
Abstract
Cardiovascular diseases, considered the deadliest worldwide by the World Health Organization (WHO), lack effective therapies for regenerating cardiomyocytes. With their self-renewal and pluripotency capabilities, stem cell therapies are increasingly used in precision medicine. Induced pluripotent stem cells (iPSCs) are a promising alternative to embryonic stem cells. Good Manufacturing Practice (GMP) principles are not yet adapted for large-scale production of iPSCs. Additionally, the quality risk for iPSC products may not always be possible to eliminate, potentially jeopardizing the health of patients. This review aims to identify critical quality attributes (CQAs) for iPSC-derived cardiomyocytes (iPSC-CMs) for the development of cardiovascular therapy to ensure compliance with regulations and safety for patients. To attain these goals, the literature review was conducted with articles related to iPSCs and iPSC-CM therapies, legislation, and regulatory guidelines of the European Medicines Agency (EMA), Food and Drug Administration (FDA), and Pharmaceuticals and Medical Devices Agency (PMDA). In conclusion, additional regulations and guidelines are needed to monitor differentiation, maturation, and tumorigenicity. GMP-compliant cell banks and fast-track approval systems may increase accessibility for patients.
Impact statement
By identifying critical quality attributes for induced pluripotent stem cell (iPSC)-derived cardiomyocyte (iPSC-CM) therapy, this work has the potential to develop the field of regenerative medicine. Developing a high quality and safe product that meets Good Manufacturing Practice (GMP) principles and applicable regulations is crucial for the success of cell therapies. Additionally, the implementation of GMP-compliant cell banks and fast-track approval systems can significantly improve patient access to these treatments. Ultimately, this could lead to more effective treatments for cardiovascular disease, improving patient outcomes and quality of life. This work represents a significant step forward in the development of regenerative medicine and has the potential to significantly impact the field.
Introduction
Cardiovascular diseases are the leading cause of mortality worldwide, accounting for 31% of deaths. Heart failure and myocardial infarction damage cardiac cells, which limited regenerative capacity, leading to irreversible muscle loss and decreased heart function. Rapid reperfusion therapy and heart transplants are common treatments, however with limitations. Precision medicine aims to provide personalized treatments for chronic diseases. Cardiomyocytes are responsible for heart contraction, and the extracellular matrix (ECM) plays a role in heart function. The regenerative potential of cardiomyocytes declines with age, and most heart disease patients suffer from atherosclerosis, leading to fibrotic tissue formation. Stem cells, particularly induced pluripotent stem cells (iPSCs), offer the potential for regenerative medicine. 1 iPSCs can be differentiated into various cell types, including cardiomyocytes, for the treatment and regeneration of cardiac tissue.2,3
Legislation and regulatory guidelines related to cardiovascular therapy play a crucial role in ensuring patient safety and the efficacy of treatments. These regulations provide a framework for the development, approval, and safe use of cardiovascular drugs and devices. The newer the technology, the more the knowledge, and nonclinical and clinical research is required to ensure product safety and quality. 4 The risks of administration of these types of products depend on the origin of the cells used, the manufacturing process, noncellular components, and detailed therapeutic use. 5 Cell line history, sampling process, reprogramming, expansion, differentiation, purification, and transplantation steps must be considered in the cellular therapy product lifecycle. 6 In addition, guidelines have also been developed for cell and gene therapies, which therefore impact stem cell products. 7 Despite the various regulatory contexts that need to be considered, the development of a stem cell product should be approached on a case-by-case basis. 8
Efforts focused on the production of iPSCs, considering factors such as efficacy and safety, regulatory compliance, and cost. The clinical effectiveness of iPSCs depends, mainly, on the quality and efficiency of differentiation, maturation, and tissue engineering. iPSCs are a promise for precision medicine in cardiovascular diseases since safety and quality standards are assured.9–11 This study aims to identify the critical quality product profile, critical quality attributes (CQAs), and production processes for iPSC-derived cardiomyocyte (iPSC-CM) therapy, as well as the legal and regulatory framework for these stem cell-derived products.
Methods
A literary review was performed to establish critical quality product profiles and required quality controls for iPSC-CMs therapy, 12 using research platforms such as PubMed, ScienceDirect, and Google Scholar (Table 1). The scope of the study was standardized by analyzing titles and abstracts, and the inclusion criteria were (1) cardiovascular intervention and trials; (2) use of iPSC-CMs; (3) research and review studies. Exclusion criteria were (1) predictive safety pharmacology and drug-development studies; (2) pharmacogenomic studies; (3) noncardiovascular research. Keywords used included iPSCs, iPSC-CMs, cardiomyocytes, stem cells, cardiovascular, cardiac, quality, quality control, manufacturing, and engineered tissue.
Search Results on Various Platforms Related to Induced Pluripotent Stem Cell-Derived Cardiomyocyte Therapies and Clinical Trials: PubMed, Science Direct, Google Scholar and ClinicalTrials.gov, EUCTR, ICTRP, ISRCTN
n, Number of articles.
