflammatory factors in the early bone healing stage. 17 Therefore, in this review, we draw attention to the influence of fibrinolysis on osteogenesis, with purpose to support the optimization of fibrinolysis-targeting strategies to enhance bone healing.
Process of Fibrin Clot Formation and Fibrinolysis
The injury of vessel endothelia initiates extrinsic pathway by releasing tissue factor into the circulation. Meanwhile, thrombin is generated through a sequence of enzymatic cascade interactions. Finally, accompanied by thrombin and factor XIII, fibrinogen polymerizes to form a fibrin network. The fibrin network characteristics is determined by the distinctions in the fibrinogen molecules, which influences fibrin polymerization rate and its susceptibility to be dissolved. Fibrinogen molecules contain six paired polypeptide chains (AαBβγ)2, E domain, D domain, coiled-coil, and αC regions. 18 The central E region, the site where thrombin cleaves, includes the amino acid termini of polypeptide chains.
The D domain is formed by the carboxyl termini of the Bβ and γ chains and attaches to the E region by 2 α-helical coiled-coil domains. αC-regions are composed of the carboxyl termini of the Aα chains. In the presence of thrombin, fibrin fiber is formed by fibrinogen polymerization through the lateral and vertical aggregation, accompanied by the release of fibrinopeptide A (FpA) and fibrinopeptide B (FpB). 19 This leads to the formation of fibrin monomers and initiation of polymerization. Then, fibrin fiber further crosslinks γ chains or α chains to from neighboring fibrin molecules through factor XIII crosslinking catalyzed by thrombin, which results in increased stability of the fibrin network.20,21
Fibrinolysis is an opposite process to fibrin polymerization, which degrades fibrin. In the initial stage of fibrinolysis, PLG is converted to plasmin by both tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). Fibrin acts as major substrate in fibrinolysis, being both a binding surface for PLG as well as t-PA, and also a substance boosting plasmin generation. The affinity between t-PA and PLG is low without fibrin, but increases notably in fibrin existence. 22 Studies further found that the major fibrin-binding sites lie in the finger domain and Kringle 2 of t-PA, and one or more of the five Kringle domains lie in PLG. 22 Those binding sites contain lysine-binding sites, thus can be blocked by lysine analogs, such as ɛ-ACA and thrombin-activatable fibrinolysis inhibitor (TAFI). 23 Conversely, components of the fibrinolytic inhibition system, such as PLG activator inhibitor (PAI) and inhibitors of plasmin, namely α2-antiplasmin (α2-AP), inhibit the fibrinolysis process. 24 PAI has two subtypes, PAI-1 inhibits fibrinolysis by suppressing both t-PA and u-PA, PAI-2 exhibits equivalent inhibitory efficiency on both two-chain t-PA and two-chain u-PA, but not single-chain t-PA and pro-urokinase (Fig. 1). 25
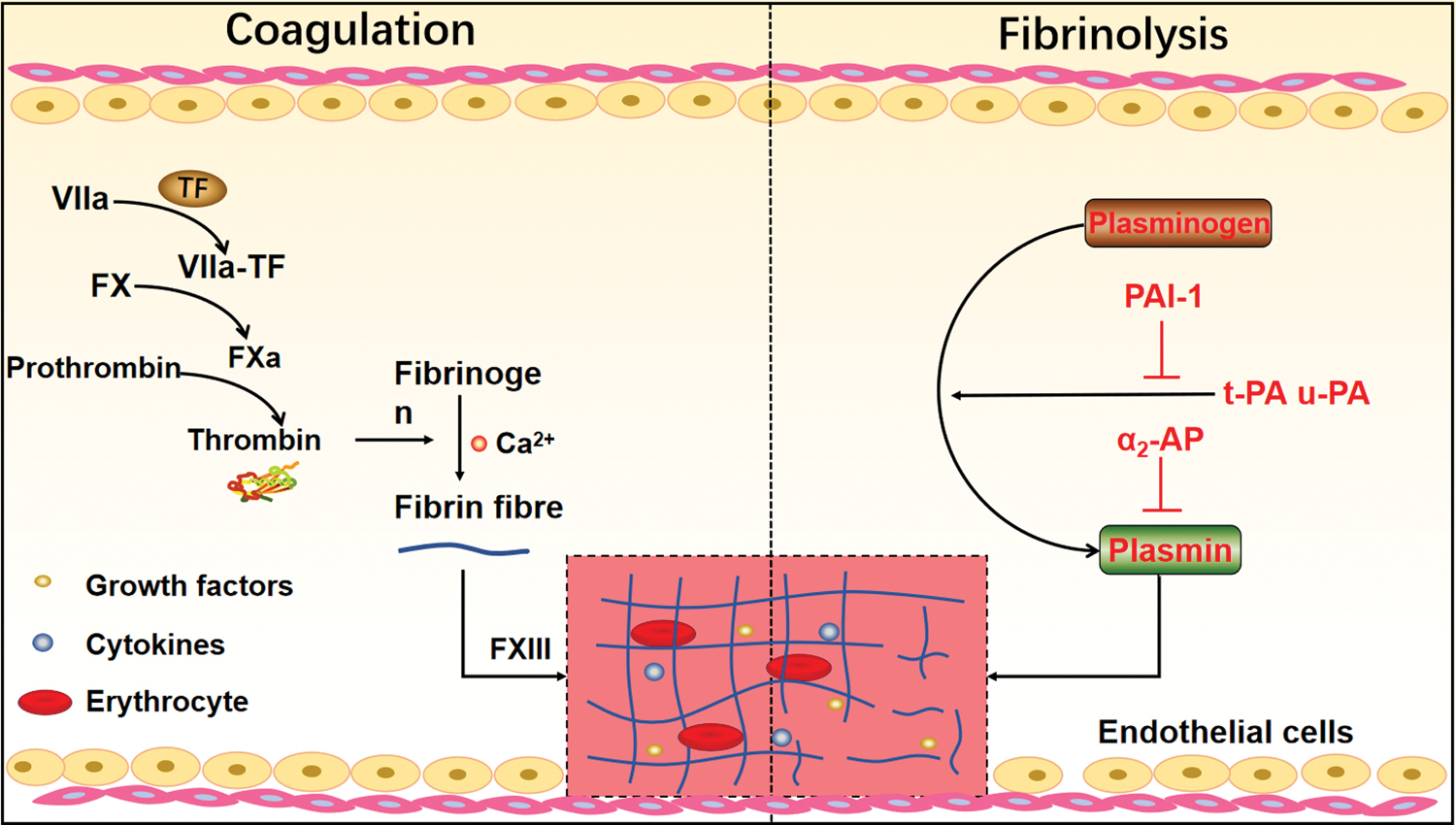
The formation of fibrin network and fibrinolysis. The extrinsic coagulation pathway is triggered by the release of TF, when endothelial vessels are damaged. Then, TF binds to FVII and converts FVII into FVIIa to form the extrinsic tenase complex (TF-FVIIa), which activates FX into FXa. In the presence of Ca2+, FXa further combines with the activated factor Va to from the prothrombinase complex (FXa-FVa), resulting in the conversion of prothrombin to thrombin. Eventually, fibrin network is formed through a sequence of enzymatic reactions. On the other hand, plasminogen is converted to plasmin mediated by t-PA and u-PA on the surface of fibrin to degrade fibrin during fibrinolysis, and the inhibitor of fibrinolysis, including PAI-1 (inhibit t-PA and u-PA) and α2-AP. AP, antiplasmin; Ca2+, calcium ion; FVII, factor VII; PAI-1, plasminogen activator inhibitor; TF, tissue factor; t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator. Color images are available online.
