
Abstract
Cardiovascular disease stemmed from atherosclerosis (AS) is well recognized to be the predominant cause of global death. To comprehensively clarify the pathogenesis of AS, exploit effective drugs, as well as develop therapeutic solutions, various atherosclerotic models were constructed in vitro and widely utilized by the scientific community. Compared with animal models, the in vitro atherosclerotic models play a prominent role not only in the targeted research of single pathological factor related to AS in the human derived system, but also in the combined study on multipathological factors leading to AS, thereby contributing tremendously to the in-depth elucidation of atherosclerotic pathological process. In the current review, a variety of pathological factors incorporated into the existing atherosclerotic models in vitro are broadly elaborated, including the pathological mechanism, in vitro simulation approaches, and the desired improvement perspectives for reproducing each pathological factor. In addition, this review also summarizes the advantages and disadvantages of current atherosclerotic models as well as their potential functionality. Finally, the promising aspects for future atherosclerotic models in vitro with potential advances are also discussed.
Impact statement
The present in vitro models in relation to atherosclerosis (AS) have played a pivotal role in the extensive study of AS pathogenesis and downstream drug screening. This review mainly summarizes each pathological factor incorporated into the current in vitro models of AS, its pathogenic mechanism and in vitro simulation methods, as well as possible improvement aspects, thus providing new insight for constructing pathophysiologically relevant in vitro atherosclerotic models.
Introduction
Cardiovascular diseases are well known to be the leading causes of global death,1–3 which are principally derived from atherosclerosis (AS), which is a multifactorial and chronic inflammatory disease.4–6 Prevailing evidences support that the initiation and progression of AS is not only related to aging, but also connected with various diseases, such as hypertension, hypercholesterolemia, hyperglycemia, and oxidation stress. In addition, vascular endothelial cells (ECs) dysfunction and thrombus formation due to coagulation factor aggregation also serve as the potential risk factors of AS.7,8 Endothelial dysfunction occurs when the fine balance between relaxing factors and contracting factors of the vascular endothelium is broken.
Inflammation and oxidative stress as well as high levels of low-density lipoproteins (LDLs) are regarded as the predominant factors that cause endothelial dysfunction followed by a series of atherogenic events. 9 Lesions with high inflammatory rate become vulnerable and prone to rupture, which renders the tissue factor exposed to the flowing blood, thus inducing coagulation cascade activation, platelet aggregation, and finally thrombus formation that leads to clinical manifestations of AS. 9
AS occurs based on the structural characteristics of vasculature, which works typically as a closed vessel system, gradually delivering blood to capillaries, thereby promoting the blood circulation in the body. ECs in the tunica intima, vascular smooth muscle cells (VSMCs) in the tunica media, and fibroblasts in the adventitia of arterial wall contact with each other and undertake crucial structural and biological functions in different spatial regions of the vasculature. The unique and complex vasculature of microenvironment determines the fate and function of these resident vascular cells, and the development of in vitro models that can precisely recreate the key structural and functional characteristics of human blood vessels remains a major challenge, 10 which is also believed to be the prerequisite for constructing in vitro AS models that can replicate the complex pathophysiological process in vivo.