To determine regulatory requirements, current legislation, and guidance from the European Medicines Agency (EMA), US Food and Drug Administration (FDA), Pharmaceuticals and Medical Devices Agency (PMDA), National Medical Products Administration (NMPA), Central Drugs Standard Control Organization (CDSCO), and Ministry of Food and Drug Safety (MFDS) were considered. Clinical trials for iPSC-CM therapy were searched without a start date limit, considering all registered trials due to the originality of the technology. ClinicalTrials.gov, EUCTR, ICTRP, and ISRCTN were used, providing information on study type, condition/disease, treatment, and result status. Table 1 details the search results.
Experiment
iPSCs and tissue engineering
iPSCs can differentiate into various human cell types, including cardiomyocytes, holding promise for cardiac tissue regeneration.13,14 However, uncertainties remain regarding cell types, differentiation methods, and quality standards. 15 Unlike embryonic stem cells (ESCs), iPSCs may exhibit genomic instability and epigenetic differences. 16 Cardiovascular tissue engineering encompasses methodologies that allow the recreation of the microenvironment found in cardiac tissue and enable the improvement of myocardial functionality after implantation, with no serious adverse effects. 13 iPSCs had been cultured in two-dimensional (2D) structures that are not capable of replicating the complex tissue environment and interactions necessary for the correct cellular maturation, which occur between cells and ECM. 13 In fact, in 2D culture, cells are subdued to only a few cell–to–cell interactions, with limited options of placement in the substrate and forced polarity and organization, as seen in Figure 1.17,18
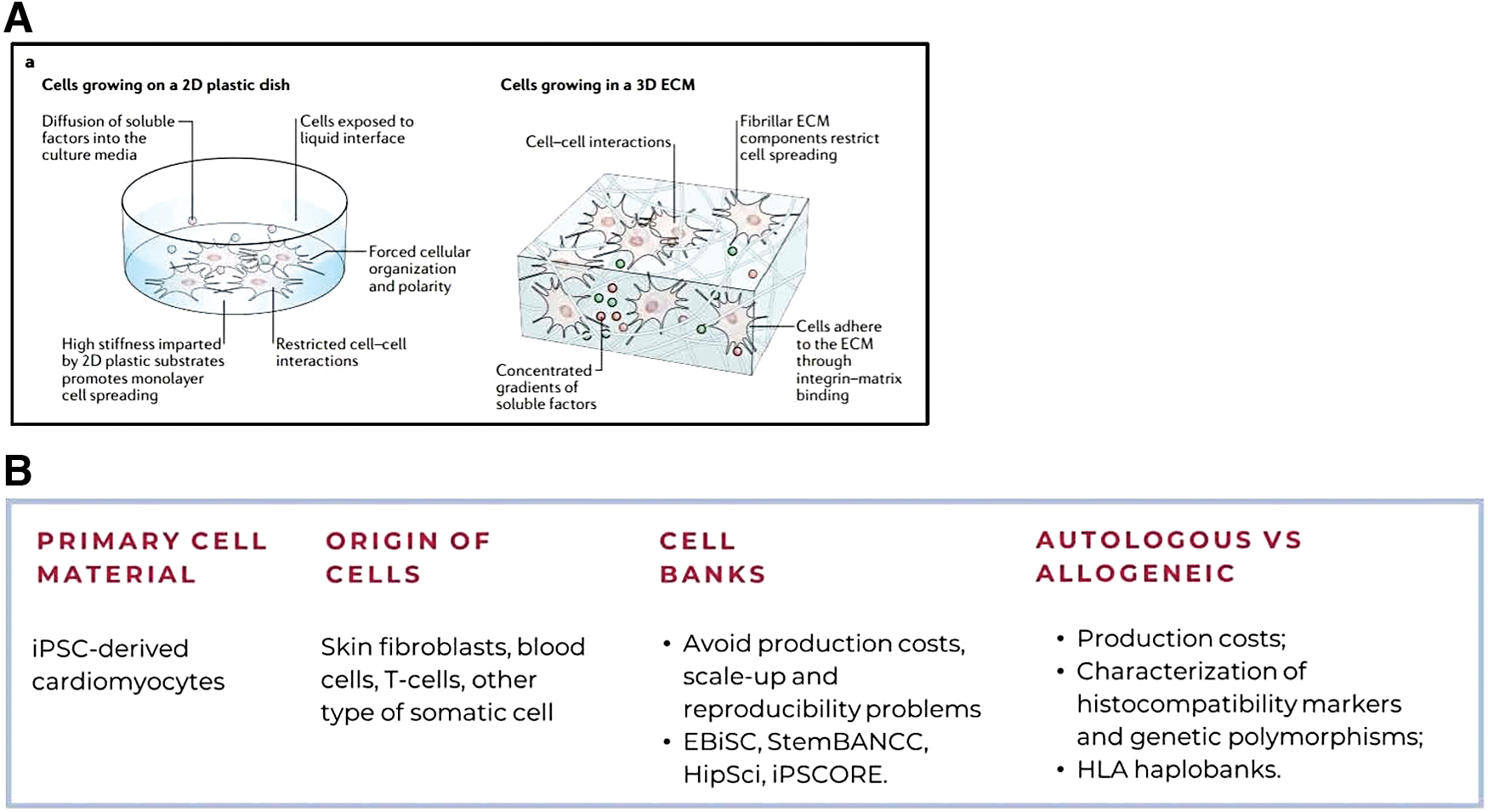
First, it is necessary to address the evident obstacles to the large-scale implementation of iPSCs technology, which is the (1) heterogeneity of iPSC-CMs population, (2) deliver a good engraftment rate by providing a high number of iPSC-CMs, and (3) choosing the best transplantation method. 12 Moreover, iPSC-CMs alone display arrhythmogenic potential, reduced contractile capacity, abnormal electrophysiological properties, and low cell survival after engraftment in vivo, having hindered the application of these models for cell replacement therapy. 13 To enhance the therapeutic efficacy of iPSC-CMs, three-dimensional (3D) environments supported by the ECM have been implemented (Fig. 1A, B), but challenges such as heterogeneity of iPSC-CM populations and low engraftment rates continue.