The Role of Fibrinolysis in Osteogenesis
Immediately after fracture, blood coagulation cascade is activated, developing a hematoma that resulted from fibrin polymerization. The physiological processes of bone healing include three consecutive phages: reactive phase, reparative phase, and remodeling phase. 26 During reactive phase, the formation of hematoma is recognized as a crucial part for early bone healing, as it constitutes the early healing microenvironment at the bone injury site. 27 Hematoma (fibrin clot) then results from the coagulation cascade to seal blood vessels thus preventing further blood loss, which also stimulates the local inflammatory response and affects the recruitment of mesenchymal progenitor cells in the early bone regeneration process. 9
Importantly, the intrinsic conformation of fibrin network at fracture sites is crucial for the subsequent fibrinolysis, thus bone healing. There are two different phenotypes of fibrin network defined by researchers, one is described as loose and porous fibrin structure, which is made of thicker fibers, the other is dense and impermeable fibrin structure formed by thinner fibers.10,28 Studies have shown that, compared with thicker fibers, thinner fibers reduce the generation of plasmin mediated by t-PA, thereby hampering the whole fibrinolytic process. 29 The degradation of fibrin clots is a complicated process, where not all fibrin fibers are cleaved by plasmin. There is a sequence of pathways where individual fibers are cleared, including transverse cleavage, bucking, fiber bunding, and fiber collapse. 30 Thicker fiber undergoes an alteration of shape before it disappears, such as an increase in diameter and bending, whereas thinner fibers stay in the same shape. 31 On the whole, thicker fibers have a slower dissolution rate compared with thinner fiber, although there seems to exist more t-PA-binding sites on thicker fibers.
At the network level, the fibrinolysis process was proceeded by transverse cutting (layer by layer) rather than by progressive cleavage uniformly around the fiber, and the lysis front (a dynamic line observed when fibrins are being degraded) of tissue PLG distribution is broader as a result of binding more plasmin in loose fibrin network structure and moves quickly compared with dense fibrin network structure, leading to loose fibrin network susceptible to fibrinolysis.30,31
Consequently, the porous fibrin structure is “optimal” as it is susceptible to fibrinolysis by plasmin, hence sustaining the constant supplying of growth factors from surrounding tissues and supporting cellular infiltration, whereas dense fibrin structure is “suboptimal” since it is relatively resistant to fibrinolysis thus hindering tissue regeneration.31–33 This may be one of the explanations that small bone defect (diameter of 1 mm) is capable of self-healing without any intervention, but large bone defect (diameter of 5 mm) is unable to completely self-heal in the native state (Figs. 2 and 3).34,35
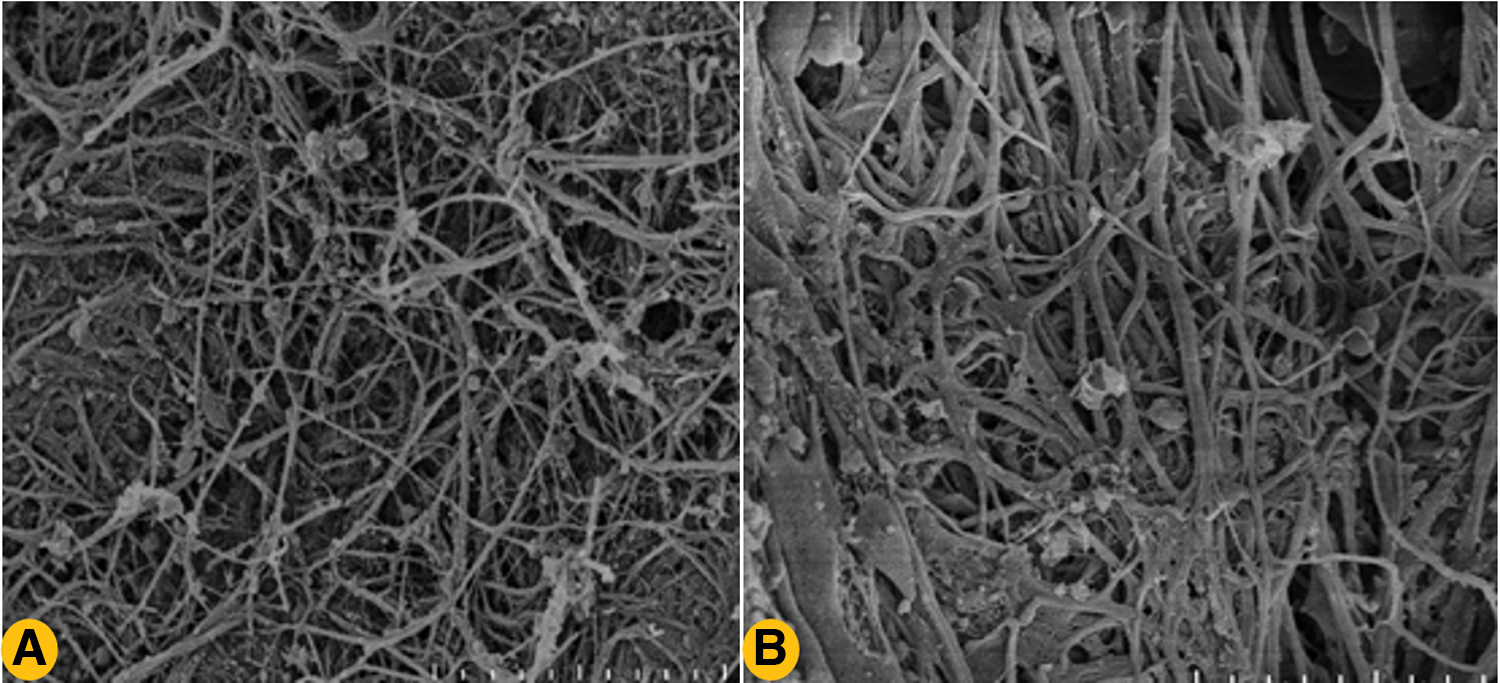
Different phenotypes of fibrin network. Clots composed of thinner fibrin fibers result in hematoma with a dense fibrin network, which has poor permeability and resistant to fibrinolysis, thus delaying bone healing

The dissolution of fibrin clot is layer by layer but not progressive cutting. When fibrinolysis starts, lysis front forms and move laterally. Fibrin clots consist of thick fibers, which present loose conformation that have a broader (bind more plasmin) and quickly moving binding front, resulting in fibrin clots susceptible to fibrinolysis, thus releasing more growth factors to facilitate bone healing. On the contrary, clots composed of thin fibers, which presents dense structure have a narrow (bind less plasmin) and slowly moving lysis front, resulting in slowly dissolved clots thus releasing less growth factors and damaging normal bone healing. Color images are available online.
Besides, the sustained fibrin can damage the normal bone healing process by inhibiting normal cell migration, and a failure of efficient fibrinolysis was found to hamper normal bone regeneration.6,36 Moreover, normal bone healing occurred after the removal of fibrin in the fracture site, which suggests that the timely fibrinolysis is vital for bone healing. 7 Under the circumstance of PLG deficiency, fibrin remains in the cartilage callus, and no vascularity exists in cartilage callus, thus primary bone healing is ruined; whereas the removal of fibrin clot in the PLG-deficient bone defect area allows for vascular invasion and the completion of primary bone regeneration (Fig. 4). 7 This indicates that delayed fibrinolysis is harmful for normal bone healing.