AS is characterized by massive lipid deposits within the arterial wall, forming fibrofatty plaques that lead to narrowing of the arterial lumen, which in turn causes arterial occlusion or plague rupture (Fig. 1). 11 The pathological process of AS is highly complex. First, excess LDL gradually accumulates in the subendothelial space to become oxidized LDL (Ox-LDL), further causing inflammation initiation that induces ECs to secrete and release plethora of chemokines. Due to the chemotaxis of these chemokines, monocytes migrate and adhere to the lining of blood vessels, subsequently differentiating into macrophages upon the stimulation of macrophage colony-stimulating factor (M-CSF). Macrophages then engulf Ox-LDL and transform into foam cells, which release multiple factors that induce VSMCs migration into tunica intima and the formation of fibrous caps. Afterward, VSMCs gradually disappear from the fibrous cap, while infiltrated macrophages degrade the collagen-rich cap matrix, which thins the fibrous cap and eventually leads to plaque rupture and thrombosis (Fig. 2). 12
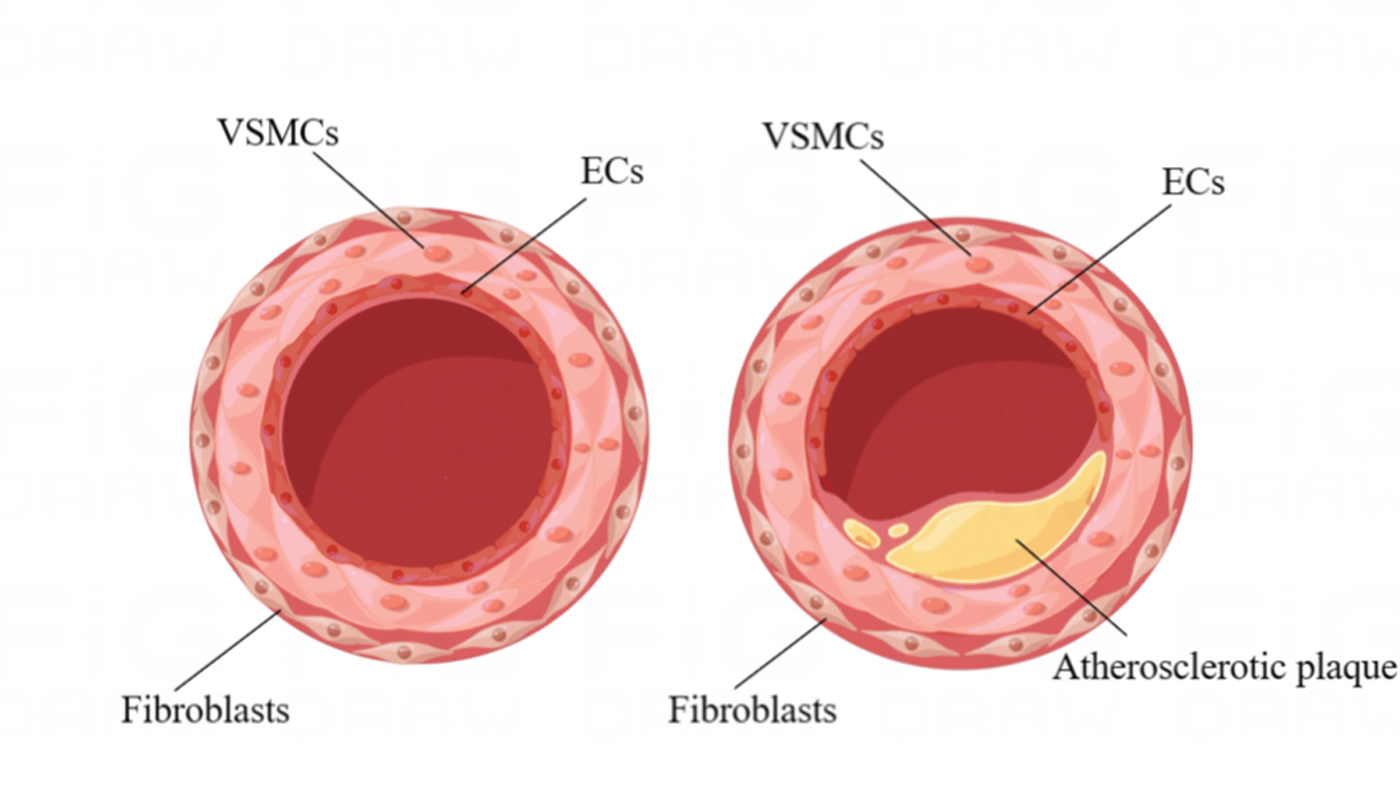
Physiological and atherosclerotic vascular structure. Designed by Figdraw (www.figdraw.com). Color images are available online.

The pathogenesis of atherosclerosis. Designed by Figdraw (www.figdraw.com). Color images are available online.
Due to the complex vascular structure and the intricated pathological factors involved in AS, as well as the difficulty in isolating and culturing intact human blood vessel tissue, establishing pathological models hold great promise in studying the structural and functional alterations of atherosclerotic blood vessels, as well as clarifying the role of various pathological factors and their underlying mechanism, thereby contributing to the detailed definition of atherosclerotic pathogenesis and development of clinical therapeutic solutions for AS-derived cardiovascular diseases.
Existing AS pathological models consist of both in vitro models and animal models, such as rabbits, mice, rats, pigs, and nonhuman primates, which have been widely used in pathological mechanism study and drug screening owing to their broadly acceptable construction protocol and convenient detection of pathogenesis. 13 Compared with animal models, the tunable characteristics of in vitro models, especially for AS biochip models facilitates the precise application of single pathological factors to specific vascular cell types, allowing the study of isolated factors rather than observing systemic effects of multiple factors.14,15
Therefore, the present in vitro models in relation to AS have played a pivotal role in the extensive study of AS pathogenesis and downstream drug screening. So far, a large number of in vitro AS vascular tissue models based on human-derived vascular cells have been constructed for studying AS. 16 There are not only soluble chemical factors and mechanical factors in tissue-engineered AS models, but also different types of interactions, including cell-to-cell and cell-to-extracellular matrix (ECM) interactions. 16
This review mainly summarizes each pathological factor incorporated into the current in vitro models of AS, its pathogenic mechanism and in vitro simulation methods, as well as possible improvement aspects. Overall, the existing in vitro models of AS accommodate not only biochemical pathological factors, including Ox-LDL, inflammatory factors, oxidative stress factors, and high glycemic factors, but also biomechanical pathological factors, including matrix stress, flow shear stress, and vascular tensile stress (Table 1).