Matrices and scaffolds
The incorporation of matrices and scaffolds proved to be an advantage for cardiomyocyte differentiation and maturation, cell functionality and survival, and engraftment. In fact tissue engineering is crucial in cardiovascular regenerative therapy, with 3D environments supported by the ECM playing a key role. 17 Biomaterials used in tissue matrices range from natural options like collagen and alginate to synthetic alternatives such as poly(ethylene glycol) and poly(caprolactone). 13 The choice of biomaterial depends on tissue-specific requirements. 13 Hydrogels have proven useful in accommodating multiple cell populations through time-controlled protein release. Collagen, the most abundant cardiac ECM polymer, supports cell attachment and growth but can be rigid, hindering integration into the heart. 13 To address this, chitosan or elastin can be incorporated, reducing rigidity and enhancing heart stability. 13 Fibrin and alginate are other viable natural options for engineered tissue construction. 13
Synthetic polymers with electroconductive properties, such as polyaniline and polypyrrole, have improved cardiac conduction and electrical signaling preservation in iPSC-CMs. 13
Natural biomaterials offer better compatibility with the host immune system due to their ECM-derived components, while synthetic materials allow greater customization of the matrix.13,14 However, synthetic materials may induce a proinflammatory host response, especially nondegradable ones. 13
Additionally, matrices can be obtained through decellularized ECM (dECM) structures, supporting cell attachment and re-cellularization. 18 VentriGel, derived from decellularized porcine myocardium, has shown promising safety results in post-myocardial infarction patients. 18
Biodegradable scaffolds facilitate the integration of implanted cells within the host environment, offering the possibility of creating constructs with iPSC-CMs, cardiac fibroblasts, and endothelial cells, along with proper vasculature. 18
Cardiac tissue engineering structures can be spheroids/organoids or engineered tissue constructs. Spheroids are spherical structures formed by multiple cell populations, while organoids mimic native organs but remain challenging for human cardiac tissue.19,20 Engineered cardiac tissue constructs embed iPSC-CMs in a 3D collagen matrix, showing the potential for restoring function in damaged myocardium.19,20
Reprogramming
Reprogramming iPSCs involves transforming adult cells into pluripotent cells by activating pluripotency-related genes21,22 (Fig. 2). Alternative reprogramming factors have been used based on the cell of origin, and surface antigens and cellular signaling pathways influence the process.21,22
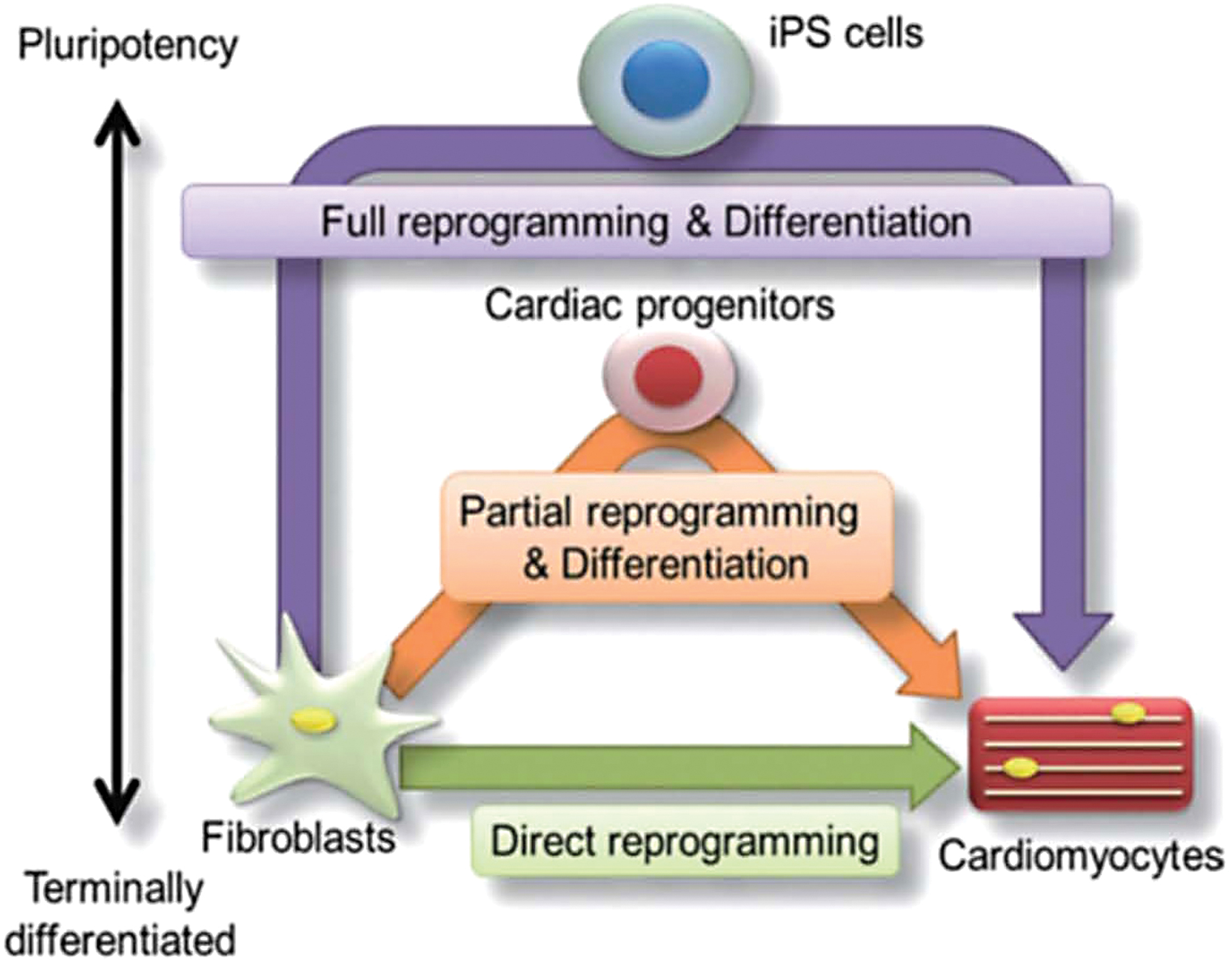
Three different pathways of emerging cardiomyocyte regeneration which include a full reprogramming approach (purple line), a partial reprogramming approach (orange line), and a direct reprogramming approach (green line). Color images are available online.
Differentiation methods include coculture with visceral endoderm-like cells, embryoid body-based differentiation, and 2D culture. The monolayer-based approach has shown higher yields of differentiated cells with defined characteristics of nodal, atrial, and ventricular cardiomyocytes.21,22