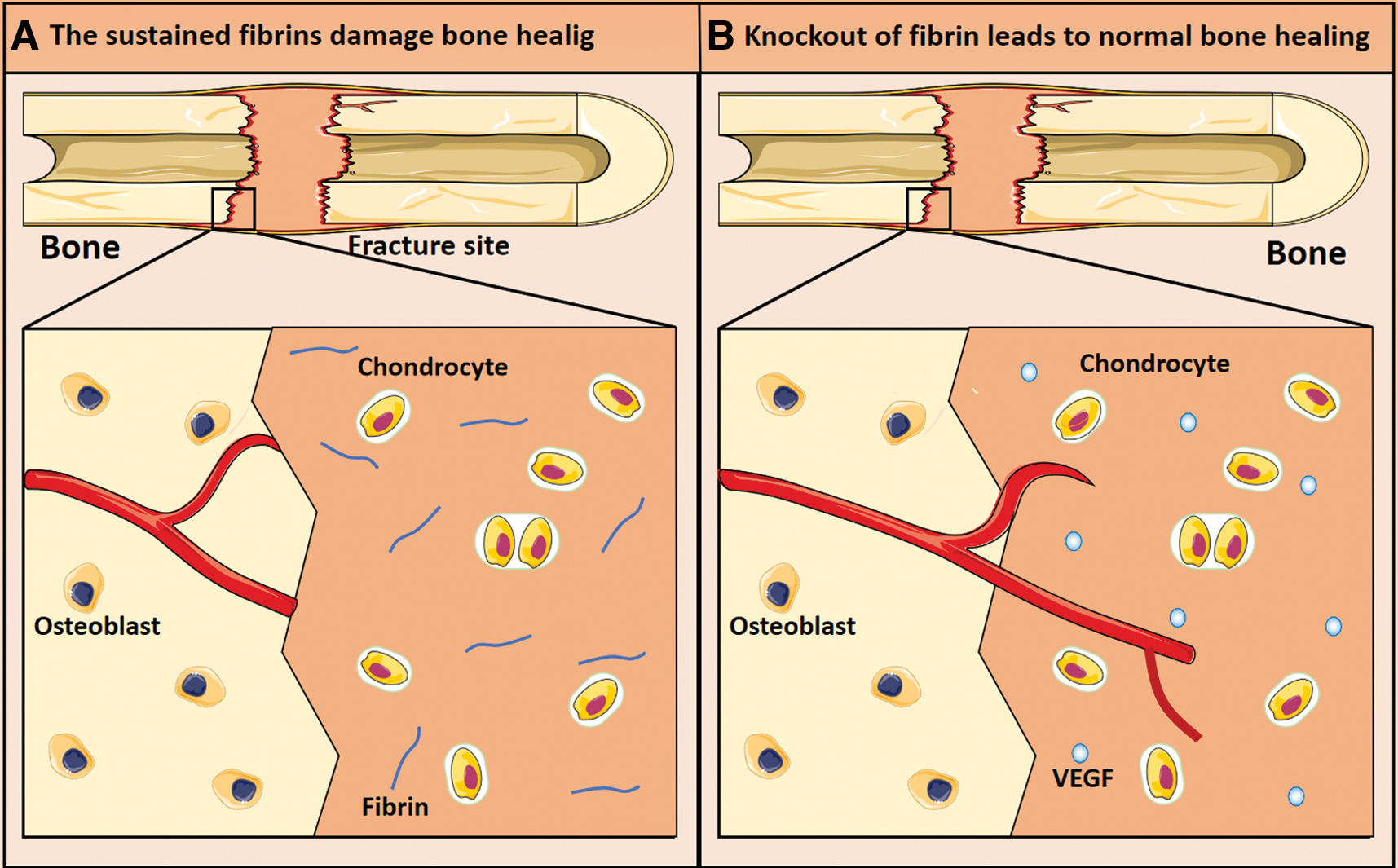
The sustained fibrins delay bone healing. In the event of plasminogen deficiency (fibrin persists in the fracture site) in mice, fibrin remains in the cartilage callus, leading to blood vessels failing to invade into the cartilage callus thus impairing bone healing
Wong et al. found that the use of low-magnitude high-frequency vibration (LMHFV) to accelerate fibrinolysis can further enhance the vascular formation, thus promoting fracture healing in animal metaphyseal fracture model. 37 Moreover, the application of highly effective fibrinolytic inhibitors, such as aprotinin and ɛ-ACA, resulted in excessively concentrated fibrin, thus severely injuring the regeneration of skin, liver, and bone. 8 Therefore, fibrinolysis is vital for osteogenesis, and factors facilitating fibrinolysis are considered to enhance bone regeneration.
Fibrinolysis System Components Modulate Osteogenesis
PLG and plasmin
PLG is converted to the active serine protease plasmin by PAs at the early stage of fibrinolysis, which participates in the removal of fibrin. The deficiency of PLG led to decreased bone mineral density and osteoprotegerin expression in osteoblasts, as well as an increased osteoclastogenesis in vitro. 15 In a mouse femur defect model, the deficiency of Plg gene declined cartilage and bone formation, damaged bone formation, reduced osteoblast cell numbers, and the transforming growth factor-β (TGF-β1) levels on day 4, by suppressing the recruitment of macrophages (a cell plays critical roles in repair of many tissues, such as bone and lung) in the fracture site. 16 Intriguingly, the local treatment of vascular endothelial growth factor (VEGF) at the bone defect significantly reversed delayed bone regeneration in Plg-deficiency mice, which further suggested that PLG affects angiogenesis by decreasing the production of VEGF. 16
PLG also enhances mesenchymal stem cell (MSC) survival and persistence, which induces paracrine effects, boosts postischemic neovascularization and tissue repair by activating cysteine-rich protein 61. 38 Plasmin regulated bone remodeling by cleaving both free and hydroxyapatite-bound osteocalcin (through binding one of the three plasmin-susceptible sites (lys19-arg20, arg43-arg44, arg44-phe45) within the osteocalcin molecule), which is secreted by osteoblast and negatively affects mineralization by directly mediating bone resorption. 39 Those findings indicate that PLG not only affects bone metabolism, but also regulates the proliferation and differentiation of osteogenesis-associated cells.