Pathological Factors Included in the Current In Vitro Atherosclerosis Models and Their Simulation Methods
EC, endothelial cell; VSMC, vascular smooth muscle cell; THP-1, human myeloid leukemia mononuclear cells; LDL, low-density lipoprotein; Ox-LDL, oxidized low-density lipoprotein; AS, atherosclerosis; M-CSF, macrophage colony-stimulating factor; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; PDMS, polydimethylsiloxane.
Biochemical Pathological Factors
Low-density lipoprotein
Vascular endothelial injury and functional changes are the initiating events for the occurrence of AS, and LDL in vascular cells undergoes oxidative stress and is converted to Ox-LDL, which works as the major contributor in damaging vascular endothelium.52–56 Ox-LDL promotes the secretion of chemokines by ECs and accumulates in recruited leukocytes through specialized receptors such as Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1).57,58 Macrophages transform into foam cells and secrete more inflammatory cytokines, which exacerbate the inflammation and oxidative stress in ECs. Ox-LDL can also stimulate the expression of adhesion molecules, such as intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), facilitating the monocyte adhesion and differentiation into macrophages so as to form AS plaques, which in turn disrupts endothelial barrier. At the same time, inflammatory factors resulting from oxidative stress and inflammation elicit fibrotic reactions in VSMCs, which destabilize plaques followed by rupture and thrombosis (Fig. 3). 59

Effect of LDL on atherosclerosis initiation and development. LDL, low-density lipoprotein. Color images are available online.
Recently, Ox-LDL-directed foam cell differentiation has been presented in a fibrous plaque microsystem by which the unique role of biofilm-based inflammation in exacerbating fibrous plaque damage was validated. 60
LDL in different forms has been used to study the pathophysiology of AS in vitro, including LDL, Ox-LDL, and enzyme-modified LDL, among which Ox-LDL is employed the most extensively because of its underlying atherogenic effect during the AS initiation stage. Dorweiler et al. established a coculture model of ECs and VSMCs in modified fibrin hydrogel, where different cholesterol levels were realized by adding Ox-LDL at different concentrations. 61 Foam cell formation and interleukin-8 (IL-8) secretion were observed in the coculture model in which human monocytes were also combined. This model is suitable for studying key events during the development of AS plaques, but the effects of culture time and lipoprotein modification were overlooked, and there is a lack of study on the interaction between vascular cells and immune cells.
During the process to explore the mechanism through which long noncoding RNA and differentiated antagonistic nonprotein-coding RNA affect the development of AS, Zhang et al. found that the expression of differentiation-antagonistic nonprotein-coding RNA increased significantly in Ox-LDL-treated ECs and VSMCs, and Ox-LDL-treated vascular cells' activity increased while cell apoptosis reduced after the differentiated antagonistic nonprotein-coding RNA was downregulated. Meanwhile, the expression level of inflammatory cytokines was also lessened and the tolerance of vascular cells to oxidative stress was enhanced. 19 To establish foam cell model, Zhang et al. employed Ox-LDL to stimulate human myeloid leukemia mononuclear cells (THP-1), and the results showed that THP-1 cell-derived macrophages are able to transform into foam cells upon Ox-LDL stimulation. As the stimulation time extends, lipid droplet levels rose up and foam cells were formed abundantly. 20
Since enzyme-modified LDL is more potential to induce foam cell formation than prototype LDL, Zhang et al. used enzyme-modified LDL to induce vascular lesions after constructing human tissue-engineered blood vessels, and found that enzyme-modified LDL treatment led to upregulation of ICAM-1 and E-selectin in ECs and obvious foam cell generation. 21
During the study on LDL-induced pathological process of AS, a variety of in vitro models have been developed, in which AS-related pathological changes have been replicated, including altered expression of key proteins and foam cell formation. However, current research on in vitro stimulation of AS based on ox-LDL treatment remain to be improved due to the difficulty in imitating constant oxidation of LDL in vivo, followed by dynamic change of ox-LDL concentrations. Therefore, a better repeat of atherosclerotic process originating from LDL in vitro might contribute to further advancement in revealing the underlying mechanism of AS.