The heterogeneous nature of iPSC-CMs, including immature cells with pluripotency capacity, poses challenges, including tumorigenesis risk and the presence of arrhythmias. Electrical and mechanical stimulation and mimicking myocardial conditions are strategies to promote maturation and functionality. 4
The choice between autologous and allogeneic cell sources is essential for iPSC-based therapies. Allogeneic cells and bioreactors are used for large-scale iPSC-CM production, necessary for treating heart failure.23,24 Bioreactors enable controlled and standardized production, reducing labor and operational costs and monitoring the environment variables.24–28
Regulation of advanced medicinal products
Regulatory science has gained importance as rapid advancements in innovation and scientific technology are made to treat a wide range of diseases, including those considered incurable until now. These advances generate vast amounts of data and information that need to be evaluated from the perspective of product quality and safety. 29
Cell-based therapies, like other areas of research, are relatively new and still not well understood.8,30–32 While they offer significant potential to surpass the efficacy of current therapies, they also carry potential risks that can seriously harm patients if not identified and controlled.30–32 In 2020, the EMA issued a warning statement on unproven cell-based therapies, 30 highlighting serious adverse reactions, including infections, unwanted immune reactions, tumor formation, and even fatalities. Regulatory framework establishes the scientific and clinical foundations for medical products, ensuring compliance and providing guidance for implementation.8,31,32 Products undergo rigorous monitoring through pharmacovigilance channels before entering the market. 5
In 2007, the European Commission (EC) introduced a regulation, No. 1394/2007/EC, specifically addressing advanced therapy medicinal products (ATMPs), 7 which are divided into gene therapy medicinal products (GTMPs), somatic cell therapy medicinal products (sCTMPs), and tissue-engineered products (TEPs). 7 This regulation also covers combined ATMPs that integrate medical devices. In 2009, the EC published Directive No. 2009/120/EC, updating definitions and scientific and technical requirements for each type of therapy. 33 However, determining the regulatory category for certain products can be challenging, especially when technologies fall within the borderline of different categories. 33 Most stem cell-derived products aim to replace damaged human tissue, making them fall under the definition of TEPs.33,34 iPSCs, in particular, undergo substantial manipulation to differentiate into mature cells with relevant biological, physiological, and structural characteristics for clinical use, placing them within the sCTMP definition.35–37 According to the ATMPs regulation, when a product falls within this borderline, the prevailing category should be TEP.6,38 However, certain characteristics and components of a product may alter its regulatory category.6,38 For example, if it includes genetically modified cells, it should be considered a GTMP, or if it incorporates a medical device (e.g., matrix or scaffold), it should be considered a combination product. An iPSC-derived product that includes a matrix, or a scaffold is a borderline product. 38 In this situation, the final product category must be considered a combination product.