Tissue-type plasminogen activator
The two PAs, t-PA and u-PA, exert their function during fibrinolysis by converting the inactive proenzyme PLG to plasmin. 40 Normal osteoblasts and osteoclasts are capable of producing both t-PA and u-PA, which play important roles in bone regeneration. 41 Both t-PA and u-PA are involved in the dissolving of noncollagen proteins from the organic matrix of bone, probably leading to the release of TGF-β1, insulin-like growth factors, and other growth factors from bone matrix.42,43 Furthermore, interleukin (IL)-1α revealed to facilitate the releasing of t-PA and as u-PA from osteoclasts, thus stimulating bone matrix turnover. 44
In a femoral bone defect model, the area of bone defect in t-PA-deficiency mice was three-fold larger compared with the controls (t-PA+/+ mice), and t-PA was found to promote osteoblast proliferation through annexin 2 and extracellular signal-regulated kinase1/2 (ERK1/2), which also facilitated angiogenesis by stimulating the production of VEGF and hypoxia-inducible factor-1α (HIF-1α) at the fracture site. 45
In addition, t-PA mobilizes CD11b+ cells from the bone marrow and upregulates the expression of neoangiogenesis-associated genes, VEGF-A, thereby locally promoting angiogenesis during tissue regeneration. 46 In a study, myeloid-derived t-PA was shown to enhance macrophage migration through focal adhesion kinase, ras-related C3 botulinum toxin substrate 1 (Rac-1), and nuclear factor kappa-B (NF-κB) signaling pathway. 47 Moreover, t-PA is conducive to the recruitment of macrophage and (macrophage inflammatory protein-1α) at the bone fracture site, thus resulting in an enhanced bone repair. 48 In a study, the topical thrombolysis with recombinant tissue plasminogen activator (rt-PA) was beneficial in preventing epidural fibrosis and arachnoiditis after spine surgery in a rat model. 49
Urokinase-type plasminogen activator
The deficiency of u-PA in mice exhibited decreased accumulation of macrophage, hence severely impaired tissue regeneration.50,51 It has been reported that u-PA boosted angiogenesis and remodeling of the fracture cartilage at the fracture site in vivo. 52 Researchers found that u-PA exerted a protective role in arthritis models with “wound healing-like” processes following local injury, probably through u-PA/plasmin-mediated fibrinolysis. 53 Delayed early bone healing accompanied with declined macrophage accumulation and phagocytic activity were detected in an u-PA gene-deficient mice model, while the deficiency of t-PA gene injured the late-stage bone regeneration.45,54
Urokinase-type plasminogen activator receptor (u-PAR) are expressed by osteoblasts and osteoclasts, and its association with β1-integrin, thus promoting the migration of human mesenchymal stem, which occurs in MSC that originated from bone marrow or adipose tissue. 55 Moreover, u-PAR has been revealed to promote osteoclast differentiation through increasing macrophage colony-stimulating factor secretion from osteoblasts, as well as initiating the phosphatidylinositol 3-kinase/Akt/NF-κB (PI3K/Akt/NF-κB) pathway. 56 In a uPAR-deficiency mice model, osteoblast was found to have increased proliferative capacity, alkaline phosphatase (ALP) activity, bone matrix protein expression level, and a promoted mineralization, indicating that u-PAR may inhibit osteoblast functions. 57 Furthermore, u-PAR has also been suggested to mediate osteogenic differentiation of MSCs through the complement C5a receptor. 58
Angiogenesis is vital for bone tissue engineering as the regeneration of bone requires vascularity to provide essential nutrition and eliminate waste products, along with establishing biocommunications. 59 The migration of endothelial cells is regarded as an essential step in angiogenesis process, which demands the proteolysis of the underlying basement membrane matrix.60,61 It is, therefore, of considerable interest for cells to produce proteolytic enzymes allowing cleavage of matrix proteins for favoring cell migration. Substantial evidence has shown that t-PA, u-PA, and matrix metalloproteinases (MMPs) are involved in the degradation of matrix proteins.62,63 The t-PA modulates fibrin homeostasis while the u-PA primarily affects cell migration and tissue remodeling. 64 Moreover, u-PA can modulate the construction of self-assembled network, which contributes to the process of prevascularization. 62
MMPs are known to perform specific functions during bone repair, and MMPs at low or high levels can result in abnormal bone remodeling. 65 The deficiency of u-PA was shown to additionally exacerbate the influence of MMP9 deficiency on bone regeneration, and compensatory upregulation of u-PA bioactivity has been observed in MMP9-deficient mice, which indicates that u-PA regulates bone regeneration by controlling MMP9 production. 66 Intriguingly, the absence of both u-PAR and MMP9 did not delay the wound repairing process, suggesting that u-PA and u-PAR function independently during wound healing in the case of MMP9 deficiency (Fig. 5). 66
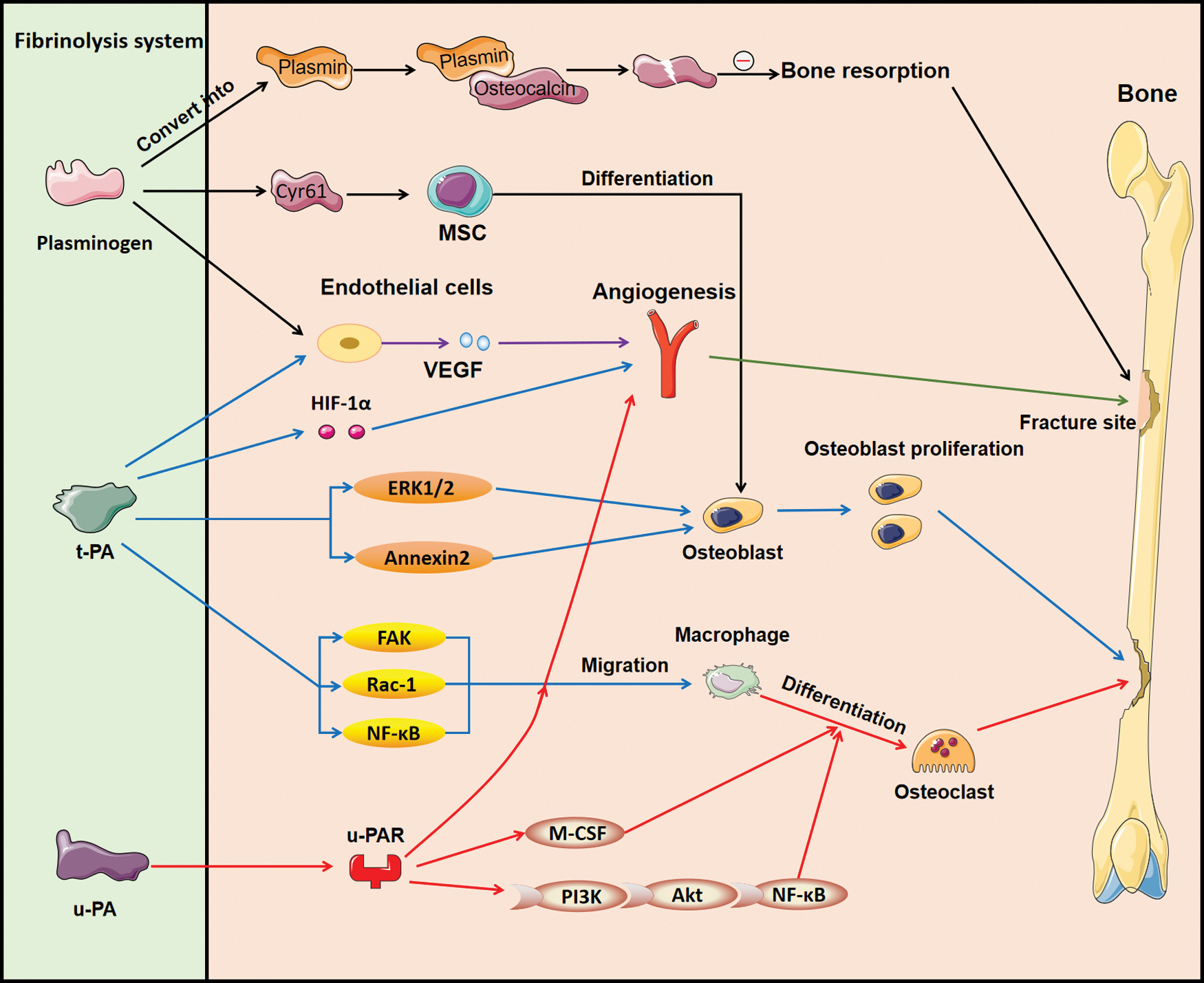
The regulatory role of fibrinolysis system in osteogenesis. Plasminogen can be converted into plasmin, which binds with osteocalcin to decompose osteocalcin into two parts to inhibit bone resorption. Moreover, plasminogen contributes to MSC differentiation through Cyr61 and promotes the release of VEGF, thus enhancing the formation of blood vessels at the fracture site. On the other hand, t-PA boosts angiogenesis by VEGF and HIF-1α, facilitates osteoblast through ERK1/2 and annexin 2 signaling pathway, and participates in the migration of macrophage through FAK, Rac-1, and NF-κB signaling pathway. u-PA exerts its role predominantly through its receptor u-PAR, which influences angiogenesis and macrophage migration as well as the formation of osteoclast through M-CSF and PI3K/Akt/NF-κB signaling pathways. Cyr61, cysteine-rich protein 61; ERK1/2, extracellular signal-regulated kinase1/2; FAK, focal adhesion kinase; HIF-1α, hypoxia-inducible factor-1α; NF-κB, nuclear factor kappa-B; MSC, mesenchymal stem cell; M-CSF, macrophage colony-stimulating factor; u-PAR, urokinase-type plasminogen activator receptor; VEGF, vascular endothelial growth factor. Color images are available online.