Inflammatory Factors
AS is recognized to be an inflammatory disease that is mainly mediated by the infiltration of macrophage and T cells into the vascular wall. Once the endothelial layer is damaged, monocytes penetrate into the subintimal layer and differentiate into macrophages that can amplify inflammatory response through the secretion of growth factors and cytokines. Besides, a group of cytokines are released from stimulated T cells and contribute to the inflammatory response of atherosclerotic tissue.62–64 A variety of inflammatory factors exhibit the potential to facilitate the occurrence and progression of AS, such as tumor necrosis factor-α (TNF-α), which inhibits the production of nitric oxide synthetase, induces ECs apoptosis, and stimulates the expression of VCAM-1, 65 leading to ECs dysfunction and promoting the formation of AS plague. 66 Exploring the role of these inflammatory factors in inflammatory and immune response is pretty helpful to deeply understand the pathogenesis of AS. Furthermore, to elaborate the interaction between inflammatory factors and various pathogenic factors might provide new perspectives to the clinical therapy of AS.
TNF-α and interleukin-1β (IL-1β) are applied extensively in the existing in vitro models of AS with inflammatory factors' incorporation owing to their significant impact on vascular cell morphology and function, as well as strong association with atherosclerotic pathophysiology. Venugopal et al. evaluated TNF-α-triggered endothelial activation in a microfluidic chip that consists of two channels separated by a semipermeable membrane, which are used to support cell growth and activate ECs, respectively. 23 The results showed that the adhesion of leukocytes to ECs exhibited dose dependency on TNF-α. Although this model can be observed in real time, it is still limited in fully simulating atherosclerotic microenvironment and studying complex processes such as cell–matrix interaction and foam cell formation due to its lack of other kinds of vascular cells. Along similar lines, Poussin et al. constructed a tubular vascular model by perfusing hydrogel mixed with ECs into a microfluidic chip, observing that higher ICAM-1 level was expressed and more monocytes were recruited in the model after TNF-α treatment (Supplementary Fig. S1). 24
In the model to study pathological effect of ox-LDL described above, Zhang et al. found that both enzyme-modified LDL and TNF-α caused elevated expression levels of TNF-α and IL-1β, and the two pathological factors showed synergistic effect (Fig. 4E). 21 The model is potential to present the fact that pretreatment with lovastatin or the P2Y11 inhibitor NF157 reduces monocyte accumulation and blocks foam cell formation, which can identify the role of drugs on specific vascular functions that cannot be assessed in vivo. 21 Moreover, perfusion with blood leads to increased monocyte adhesion in the model, supporting the role of this AS model in simulation of AS progression. 21 Since the static coculture system lacks the tubular structure of the artery, Robert et al. used human-derived vascular cells to develop a three-dimensional arterial model that mimics the structural and functional characteristics of natural arteries (Supplementary Fig. S1). 43

Construction of some shear stress platforms. Reproduced with permission.
This model was used to study the initial events of AS, including accumulation of LDL and high-density lipoprotein in the intima, as well as binding and transport of monocytes under dynamic pulsatile flow, demonstrating that monocytes' adhesion was remarkably augmented followed by TNF-α or LDL stimulation. 43 Vasudevan et al. developed a coculture system to study macrophage activation and its adhesion to VSMCs. Their findings showed that after M-CSF induction, the adhesion and aggregation of macrophages on VSMCs increased dramatically, and the apoptosis rate of VSMCs can reach 60%. 22
A plethora of inflammatory factors are participated in the entire pathological progress of AS, whereas there is only one or two kinds of inflammatory factors incorporated into the current in vitro AS models, leading to the fact that the combined effects of two or more inflammatory factors have not been fully explored. Furthermore, there exist many other factors that are able to interact with inflammatory factors, such as Ox-LDL discussed above, enhancing their respective pathological effect and finally promoting the development of AS, which remain crucial knowledge gaps that require to be filled.