The FDA also faces challenges in classifying stem cells under specific product descriptions, as they can fall under the regulatory definitions of human cells, tissues, or cell and tissue products (HCT/Ps), biologics, and drugs.6,38 Guidelines have been developed for cell and gene therapies, which also impact stem cell products.38–41 Despite these regulatory contexts, the development of a stem cell product must be assessed on a case-by-case basis. Certain criteria must be met for all products, including safety, identity, purity, potency, and clinical efficacy, with analytical methods tailored to each product.42–44 Safety Act and the Act on Pharmaceuticals and Medical Devices (PMD) regulate regenerative medicine in Japan.45,46 The Safety Act applies to regenerative technologies where safety and efficacy have not been established, while the PMD Act applies to products with known safety and efficacy profiles that are in the process of acquiring MA for commercialization, regardless of whether they are drugs, medical devices, or regenerative medicinal products.47–50 These acts were implemented following reported cases of patient deaths after stem cell therapy interventions, particularly in China and South Korea. The PMDA recognized the need for a regulatory framework for the development of stem cell products to prevent medical practice and interventions without sufficient knowledge of their safety profiles in humans. 45
Quality
It is widely recognized that every health care product carries inherent risks. According to the International Council for Harmonization (ICH) Guideline Q9 on quality risk management. Risk is defined as the probability of harm occurring and the severity of that harm. 51 As technologies advance, increased knowledge, nonclinical research, and clinical research are required to ensure the safety and quality of health care products. Quality risks must be identified and controlled throughout the entire product lifecycle, from the selection of starting materials to the final product, to consistently deliver high-quality products while safeguarding patients and maintaining the confidence of regulators.52–54 The risks associated with the administration of cell-based medicinal products, as outlined in the EMA Guideline on Human Cell-Based Medicinal Products (EMEA/CHMP/410869/2006), depend on factors such as the origin of the cells used, the manufacturing process, noncellular components, and the specific therapeutic use.55–59 For iPSC cell therapy, there are well-known risks associated with its clinical use, including uncontrolled proliferation and differentiation, teratoma formation, tumorigenicity, and migration to ectopic locations.58–62 It is important to note that the quality risks associated with these types of products differ from those of biologic products, and therefore eliminating the risk entirely may not always be feasible. In assessing quality risks, factors such as cell line history, sampling process, reprogramming, expansion, differentiation, purification, and transplantation steps in the cellular therapy product lifecycle should be taken into consideration. 6
According to the European Directive No. 2009/120/EC, the risk analysis should require care of risk factors that include “the origin of the cells (i.e., autologous, allogeneic, xenogeneic), the ability to proliferate and/or differentiate and to initiate an immune response, the level of cell manipulation, the combination of cells with bioactive molecules or structural materials, (…) the extent replication competence of viruses or microorganisms used in vivo, (…) the long time functionality, the risk of oncogenicity, and the mode of administration or use.” 33 Additionally, the application of a quality risk management process is crucial in identifying, assessing, and analyzing potential risks, allowing for science-based decisions to either mitigate or accept those risks. Product design and the manufacturing process should align with this risk-based approach, taking into account factors such as donor eligibility, starting materials, facilities, equipment, utilities, and more.63–67 Process validation and verification are essential to ensure the quality of the final product. Process validation certifies the control and monitoring of the manufacturing process through parameters and in-process controls, while verification confirms that a specific batch of the product meets the established quality requirements through quality tests conducted during and after manufacturing. 59
As the understanding expands, the process of risk assessment undergoes continuous updates throughout the entire product life cycle, forming the bedrock for crafting a comprehensive risk management plan, in accordance with the Guideline on Risk Management Systems for Medicinal Products for Human Use (EMEA/CHMP/96268/2005).68–70
The PMDA recognizes the importance of quality risk management, knowledge management, training, and control strategies for cell therapy products. 45 The establishment of a consensus within the scientific community regarding quality assurance, manufacturing process control, and quality control of investigational products will facilitate communication with regulators and expedite the development and approval of cell therapies for the market. According to the PMDA's risk classification, tissues derived from allogeneic iPSCs intended for transplantation into patients are classified as Class I Regenerative Medicine (High Risk) regenerative medicine technologies. 21 Therefore, it is crucial to implement quality assurance principles specific to these therapies, which have distinct characteristics and specifications compared to those required for the release of a product batch. Continuous quality control, including in-process controls during research and development, should be maintained throughout the manufacturing process, following a product specification control strategy. Addressing variability factors and quality risks will be ensured which impacts the effectiveness of the manufacturing process, quality, and consistency.