The Crosstalk Between Fibrinolytic Inhibitory System and Osteogenesis
The fibrinolytic inhibitory system consists of PAI (for example, PAI-1) and inhibitor of plasmin (such as α2-AP), which is essential for bone repair. PAI-1, an inhibitor for t-PA and u-PA, exerts an array of biological functions, which is involved in the regulation of extracellular matrix degradation, cell migration, apoptosis, and cell proliferation through fibrinolysis-dependent or independent pathways. 67 PAI-1 has been shown to be essential for bone metabolism. It induces bone loss and delays bone healing as a result of hampering osteoblast differentiation in a streptozotocin (STZ)-induced diabetes model in mice. 68 In another research, PAI-1 deficiency not only defended STZ-induced bone loss, but also reduced the fluctuation of Runx2, osterix, and ALP in tibia as well as serum osteocalcin levels suppressed by the diabetic state in female mice. 69
Besides, the addition of small-molecule PAI-1 inhibitor (iPAI-1) into an estrogen deficiency-induced osteoporosis model significantly increased the trabecular bone volume compared with the control group (without adding iPAI-1). 70 Moreover, the absence of PAI-1 caused a larger fracture callus at 7 and 14 days through boosting extracellular matrix remodeling in a fractured femur model of mice. 71 One of the possible mechanisms was that PAI-1 can cause bone loss through decreasing accumulation and phagocytosis of macrophages at the fracture site during early bone healing. 72
In addition, PAI-1 affects the osteogenesis-associated cells (such as MSCs, osteoblasts, osteoclasts, etc.) directly or indirectly. In a diabetic mouse femoral bone defect model, the deficiency of PAI-1 induced a significantly increasing number of osteoblasts and facilitated bone formation. 73 Besides, the impaired osteoblast differentiation caused by the upregulation of PAI-1 is involved in the pathogenesis of congenital kyphoscoliosis. 74 Chondrogenic differentiation of naive induced pluripotent stem cells (iPSCs) was suppressed by adding a PAI-1 inhibitor in the process of MSC induction. 75 PAI-1 exerts vital role in the differentiation of MSCs into osteoblasts, as the deficiency of PAI-1 can significantly blunt the expression of osteogenic genes in MSCs, and the same results can be obtained by a reduction in endogenous PAI-1 levels by siRNA in vivo. 76 Furthermore, the mutation of activin type I receptor/ALK2 in fibrodysplasia ossificans progressiva patient-derived iPSCs induced the expression of PAI-1, resulting in the phosphorylation of Smad1 and Smad5, and thus activating bone morphogenetic proteins (BMP) signaling without ligand stimulation.75,77
α2-AP, another physiological blocker of fibrinolysis system, was shown to contribute to wound healing. The α2-AP-deficiency mice displayed an accelerated wound repairing compared with the wild-type mice. 78 Intriguingly, significantly upregulated VEGF mRNA level was observed in wound lesions compared with the wild-type mice. 78 Those findings suggest that α2-AP promotes the expression of VEGF, thus resulting in an accelerated wound closure. In addition, α2-AP stimulates the generation of type I collagen (which is crucial for bone mineralization by not only providing structural sites for osteocalcin, but also combining with noncollagenous proteins, such as osteocalcin to form a network scaffold) and is involved in the synthesis of TGF-β1 (which facilitates bone regeneration through coupling osteogenesis with angiogenesis).79,80 Furthermore, α2-AP negatively affects osteoblast differentiation by suppressing the Wnt/β-catenin pathway. 81
Fibrin(ogen) regulates migration and adhesion of leukocyte
Blood clot acts as a provisional matrix for leukocyte adhesion and migration during bone healing process. 82 Leukocyte migration is a process where leukocytes are recruited from the circulation to the injured tissue, which is an integral part of the inflammatory response. 83 Countless evidence revealed that fibrin(ogen) exerts essential roles in the migration and adhesion of leukocytes.84,85 There are two pathways involved in leukocyte migration induced by fibrin(ogen). One pathway is that fibrinogen bridges leukocyte to the endothelium through binding to leukocyte integrin receptor Mac-1 and endothelial cell receptor intercellular adhesion molecule, and the site of Mac-1was related to the fibrin gamma chain residues 390–396.85–87 It has been shown that the existence of fibrinogen increases two- to five-fold of the adhesion of leukocyte to endothelium. 86
The other pathway is through the very-low-density lipoprotein receptor (VLDLR), a binding site that exists in vascular endothelium.86,88,89 However, the existing phenomenon is that the specific inhibitors of fibrin–VLDLR interaction could not completely obstruct leukocyte transmigration induced by fibrin, indicating that other pathways participate in this process, whereas the effects of them are much lower. 90
Fibrin(ogen) regulates the releasing of cytokines and chemokines
Fibrinogen and fibrin are recognized as proinflammatory factors. A significant body of basic science literature found that fibrinogen and fibrin contribute to inflammation not only by indirectly inducing leukocyte migration, but also by directly modulating the expression of cytokines on monocytes and chemokines on leukocytes.91–93 For example, fibrin was demonstrated to promote the expression of IL-1β by human peripheral blood mononuclear cells mediated partly by the integrin receptor CD11b/CD18. 94 The coculture of fibrin(ogen) with human synovial fibroblasts led to the upregulation of IL-1 (IL-8). 95
Moreover, culturing macrophages on fibrin gels resulted in an increasing secretion of IL-10, and exposure of macrophages to soluble fibrinogen led to the upregulation of tumor necrosis factor alpha (TNF-α). 96 Fibrin(ogen) can directly stimulate the release of IL-6 and monocyte chemoattractant protein-1 (MCP-1) in vivo. 93 Fibrin prevents the activation of macrophage inflammation stimulated by fibrinogen, lipopolysaccharide, and interferon-γ. 96 Research recommended that Mac-1 on the monocytes may act as a fibrin(ogen) receptor in the release of cytokines chemokines. 97
Another study has shown that fibrin(ogen) stimulates macrophage chemokine secretion through toll-like receptor 4, 98 but the conclusion is controversial. 99 Importantly, in the case of fibrin(ogen) knockout, macrophage adhesion capacity was suppressed and the production of IL-6 was reduced. 93 On the other hand, fibrin(ogen) can upregulate the secretion of metalloproteinase-9, which promotes monocyte coagulation through an autocrine mechanism. 100 Fibrin(ogen) may act as a crucial switch in regulating macrophage polarization, which has the potential to supply valuable immunomodulatory strategy for tissue regeneration.
Other Fibrinolysis Regulators Promote Osteogenesis
There are many factors that contribute to the architecture and properties of fibrin network. Shiu et al. used carboxyl and alkyl functional groups to modulate the surface chemistry of biomaterial, thereby altering the properties of blood clot formed on biomaterial surface and making the clot susceptible to fibrinolysis thus enhancing bone healing in vitro. 13 Evidence showed that the implantation of β-tricalcium phosphate (bone substitute biomaterial) into rat mandibular bone defect model resulted in dense fibrin network formed around the implanted substitute, leading to delayed bone healing; on the other hand, the use of s-nitrosoglutathione (GSNO) to modify fibrin structure contributed to bone regeneration in vivo. 9 There are other factors that regulate fibrin structure, such as ionic strengths, thrombin concentration, and dextran.101,102 The increasing of plasma ionic strengths leads to a shift from thicker to thinner fiber, especially at an ionic strength of 0.35, and hematoma cannot be degraded for hours in vitro. 103 Thrombin concentration has been shown to influence fibrin thickness and density.