Oxidative stress
Oxidative stress is mainly triggered by the abundant generation of reactive oxygen species (ROS) and reactive nitrogen species, which can cause long-term imbalance between the production of highly active reaction molecules and antioxidant effect, resulting in vascular endothelial remodeling, tissue damage, and the final occurrence of AS. 4 A large number of studies have shown that mitochondrial dysfunction and nicotinamide adenine dinucleotide phosphate oxidase can cause excessive ROS production, leading to impaired EC function, elevated VSMCs proliferation activity and migration ability, increased macrophage-derived foam cells, as well as inflammatory response through multiple pathways, thus accelerating the progression of AS. 31
Existing in vitro models for simulating oxidative stress-derived AS depend heavily on induction by H2O2. For example, Salmon et al. constructed a tissue-engineered vascular model in vitro, which is induced with H2O2 at high concentrations, and studied the effect of ECs' or VSMCs' aging on vascular functionality. The results indicated that the expression level of cell cycle inhibitor p21 in ECs increased after H2O2 treatment, while the expression level of endothelial nitric oxide synthase in ECs decreased, reflecting the evident aging of ECs in the vascular model. 14 Besides, the effect of H2O2 on vascular function is endothelium specific, which is manifested as increased VCAM-1 expression in ECs and weakened endothelial-dependent vasodilation. 14 To explore the relationship between ferroptosis and AS, Hou et al. also used H2O2 to treat ECs in which ferroptosis is caused. 25
Moreover, cell viability, lactate dehydrogenase, oxidized glutathione, malonaldehyde, and lipid ROS levels were significantly increased in H2O2-treated cells, and reduced glutathione and glutathione peroxidase 4 were significantly decreased, indicating that ferroptosis-induced ECs' damage played an indispensable role in the occurrence and progression of AS. 25 However, the underlying molecular mechanism remains to be explored due to lack of functional validation. Although pathological state derived from oxidative stress has been successfully replicated by using H2O2, there still lacks natural approaches to realize oxidative stress in vitro. Actually, existing microsystems are potential to trigger ROS without H2O2 by modulating flow patterns and magnitude of shear that determine the amount of ROS produced by ECs, usually an irregular flow pattern (disturbed or oscillatory) producing higher levels of ROS than a regular flow pattern (steady or pulsatile).67,68
There are few differences between the responses of ECs derived from different individuals to oxidative stress, but the distinct interspecific difference for VSMCs' response to oxidative stress has been recognized. 31 Furthermore, the proliferative capacity of primary VSMCs after isolation is pretty limited despite their potential advantage in studying oxidative stress-derived AS. Therefore, it is extremely difficult for present in vitro models of oxidative stress to accurately imitate pathological process of AS based on oxidative stress in a specific individual.
Hyperglycemia
Since it is well known that there is a strong correlation between hyperglycemia and AS, glycemia is also regarded to be an important pathological factor while constructing in vitro AS models. Hyperglycemia-induced EC dysfunction is clinically considered a hallmark of vascular complications of diabetes.69,70 Under high glycemic level, ECs are able to ingest plenty of LDL, which is transformed to ox-LDL upon the increased level of ROS in vascular tissue due to reduced antioxidant ability of vascular cells undergoing hyperglycemia, finally resulting in the occurrence of initial AS events.71–74
To explore the interaction between ECs and VSMCs under high glycemic level, Zitman-Gal et al. constructed the in vitro coculture model of ECs and VSMCs at high glucose concentration, and found that the genes related to VSMCs' arrest and decelerated cell cycle were upregulated, while the damage response regulators in ECs that promote tissue remodeling were downregulated. 75 Wang et al. used Ox-LDL and high-concentration glucose to treat ECs, respectively, so as to study the relationship between hyperglycemia and oxidative stress, and the results showed that hyperglycemia caused decreased ECs activity, increased cell apoptosis, endothelial–mesenchymal transition and inflammation. Besides, ginsenoside Rb1 exhibited the ability to reduce Ox-LDL level in ECs and glucose-induced ROS production, thereby probably inhibiting AS development based on diabetes mellitus. 26 Similarly, Zheng et al. developed a microfluidic AS model to study the effect of shear stress, glucose, and LDL on ROS production as well as EC function. 11
To mimic hyperglycemic or hyperlipidemic regions susceptible to AS, ECs monolayer in the microfluidic chip are treated with glucose and LDL, respectively, under low fluid shear stress and cyclic stretch, identifying the fact that high concentration of glucose potently induces ROS production by ECs, thereby inducing expression of chemical factors associated with AS, such as TNF-α, growth factors, and vasoactive peptides. The above model is capable of simulating the vascular biomechanical and biochemical microenvironment in vivo partly, thus appropriate for studying AS-related pathological factors and drug screening. 27
During the study on exploring the effect of hyperglycemia on AS initiation and progression, much higher glucose concentrations than the in vivo glycemic concentration in diabetic patients are usually applied because of the purpose to shorten the term of constructing model and achieving significant inflammatory response as soon as possible. Furthermore, the fluctuation and gradual rise of glycemic concentration in vivo has not been completely considered in the current in vitro models simulating hyperglycemia. Therefore, future models that employ glucose concentrations close to glycemic concentrations in diabetic patients and investigate the long-term hyperglycemic effect on AS occurrence would be well encouraged.