Discussion
Quality target product profile
Pharmaceutical development necessitates the consideration of quality throughout the developmental process rather than solely relying on the final product, particularly in large-scale cell-based therapy production. The ICH guideline Q8 (R2) proposes the design of a quality product and manufacturing process that consistently delivers the intended performance2,37 (Table 2). However, the use of living cells as starting materials in cell-based therapies poses challenges due to their variability and to the incomplete understanding of their mechanism of action. Recently, the biotechnology industry has adopted a Quality by Design (QbD) approach, which involves the systematic development of a high-quality product based on scientific knowledge and risk management. In the regulatory framework for stem cell products, compliance with Good Manufacturing Practice (GMP) requirements, including Directive 2003/94/EC, and relevant regulatory standards such as the EC “Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products,” is mandatory.
The Main Quality Target Product Profile (i.e., Release Criteria) for Induced Pluripotent Stem Cell-Derived Cardiomyocytes Defined by Prowse et al.
CMs, cardiomyocytes.
Source: Prowse et al. 2
CQAs and control strategy
CQAs are the physical, chemical, biological, and microbiological measurable properties that influence the quality and safety of the product, and they are decisive in ensuring the best quality of the intended process and product design. 58 CQAs, during the manufacturing process, must be maintained within a certain limit or range to ensure the product quality of iPSCs and their derivatives (e.g., cardiomyocytes). These quality controls can interfere with raw materials, intermediates, container closure systems, and the final product, through critical process parameters (CPPs) and raw material attributes. CPPs are the process endpoints that impact CQAs and, consequently, need to be equally monitored and controlled. The control strategy may involve interventions in starting materials, intermediates, the container closure system, and the final product through CPPs and raw material attributes.
To ensure the quality of the final product in regenerative cell therapy, the selection of appropriate starting materials is crucial. iPSCs are commonly used as active substances, and it is imperative to preserve their safety, sterility, and CQAs.71–73 Although there is no regulatory guidance on the choice of somatic cell type for generating iPSCs, evidence suggests that CD34+ peripheral blood cells exhibit higher reprogramming efficiency compared to differentiated blood cells.21,22 Cell banks, such as the European Bank for induced pluripotent Stem Cells (EBiSC), offer advantages in terms of cost-effectiveness, scalability, and reproducibility in iPSC development. Allogeneic iPSC-derived therapies may provide a potential solution to serve a larger number of patients; however, it is crucial to characterize histocompatibility markers and genetic polymorphisms relevant to the intended use due to potential immunogenicity issues associated with host-donor variability. Additionally, it is advisable to establish haplobanks of iPSCs that encompass a substantial proportion of the population. 37 The International Stem Cell Banking Initiative (ISCBI) provides guidance on the required assays for the registration of clinical-grade iPSCs, including pluripotency tests, karyotype analysis, cell identity, gene expression profiling, and microbiological tests. Gene editing is a significant technology enabling the modification of specific genes to create more effective cell lines. 74 Commonly used nucleases for introducing desired genomic modifications include Zinc Finger Nucleases (ZFN), Transcription Activator Like Effector Nucleases (TALENs), and the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (CRISPR/Cas9). 56
The recommended dosage for stem cell transplantation is not specified and an abnormal number of cells can potentially jeopardize patient safety. The healing process of the cardiovascular system relies on multiple signaling factors, including growth factors, ECM components, and modulators of Wnt and Hippo pathways. 35 Buikema et al. 35 discovered that the expansion of iPSC-CMs and the delay in maturation can be achieved by removing cell-cell contact and inhibiting GSK-3b. Furthermore, prolonging the culture period can lead to the development of multinucleated iPSC-CMs with mature sarcomeres and improved electrophysiological characteristics, particularly when cultured in 3D structures with electrical and physiological stimulation. The regulation of pluripotency is governed by various pathways, such as TGF-β superfamily-activated cascades, receptor tyrosine kinase signaling, and insulin-like growth factor pathways. 35
Following multiple rounds of mitosis, cells undergo aging, acquire instability, and develop genetic abnormalities that block cell differentiation and compromise purity.
Manufacturing methods
iPSC-CMs can be produced in flask-based cultures and spinner flasks as aggregates using adherent culture methods like microcarriers, and they can be scaled up in closed bioreactor systems. The use of stirred tank bioreactors (STR) allows for iPSC culture without the need for an attachment substrate, simplifying downstream processes and enabling the integration of monitoring probes for better control of culture variables24,28 (Fig. 3). Various bioreactors were used for scale-up, with studies demonstrating iPSCs expansion by a maximum of 32-fold. The hydrodynamic environment within bioreactors plays a crucial role in controlling cell phenotype, quality, and differentiation potential. Computational Fluid Dynamics (CFD) can be employed to calculate hydrodynamic parameters for scale-up equations.24,28

Manufacturing process of induced pluripotent stem cell-derived cardiomyocytes. Color images are available online.