Low thrombin concentrations (<1 nM) lead to fibrin meshwork consisting of thicker fibrin fibers and can be easily degraded, whereas high thrombin concentrations result in fibrin meshwork composed of thinner fibrin fibers resistant to fibrinolysis. 104 Blood clots are more turbid and consists of thicker fibers in the presence of dextran, thus facilitating its fibrinolysis. 103 Additionally, calcium ion (Ca2+) boosts fibrin monomer polymerization, thus increasing fiber thickness. 105 The fibrinogen molecule contains 3 high-affinity Ca2+-binding sites and 10 low-affinity Ca2+-binding sites.106,107 Evidence suggested that the binding of Ca2+ to the high-affinity calcium-binding site of γ-chain was a guarantee of pocket “a” working normally during fibrin polymerization, whereas the mutations of high-affinity calcium-binding site severely damaged the function of pocket “a” thus causing fibrinogen failing to polymerize to form fibrin fiber.108,109 Whereas chlorine antagonizes lateral aggregation, leading to form thinner fiber; zinc (Zn2+) increases fiber diameter and alters overall clot structure.110,111
Fibrinolysis-related physical factors
The degradation of fibrin clots involves in the clearance of individual fibrin fiber. There are several pathways by which fibrin clots are dissolved, such as transverse cleavage of fibers, fiber bundling, buckling, and collapsing, and those pathways are regulated by the concentration of plasmin.30,112 Transverse cleavage happens when plasmin cuts fiber at a single site. Buckling is a process where fibers are bent, ascribable to tension change in the fibrin network, as a result of which surrounding fibers are broken. The bunding process is where two or more uncutting fibers move together to form fresh and thicker fiber. Fiber collapsing is characterized by the disappearance of uncutting fibers (folded to the edge) on account of elastic recoil force caused by transverse cleavage of surrounding fibers.
The polymerization of fibrin is under tension, leading to tension distributed on fibrin network in the native state. Studies have shown that if there was no tension on the fibrin network, only ∼60% of the fibrin can be degraded eventually. However, the existing tension (in natural state) results in ∼95% clearance of fibrin finally. 30 Under normal circumstance, the tension of the fiber exceeds its equilibrium length by ∼23% during polymerization. 30 Those findings indicate that fibrin tension probably acts as one of the targets to modulate fibrinolysis (Fig. 6).
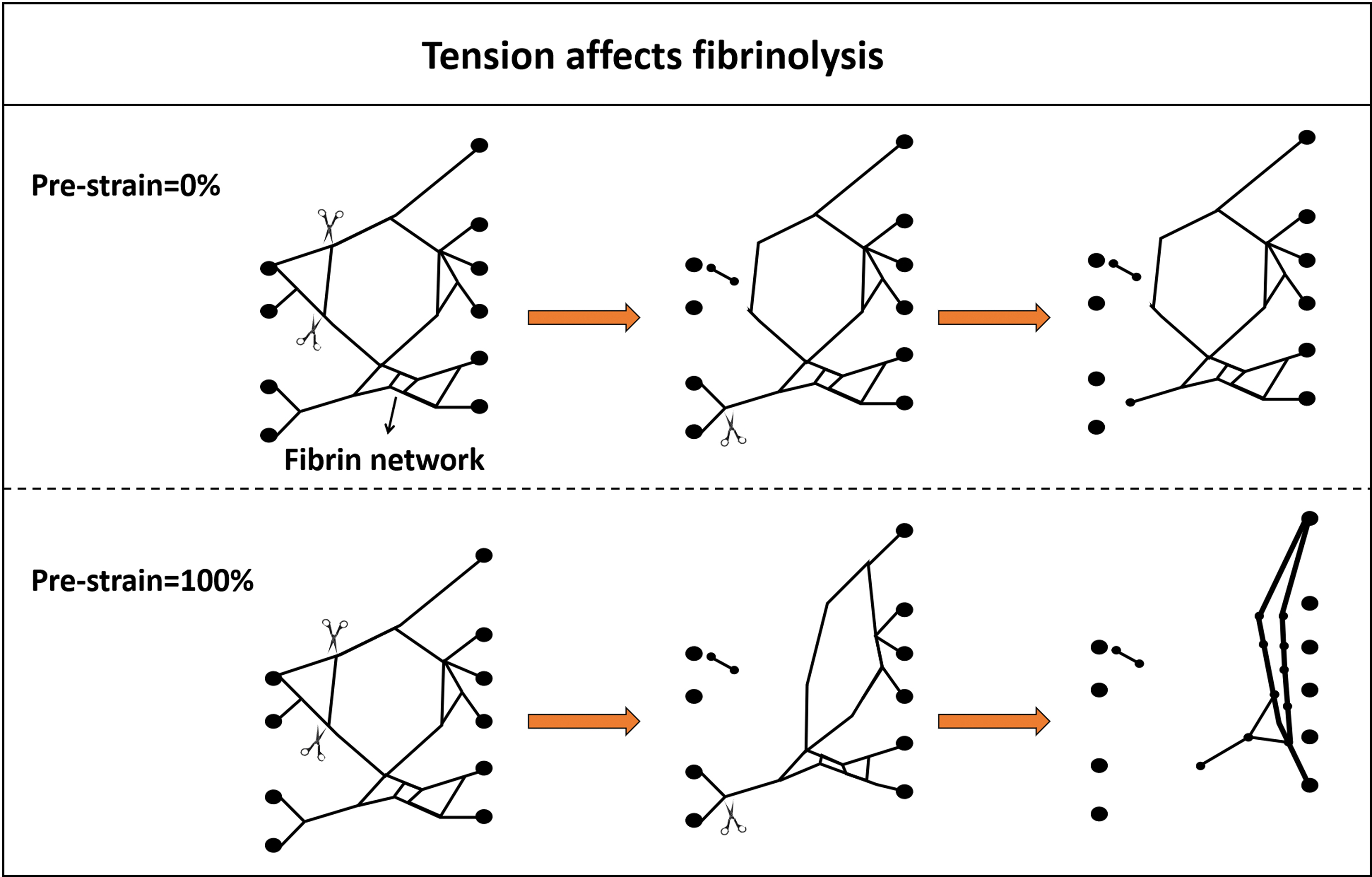
Tension has a profound influence on the dissolution of fibrin network. In the case of pre-strain = 0% (no tension) distributed on the fibrin network, the uncutting fiber would not alter its shape when transverse cleavage occurred in the surrounding fibers; but when pre-strain = 100% (a state of tension), fibrin recoil force as a result of the cutting of surrounding fibers leads to uncutting fibers folded to the edge to form a new and thick fiber. Color images are available online.
After being implanted into fracture, biomaterials come into contact with blood to form clots on the surface of the implanted materials. Therefore, the properties of the material surface play crucial roles in the formation of blood clots. The hydrophobic surface of polystyrene material was observed to reduce the volume of blood clots, and the inner conformation of the formed clots are looser compared with clots formed on the hydrophilic polystyrene surfaces, and fibrin clots formed on the hydrophobic surface of polystyrene material are easier to dissolve compared with those developed on the hydrophilic polystyrene surfaces. 113
Fibrinolysis-related chemical factors
The regulation of fibrin clot characteristics by modulating fibrinogen molecule facilitates the controllable fibrinolysis, and the approaches, including oxidation, glycation, acetylation, and carbamylation.