Biomechanical Pathological Factors
Matrix stress
Vascular ECM works as an important factor for mediating cell–cell interplay, maintaining structural integrity of blood vessel, regulating cell adhesion, survival, apoptosis, controlling vasodilation, and inflammatory remodeling etc. Recently, ECM, in the form of different hydrogels, has been reported to be potential to regulate vascular morphogenesis and vascular regeneration.76–78 Vascular ECM stiffness potently regulates the integrity of EC monolayer, endothelial permeability, and leukocyte migration.
As the hardness of blood vessel wall gradually changes during the progressive process of AS, vascular ECM stiffness is identified as a pivotal factor linked with the pathological progression with the advancement in the knowledge of atherosclerotic mechanism. Previous studies demonstrate that arterial stiffness in rabbits fed with high-fat diet decreases in early stage, but increases dramatically in later stage, 79 and arterial stiffness in monkeys also increased with age. 80 Moreover, different regions of plaques in apolipoprotein E-deficient mice exhibit varied stiffness that is from a low average of 5.5 kPa in lipid-rich regions to an average of 59 kPa with high values of 250 kPa in areas of hypocellular fibrosis.81,82
Thus, matrix stress is commonly taken into account and evaluated when constructing in vitro AS models.
Dorweiler et al. established an in vitro model of multilayer VSMCs covered by endothelial monolayer, which aims to simulate the structural characteristic of arteries to study the initial events of AS. Although this model was used to study vascular cell–ECM interactions and key events in the development of AS plaques, 17 the effect of ECM on monocytes and macrophages was not explored. Garcia-Sabaté et al. used collagen scaffolds of increasing density to reproduce the different porosity of vascular ECM in early and late AS stage, respectively, thus simulating ECM stiffness during AS process, and concluded that collagen density in plaques may be one of the main factors driving AS progression by regulating monocyte and macrophage behaviors. 31 Zamani et al. compared functional activity of ECs cultured on polyacrylamide hydrogel of physiological hardness (4 kPa) with that on tissue culture plate that simulated pathological stiffness, both of which were treated with fibronectin, due to its favorable adhesion of ECs and identified the role as ECM protein deposited in AS-prone sites, as well as its positive effect on AS development. 32
They found that rigid matrix induces endothelial–mesenchymal transition, while soft matrix close to physiological condition is beneficial for maintaining ECs phenotype, and stiff ECM promotes ECs heterogeneity and phenotypic alteration. 32 The mRNA expression analysis of adhesion molecules in ECs also confirmed the interaction of ECs with two different matrices, strongly suggesting that matrix hardness is one of the main potential regulators of ECs phenotype. 32 Garcia-Sabaté et al. employed monocytes incorporated into collagen hydrogel with different densities to simulate early or late atherosclerotic vascular tissue, studying the effect of ECM on macrophage behaviors. The authors found that both monocytes derived proinflammatory (M1) and anti-inflammatory (M2) macrophages, as well as monocytes cultured on collagen hydrogels with different densities exhibited markedly different responses. 31
The current in vitro AS models usually adopt biomaterials at a couple of specific hardness gradients to study the influence of matrix stress on AS pathological development, which is distinctly different from in vivo microenvironment, where vascular ECM stiffness shows continuous change with AS progression. Moreover, during the process of AS development, blood vessels are first softened due to the attachment of fat, and then harden, which has not been simulated in vitro exactly until now. Notably, dynamic properties of the matrix, such as stress relaxation and matrix remodeling, also influence vessel formation, which not only facilitate future studies of vascularization and tissue engineering applications, but also provide new insight to better replicate atherosclerotic matrix microenvironment in vitro. 76
Shear stress and hemodynamics
Numerous studies have shown that AS plaques do not appear randomly anywhere in the arterial system, but preferentially in specific arterial sites, such as arterial regions with bifurcation, stenosis, and curvature, where disturbed blood flow and low-magnitude oscillatory shear stress exist, resulting in increased permeability and thickening of the local intima. The stability of the blood flow, its velocity and direction, as well as the magnitude of flow shear stress on the vascular cells all vary depending on the geometries of blood vessel. 83 Fluid flow is another major factor in determining the phenotype and behavior of ECs, and its influence is highly dependent on flow characteristics. 84 When ECs are subjected to specific shear stress, their integrity, phenotype, and functional activity may change, leading to endothelial dysfunction that causes pathophysiological alterations of blood vessels.85–87 Therefore, there exists a considerable number of in vitro AS models incorporated with shear stress, which are potential to mimic in vivo mechanical microenvironment and helpful for revealing further mechanism by which AS occurs and progresses (Table 2).