Identity, purity, and dosage
The number and viability of cells within a product are essential in determining the appropriate dosage for administration to patients and ensuring the product's efficacy. Various validated methods can be utilized to assess these parameters, including automated live cell count machines, flow cytometry, and imaging techniques. 65 The expression of surface markers, such as cardiac troponin T, can aid in the identification of specific cells. Flow cytometry is commonly employed for this purpose. 65 Additionally, ensuring purity is crucial to prevent the presence of undesirable cells that could pose safety risks. Cell purification techniques, such as isolation based on cardiac markers, have been developed to mitigate adverse events. The risk of tumorigenicity must also be evaluated, taking into account factors such as product design, the use of immunosuppressants, and the engraftment microenvironment to ensure product safety.4,50
Product potency refers to its capacity to effectively impact disease effects. 42 Uncontrolled potency can lead to ineffective products, tissue chimeras, and altered cell function. 41 For complex products, the FDA recommends employing complementary assays to measure different CQAs. 81 Park et al. 54 observed that the maximal contraction force and maximal compression of cardiac constructs were 452 ± 215 pN and 0.595 ± 0.283 Pa, respectively. They proposed three functional conditions for these constructs before implantation, 54 including appropriate dimensions, shape, and cellular content; assessment of contractile force and beating frequency; and evaluation of indicators of stress during heart contraction. 54 Mechanical measurements can also guide decisions regarding the utilization of ECM proteins to enhance maturation and support, as they may trigger immunogenicity after implantation. 54
iPSC-CMs often exhibit an immature structure and function, characterized by a poor calcium cycle and a rounded or multiangular shape with a smaller size compared to the adult phenotype. 48 Fluorescent staining and image analysis are frequently employed to assess morphology. 60 Electrophysiology assays are commonly used to measure rhythmic contractility capacity in iPSC-CMs, but they do not provide a comprehensive assessment of cell potency. 39 Techniques such as tissue contraction force microscopy and tissue gauge assess mechanical forces and/or stress in cardiac tissues but lack precision and reliability for clinical applications. 54 Other methods of force measurement include video edge detection, single particle tracking, and particle image velocimetry (PIV) quantification, which measure the particle displacement of spheroids or patches labeled with particles containing an antibody.54,65,66 Sharp electrode electrophysiology can be used to differentiate between atrial, ventricular, or nodal cells based on action potential shape, but the immature phenotype of these cells may lead to misleading results. 54
Ribeiro et al. 65 have proposed a novel method to enhance parameter comparison by integrating several less invasive image-based methods for quantifying mechanical forces from videos, including bright-field videos of single cells, videos of fluorescent microbeads, and videos of fluorescent myofibrils to measure sarcomere length. 65
To ensure the safety and quality of cell-based products, it is necessary to assess the eligibility of donors and perform screening for microorganisms. Donor eligibility, in terms of identifying risk factors, should be established based on characteristics such as age, sex, health status, and ethnic origin. Donors of cells or tissues must be tested for infectious agents such as HIV, HBV, HCV, syphilis, and TSE. Three approaches to address contamination include selecting and testing cell lines and raw materials, defining production processes for viral infection clearance, and implementing multiple control checkpoints. Microbiological screening should be conducted to detect human infectious agents, and living cells of allogeneic origin should undergo comprehensive characterization. Screening for bacteria, fungi, and mycoplasma should be performed on finished products. Genomic instability resulting from genetic changes in iPSCs may occur, and the safety of the cell line must be assessed through karyology testing. The European, United States, and Japan Pharmacopoeias46,59 have harmonized methodologies for microbial testing and sterility.
In 2014, iPSCs were evaluated in clinical trials for the treatment of macular degeneration using iPSC-derived retinal pigmented epithelial cells. Since then, clinical trials have been conducted for the treatment of Parkinson's disease and solid tumors, and attention is now shifting toward the cardiovascular field. Improvements in the left ventricular ejection fraction (LVEF) serve as the primary focus for evaluating clinical outcomes in this area. In Japan, iPSC-CMs have already been used since 2018 to showcase their potential for cardiac regeneration. While ongoing clinical trials are testing iPSC-CMs for the treatment of heart failure, the highly restrictive exclusion criteria limit access to a significant portion of the target population (Table 2). The development of a certified quality system and continuous monitoring of CQAs and CPPs are necessary to ensure the safety and efficacy of these therapies. With the implementation of a QbD approach, regulators can gain assurance regarding the quality of the manufacturing process (Fig. 4).

Regulation of induced pluripotent stem cell-derived cardiomyocytes development. Color images are available online.