Oxidation
The oxidation of specific methionine residues on the α, β, and γ chains of fibrinogen molecule by hypochlorous acid (probably hinder lateral aggregation of protofibrils in fibrin gels) leads to the formation of denser fibrin network of thinner fibers, inducing delayed fibrinolysis. 114 Similar results have been found in other studies, which further elucidated that the hypochlorite oxidizes the D region rather than E region of fibrinogen thus affecting the protofibril aggregation leading to denser fibrin network.115,116 However, there are contrary findings that hypochlorous acid was observed to reduce fibrin clot formation and attenuate clot retraction and fibrinolysis under normal circumstances. 117
Moreover, the fibrin susceptibility to plasmin degradation was reduced after oxidation by concentrations of peroxynitrite higher than 100 μmol/L, whereas a low extent of nitration and oxidation showed no effect on these properties. 118 Except for the above oxidation caused by reactive oxygen species, the application of ozone, or illumination to modulate fibrinogen achieved consistent results, resulting in a decreased stiffness, lower permeability, and higher density of fibrin clots.115,119–121 Overall, oxidation contributes to denser fibrin meshwork with thinner fibers, which are impermeable, leading to a fibrin network resistant to be dissolved by plasmin, as compared with nonoxidized fibrins.
Acetylation
The acetylation of fibrinogen by applying aspirin showed a significantly decreased rate of polymerization as well as maximum turbidity compared with nonacetylated fibrinogen.122–124 The diameter of individual fibrin fiber modulated by acetylation was increased, and the formed fibrin network shower higher permeability and lower density, as well as increased susceptibility to fibrinolysis.122–125 Studies further found that mounts of lysine residues existing in the fibrinogen peptides were involved in the crosslinking of fibrinogen, and those lysine residues could be easily acetylated during fibrin polymerization, which is probably the mechanism by which aspirin enhances fibrin fibrinolysis. 126
Homocysteinylation
The homocysteinylation of fibrinogen affects fibrin clot structure. Animal study showed that rabbits treated with chronic injections of homocysteine or dietary manipulation (to make hyperhomocysteinemia) were observed to develop fibrin clots that consisted of thinner, more tightly packed fibers compared with the control group. 127 The adding of homocysteine in the reduced form (0.01–1 mM) into whole human plasma delayed the fibrinolysis process, and the adding of homocysteine thiolactone presented similar results. 128 In general, the homocysteinylation of fibrinogen not only reduces polymerization rate, but also leads to denser fibrin clots composed of thinner fibers.127,129,130
Glycation
Studies have shown that glucose molecules are able to form strong H-bonds with fibrinogen, and further affect the fibrin structure and morphology. 131 The glycation of fibrinogen leads to altered fibrin clot structure with smaller pore size and delayed degradation rate during fibrinolysis, and deglycosylation of fibrinogen promotes fibrin clot permeability and degradation rates.132,133 In addition, nonenzymatic glycosylation of fibrinogen impairs the clot susceptibility to fibrinolysis by plasmin. 134 The accumulated evidence suggests that the glycosylated fibrinogen results in delayed fibrinolysis process.131–136
Carbamylation
Additionally, although it has been studied only marginally, the carbamylation of fibrinogen was found to alter fibrin polymerization kinetics and impair crosslinking, forming fibrin network with thinner fibers, higher density, and delayed fibrinolysis compared with the control group. 137
Fibrinolysis-related drugs
Tetranectin is a protein consisting of four identical, noncovalently bound and 181-amino-acid polypeptide chains, which participates in fibrinolysis and proteolysis by binding to PLG and fibrin. 138 Although its specific mechanism still remains unclear, it is considered to play important roles in mineralization during osteogenesis in vivo and in vitro. 139 In addition, pentamidine inhibits fibrinolysis by interacting with plasmin in a dose-dependent manner. 140
Platelet regulates fibrinolysis
Platelets play a central role in hemostasis by affecting fibrin polymerization to safeguard vascular integrity as well as accelerating wound healing. Platelets are previously known as an antifibrinolytic regulator by inducing clot retraction of the formed fresh fibrin meshwork and unleashing high concentrations of PAI-1 from their α-granules.141,142 Clot retraction is a process where clot volume is compressed and excess fluid is extruded mediated by platelets, and platelets tighten the fibrin meshwork surrounding the injured area through expressing integrin αIIbβ3 by α-granules on platelets. 143 The activated platelets initiate conformational change of αIIbβ3, leading to the activation of αIIbβ3-mediated outside-in signaling. 144 The retraction force that draws the edges of the wound together is generated from the fibrin–αIIbβ3–myosin complex, which is considered to act as an axis to support clot retraction. 145
Retracted clots are more resistant to fibrinolytic degradation compared with nonretracted clots, due to condensing of crosslinked α2-AP and decreased binding of t-PA to fibrin.146,147 Furthermore, the fibrinolysis of clot is divided into internal fibrinolysis and external fibrinolysis, and both are affected by clot contraction induced by platelets, as clot contraction initiated by platelets results in approximately two-fold enhancement of internal fibrinolysis rate, but approximately four-fold slower external fibrinolysis rate compared with uncontracted clots. 148
There are reports indicating that platelets exert diverse roles in regulating fibrinolysis. Under physiological flow rates, PLG associates with phosphatidylserine-exposing platelets to initiate fibrinolysis. 149 Moreover, platelets contribute to fibrinolysis mediated by single-chain urokinase-type plasminogen activator (scu-PA). 150 Activated platelet surfaces are anchored by plasma TAFI, to attenuate PLG accumulation and fibrinolysis, resulting in thrombogenicity under flow. 151 Platelets contain three types of secretory granules, namely, the α-granules, dense granules, and lysosomes. Those secretory granules secrete amounts of proteins to affect fibrinolysis, such as PLG, PAI-1, α2-AP, and polyphosphate. 152
The Important Role of Early Inflammation in Bone Regeneration
Bone has a natural ability to repair spontaneously, which is closely associated with a sequential process consisting of inflammatory, reparative, and remodeling phases similar to other tissues.153,154 Following a fracture, blood flows out of the injured vessel to form hematoma, and inflammatory cells are recruited to evoke an acute inflammation, which are known to play crucial roles in bone healing cascade. Inflammation process is thought to activate the coagulation cascade. 155 Under conditions of compromised coagulation, the deposition of fibrin and microvascular failure can enhance the inflammatory response.156,157 Inflammation is considered to affect both bone resorption and formation processes through the recruitment of inflammatory cells (neutrophils, macrophages, lymphocytes) and the release of various cytokines and growth factors. 158
Early inflammation is regarded to trigger regeneration cascade through initiating angiogenesis, recruiting and boosting differentiation of MSCs, and promoting extracellular matrix synthesis.159–161 A certain level of inflammation is necessary for normal bone repairing as the application of anti-inflammatory substances, such as cyclooxygenase-2 (a strong inducer of inflammation released after fracture) inhibitors, are able to significantly damage normal bone healing. 162
During the processes of the polymerization and degradation of fibrin(ogen), various peptides are released, including FpA, FpB, D-dimers, fragment-X, fragment-Y, fragment-D, and fragment-E, as well as minor fragments such as the peptide Bβ15–42. 163 Those peptides are recognized to influence inflammation by affecting cell response and cytokine release during early bone healing.91,164–166 Elucidating the function of fibrinogen in inflammation is warranted as fibrin degradation product are derived from fibrinogen and possess similar biological functions with fibrinogen.