Studies on the Effect of Shear Stress on Atherosclerosis
ICAM-1, intercellular cell adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1.
There are various in vitro models to study how local hemodynamic conditions regulate ECs function, including parallel plate flow chamber, vertical stepped flow chamber, cone plate viscometer, and microfluidic device (Fig. 4).21,33,41,45–47,88 Using cone plate viscometer system that opened up a new field in the mechanobiology of ECs, it was demonstrated for the first time that shear stress can modulate the shape and orientation of ECs as well as complex biological functions, such as wound repair and platelet–endothelium interactions. 34 Blackman et al. presented a novel in vitro cell shearing device based on the principles of a cone and plate viscometer that utilizes microstepper motor technology to control independently the dynamic and steady components of a hydrodynamic shear-stress environment. 35 The device that is also integrated with fluorescence microscopy system for real-time cell response monitoring has been used to replicate blood flow patterns that cause AS in vivo to observe how ECs respond to disease-related shear stress. 36
To accurately control the input concentration of reagents over time and observe the interaction between leukocytes and ECs dynamically, Schaff et al. developed a microfluidic chamber, by which they examined the relationship between hydrodynamics and neutrophil recruitment. 39 Fearon et al. constructed a microfluidic perfusion system where a series of microslides are connected to a computer-controlled pump, and the medium flows through the microslides in both directions, resulting in proatherogenic flow patterns. 41 Chen et al. exposed ECs to low shear stress for 30 min to evaluate their oxidative stress status and functional changes, and found that low shear stress can lead to oxidative stress and EC's dysfunction, including reduced NO levels and upregulated endothelin-1 (ET-1), and ultimately leading to AS plaque formation. 42 In addition, Liu et al. found that low shear stress significantly inhibited autophagy of ECs compared with physiological shear stress, which involved mitochondrial homeostasis damage and endothelial barrier dysfunction, resulting in initial events of AS. 89
Microfluidic chips display significant advantages over traditional culture systems that are utilized to explore the effects of shear stress, such as enhanced sensitivity, dynamic monitoring, combining of both flow and media supply. Patibandla et al. developed a new microfluidic EC culture model that can not only provide mechanical conditions and flow patterns similar to those in vivo, but also can be observed by microscopy in real time, demonstrating that low shear stress is able to induce ECs dysfunction. 45 Recently, 3D printing technology has been broadly applied in constructing in vitro physiological or pathological vascular models with its increasing advancement. Gao et al. developed a three-layer artery model with adjustable geometries based on a novel 3D in-bath coaxial cell printing technology, which was used to study the landmark events in the early stages of AS, and evaluate the therapeutic effect of atorvastatin, as well as signaling mechanisms of foam cells' formation regulated by coculture and turbulent flow conditionings. 46
Their results indicate that atorvastatin could inhibit foam cell formation in an obvious dose-dependent manner, which supports the excellent potential of this in vitro model for improving AS research and revolutionizing the drug screening process. 46
Menon et al. employed 3D printing technology to fabricate narrow microchannels with different shrinkage geometries, investigating disturbed flow under different degrees of occlusion and wall shear stress. Experimental results and fluid simulation confirmed the presence of pathological shear stress in the stenotic area and recirculation flow after stenosis, which induces proinflammatory response of ECs, including increased platelet adhesion in stenotic region and abnormal leukocyte–EC interactions after stenosis. 92
Although diverse dynamic culture systems mentioned above provide numerous approaches to exploring shear stress alteration-based AS occurrence, it should be noticed that the magnitudes and patterns of shear stress in in vitro models differ greatly from those in vivo due to disparity in physical and chemical properties between culture medium and blood. Moreover, shear stress levels in blood vessels vary obviously in different locations due to different hemodynamic microenvironment in different organs, probably leading to the propensity of atherosclerotic initiation in some organs, which remains a crucial area of research to be deeply unveiled.