The Quality Target Product Profile (QTPP) and manufacturing process control for iPSC-CMs, as the evaluation of the regulatory guidelines from FDA, EMA, and PMDA regarding GMP were addressed. Key concerns were addressed, including differentiation, maturation, tumorigenicity, and cell loss, emphasizing rigorous process controls for patient safety. Challenges arise due to undefined CQAs and CPPs affecting product design and manufacturing methods. Lack of validated assays for identity, potency, viability, dosage, and genomic stability requires comprehensive evaluation for product quality and safety. Standardized, cost-effective manufacturing processes require certified quality systems and continuous monitoring of CQAs and CPPs.
A QbD approach and certification of cell banks like EBiSC and StemBANCC could enhance utilization and regulatory review. Research findings on iPSCs, reprogramming, stimulation, coculture, and vascularization are essential. Clear guidance on control strategies for dosage, genetic stability, potency, and starting material characterization is necessary. Early-stage clinical trials may aid in comparing methods and determining cost–benefit ratios for wider clinical applications (Table 3). Study limitations include quality control of culture media, organ/tissue dissociation, shelf-life requirements, and cell banking.
Summary of Characteristics of the Clinical Trials Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes
iPSC, induced pluripotent stem cell.
Conclusion
This work aimed to identify the QTPP for the production of cardiomyocytes derived from iPSCs lines and the necessary controls to be implemented during the manufacturing process. At the same time, it aimed to evaluate the guidelines and reference standards for the production of these cells in the context of GMP from the main regulatory agencies (FDA, EMA, and PMDA). The major concerns of iPSC-derived cells include differentiation and maturation, tumorigenicity, cell loss, etc., which require monitoring through process controls during the manufacturing process. Failure to control these factors can cause life-threatening situations after transplantation into patients, as observed by EMA and PMDA. Without the definition of the CQAs and CPPs, it becomes increasingly challenging to design the final product and define the manufacturing methods that suit the desired clinical purpose. Additionally, the absence of validated methods for most assays, in cases such as identity, potency, viability, dosage, or genomic stability, demands a more thorough evaluation of the product's quality and safety profile and a difficult comparison with other technologies by regulators.
Therefore, the need for a certified quality system and constant monitoring and verification of these CQAs and CPPs is emphasized to develop standardized, replicable, and cost-effective manufacturing processes for large-scale distribution. Furthermore, implementing a QbD approach would reassure the regulators' trust, since it is based on scientific knowledge and risk management strategy. Certification of cell banks would also be a solution to consider for the optimal and reliable use of these cell lines, facilitating the review process by regulatory authorities during clinical trial approval or MA (e.g., EBiSC and StemBANCC).
Throughout the discussion, the research results of several authors are highlighted, with the aim of demonstrating important developments of iPSC that can be applied in the future for its production. Some of them aimed to improve reprogramming by using H3K9 methyltransferases and DNA demethylation proteins. Others highlighted electrical and mechanical stimulation, as well as the crucial role of co-culture and vascularization in improving cell maturation. Furthermore, the use of an HLA-matched donor–host system has been shown to decrease the immune response against transplanted iPSC-derived cells, allowing the possibility of allogeneic iPSCs therapy.
An analysis was made of the regulations of several regions, especially focusing on the EU, the United States, and Japan, which have the most influential regulatory agencies in the pharmaceutical industry. These agencies have made efforts to regulate the field of ATMPs, defining what they are and the subtypes of therapies. Many of the recommended methods come from the Pharmacopoeias or are standard methods used for factor identification and quantification (some not covered by the recommended guidelines—for example, ICH guidelines Q6A and Q6B). However, there is still no clear guidance on what control strategies should be applied for dosage checks, genetic stability, potency, characterization of starting materials, and integration of genetic material for iPSCs 41 and their derived cell products. There are only a few guidelines and communications related to stem cell products and, in particular, their derivatives. For example, EMA has issued a reflection paper on stem cell-based medicinal products, without any clear guidance from the QCAs. In the future, they may be an important tool for comparing different manufacturing methods, as long as quality controls are defined, and in turn for setting cost–benefit ratios, which will enable the accessibility of these precision medicine therapies in current clinical practice.
Quality control of biological active additives used in culture media (e.g., growth factors, cytokines, and antibodies), organ, or tissue dissociation and isolation of the cell population of interest, shelf-life requirements, and cell banking are points of focus. Furthermore, a fast-track system of development and approval should be implemented between applicators and regulators, to accelerate the access of this innovation to the patients, considering that cardiovascular disorders are currently at the top of the most fatal diseases worldwide.
Footnotes
Authors' Contributions
All authors have substantially contributed to the conceptualization, methodology, resources, writing—original draft preparation, writing—review, and editing. All authors have read and agreed to the published version of the manuscript.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by FCT—Foundation for Science and Technology, I.P., through National Funds, under the projects UID/DTP/04138/2021 and UID/BIO/04565/2020.