Fibrinolysis Products Regulate Early Inflammation
Fragment X
Fibrinogen is a 340-kDa plasma protein that consists of three pairs of polypeptide chains (α, β, and γ), to present an extended globular structure. 167 Fragment X originates from fibrinogen molecule by separating the COOH-terminal section from the Aα chain, which is an early fibrinogen degradation product mediated by plasmin. The clots formed from fragment X mediated by thrombin are more fragile and present a loose fibrin conformation, as compared with fibrin network that originated from complete fibrinogen molecules as a result of decreased branching of fibers and fewer interconnections. 168 Those clots, which consist of fragment X, are more susceptible to fibrinolysis. 169 Furthermore, in the company of t-PA, the combination of fragment X with fibrin meshwork results in clots susceptible to fibrinolysis. 169
There are two mechanisms involved in the phenomenon where the adding of fragment X promotes fibrinolysis, one is the presence of fragment X into clots raising maximum turbidity, leading to the formation of thicker fibers that are easily degraded, and the other is the existence of fragment X rendering clots more susceptible to plasmin.169,170 Those data indicate that fragment X may act as a regulator to promote lysis, thereby promising to enhance bone regeneration.
Fragment D and D-dimer
Fragment D and fragment E are soluble fibrin degradation products produced from the plasmin-mediated degradation of crosslinked fibrin. Fragment D holds the independently folded C-terminal domains of the Bβ- and γ-chains. Purified fragment D has the ability to boost human hemopoietic cell proliferation, which also enhances the effect of IL-3 on primitive hematopoietic progenitors in vitro, and those effects are the same as fibrinogen.171,172 D-dimer, a covalently bound dimer generated by fragment D, is considered as the marker of fibrinolysis and many fibrinolysis-related diseases. D-dimer is capable of inducing the monocyte/macrophage lineage to secrete essential components, such as IL-6, u-PA, and PAI-2, to affect blood coagulation, fibrinolysis, and inflammation. 17 Fragment D and D-dimer are capable of promoting the secretion of IL-1α, whereas only D-dimer can stimulate the release of IL-1β. 17
Fragment E
Fragment E is the central part of fibrinogen molecule that consist of the N-termini of the three polypeptide chains. Fragment E can enhance the secretion of IL-1α, which stimulates IL-6 release in rat peritoneal macrophages.17,173 Fragment E potentiates TGF-β-induced fibroblast development thus facilitating wound recovery through forming stable complexes with αVβ3 integrin. 174
Bβ15–42
Bβ15–42 is a small fragment of the N-terminal β chain produced from fibrin cleavage (by plasmin). Bβ15–42 was shown to regulate platelet and endothelial cell migration, fibroblast proliferation, and new blood vessel formation through binding to heparin. 20 It can also boost capillary tube formation and angiogenesis by interacting with vascular endothelial (VE)-cadherin. 20 Furthermore, Bβ15–42 responds to endothelial cell receptor VE-cadherin and leukocyte integrin CD11c thus promoting leukocyte transmigration.175–178 Studies have further demonstrated that Bβ15–42-regulated leukocyte transmigration is associated with the activation of the VLDLR-dependent pathway and the Scr kinase Fyn.179,180
FpB
FpB is a peptide of 14 amino acids released from N-terminal end of B-beta chains of fibrinogen molecules by thrombin in the polymerization of hematoma fibrin network.181,182 FpB has been shown to perform important functions in many diseases, for example, it can attract macrophages to the intima, which accelerates the early atherosclerotic lesion formation. 183 FPB has also been recognized as a maker of many other diseases, such as myocardial infarction, chronic liver, etc. However, the chemotaxis effect of FPB was rarely noticed until researches found FPB could act as chemokines for inflammation cells.164,184 It has been reported that human fibrinopeptide B (hFpB) contributes to the migration of neutrophils and fibroblasts at an optimum concentration of 10–8 M, and this chemotactic activity can be blocked by applying antiserum to hFpB. 182
Researchers further found that the chemotaxis of hFpB for neutrophils was similar to that of anaphylatoxin from the fifth component of human complement (C5a), leukotriene B4. 182 The injection of purified FpA and FpB in rat air pouches showed that the number of leukocytes and monocyte chemoattractant protein-1 (MCP-1) levels were increased.155,185 Researches further found that it was FpB rather than FpA attracting inflammatory cells. 183
Diseases with retarded fibrinolysis affect bone healing
Delayed fracture healing is usually found in patients with chronic diseases such as diabetes mellitus (DM) and obesity. Moreover, patients with those complications show poor capacity to remove fibrin or low fibrinolytic activity.186–188 For example, patients who suffer from DM develop fibrin clots that is composed of thicker fibers and denser fibrin network with fewer branch points, resulting in slower fibrinolysis process and significantly delayed wound healing. 189 Additionally, people with obesity showed slower fibrin lysis rate and damaged capacity to recover from long bone defect compared with the control group.190,191 Furthermore, in a model of obese mice, bone healing was notably decreased compared with the control group, and a reduction of growth factors, such as fibroblast growth factor, TGF-β1, and TNF-α, were observed. 192 One explanation for this may be that the delayed fibrinolysis led to the slow release of growth factors from the dense fibrin clots, thus injuring bone healing.
Additionally, people who are burdened with chronic kidney disease (CKD) have less clot permeability and prolong the fibrin lysis time.193,194 In addition, in calvaria and femoral defect model of animal, mice with CKD were observed to show an adverse impact on bone regeneration, 195 and patients who were affected by chronic obstructive pulmonary disease (COPD) presented decreased fibrin clot permeability and prolonged fibrinolysis time compared with healthy people. 196 Those patients with COPD showed an impaired bone metabolism and were easily afflicted with fragility fracture as well as had a delayed recovery rate from fracture, such as neck fracture.197,198 Other diseases, such as inflammatory bowel disease, 199 sickle cell disease, 200 and chronic cardiovascular disease, 201 noticeably alter patient's clot structure and damage the ability to clear fibrin. As far as those diseases, they are believed to have an unsatisfactory recovery ability from fracture as a consequence of the impeded fibrinolysis, although few studies have directly elucidated the influence of those diseases on bone healing.
Conclusion and Future Perspective
Fibrinolysis is a pivotal step for osteogenesis, and is thought to be a target to promote bone healing. The application of various drugs (such as GSNO), ion strength (such as Ca2+), and enzyme concentration (such as thrombin) to alter conformation of clots to facilitate fibrinolysis have shown positive effects on bone healing. Furthermore, modifications indicated by the material interfaces (such as change surface wettability) to generate fibrin clots that are easy to dissolve results in enhancement of osteogenesis. 113
Many chronic diseases, such as obesity and DM, which exhibit retarded fibrinolysis and damaged bone healing ability, assign meaning with regard to fibrinolysis, act as a target to regulate osteogenesis. More attention should be paid to the above chronic diseases, with a purpose to further elucidate the underlying mechanism that causes the alterations in fibrinolysis and to intervene the fibrinolysis-related and inefficient bone healing. We propose that future investigations for the research of fibrinolysis/osteogenesis-based strategies should be considered, but not be confined only to aspects that facilitate fibrinolysis, thus promoting bone repairing, such as: (i) new and efficient methods or techniques to alter the inner structure of fibrin clots thus enhancing fibrin degradation in the fracture sites; (ii) the further effects that products of fibrinolysis exert on early inflammation and bone regeneration. It is expected that the understanding of the mechanism that fibrinolysis promote osteogenesis is promising to develop new strategies (similar to LMHFV or other drugs) to alter fibrinolysis process, thus enhancing bone healing.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 86060620, 31960209, and 31760266), Guizhou Science and Technology Fund Project (Grant No. [2020]1Y093), the “Thousand” Level Project of Training High-Level Innovative Talents in Guizhou Province (Grant No. fzc20200612), the 2018 Zunyi “15851 Talent Elite Project” (Grant No. National Natural Science Foundation of China 31760266), and the Zunyi Science and Technology Fund Project (Grant No. Zun Shi Ke He HZ Zi 2021-40). The Doctoral Science Research Startup Funding of Zunyi Medical University (Grant Nos. F-934 and 2017-01).