Tensile stress
Tensile stress, which is the ratio of circumferential wall tensile force to wall thickness, is another significant biomechanical effect that vascular tissue experiences during the periodic contraction and relaxation of blood vessels. 93 Studies have shown that the tensile force increases as blood pressure goes up, facilitating the passage of macromolecules and resulting in increased intimal accumulation of LDL, which in turn promotes the occurrence of AS.94–96
The two-dimensional flexible substrate for cell support that is often composed of deformable materials, such as polydimethylsiloxane, is mainly integrated into the current in vitro models that focus on the influence of tensile force on the AS process, and the tensile force was adjusted by external devices. Gu et al. constructed an in vitro coculture system in a stretchable microfluidic device, where the elastic membrane is able to deliver uneven strain to VSMCs, ECs, and monocytes attached to ECs, studying the role of different types of cells played in the foam cell development, as well as the activities of anti-AS drugs. 18 The results demonstrated foam cell formation and altered expression of some related receptors and enzymes under LDL and stretching treatment, as well as significant inhibitory effect of atorvastatin on foam cell formation in a dose-dependent pattern. 18
Meza and others designed the devices that can simultaneously apply precise dynamic shear stress and cyclic tensile strain to ECs, achieving the result that ECs respond morphologically to combined stimulation of fluid shear stress and cyclic tensile strain differently compared with a single stimulation, including cell elongation, alignment, and actin fiber accumulation. 50
Yang et al. applied cyclic tensile strains at three levels to VSMCs and reported significantly enhanced VSMC proliferation activity and migration capability, as well as reduced apoptosis and upregulated miR-187-3p upon tensile strain treatment, indicating that miR-187-3p might be an important player involved in AS process. 51 To better characterize the response of vascular cells to different biomechanical stimuli, finite element simulation is usually applied to predict the mechanical state before in vitro experimental studies. For instance, Chu et al. developed a shearing–stretching device to mimic blood vessel, in which COMSOL simulation for different combinations of stretch and flow conditions was performed and then the effect of cyclic stretch and fluid shear stress on bovine aortic ECs was observed under microfluidics, including cytoskeleton proteins in EC's alignment with the fluid flow direction and paxillin redistribution to the cell periphery or the end of stress fibers. 97
Despite the great progress in identifying the substantial role that tensile stress has played in the entire pathological process of AS, the current in vitro models that are participated in the tensile strain-related vascular pathological studies still require modification in terms of the magnitude, frequency, and duration time, owing to the reality that stretch force of physiological blood vessel in vivo fluctuate with time and other stimuli. In addition, the potential molecular mechanism underlying tensile stress at different levels that regulate AS occurrence and progression remain to be elucidated due to the fact that the signaling molecules implicated in tensile stress regulated vascular cells' phonotype and function have not been fully identified. Furthermore, current in vitro models often ignore the interplay between tensile strain and other two kinds of biomechanical factors and their combined contribution to AS progress, thus the models accommodating three biomechanical factors are highly to be recommended.
Conclusions and Outlook
The in vitro AS models summarized above performed well in studying the influence of each pathological factor on AS and related molecular mechanisms, as well as drug screening for AS, but some extra points have to be taken into account while developing advanced in vitro AS-related models in future.
First, structural characteristics of in vitro models should be further upgraded to mimic the vascular physiological structure. For instance, the adventitia of arterial wall that also plays a critical role in vascular functionality has usually been ignored in previous studies. Meanwhile, the ECM components and properties that enormously determine mechanical environment in blood vessel deserve to be further studied. We believe the exact role played by each pathological factor mentioned above in AS process would be recognized better based on vascular models with physiological structure due to its complex cell–cell interactions and cell–ECM interactions. Second, AS is a chronic and multifactor-mediated complicated disease, thus the current pathological models still have large improvement space with regard to pathological factor combination, concentrations, and duration.
Especially, the interplay between biochemical factors and biomechanical factors during AS progression has been little known, which is not only helpful in uncovering novel mechanism underlying AS, but also potential in offering a new therapy target for AS. Lastly, most of the current in vitro models are focused on reproducing the initial events of AS due to the difficulty in simulation of the physiological microenvironment and long-term maintenance culture, thereby severely lacking the in vitro atherosclerotic models for mid and late stage. Although all the pathological factors are kept at dynamic levels during the whole process of AS, it is still practical to construct AS models for mid and late stage by employing the pathological factors at static levels for long term to simulate the natural AS progression, as long as the pathological state and each pathological factor could be monitored dynamically.
Additionally, most studies on applications of existing in vitro AS models are confined to verification of pharmacological effect of statin drugs, but the practical drug screening on active compounds as well as other applications such as simulation of AS progression, and evaluation of new treatment methods are pretty limited.
Overall, we think the in vitro models that can reflect the pathological state dynamically at each stage of AS based on the perfect simulation of physiological vascular structure, and intensive study on the pathological effect of multiple factors related to AS, as well as the implicated regulatory mechanism would be the best AS models in future, which might be highly beneficial for defining the pathogenesis of AS thoroughly and developing effective therapeutic approaches to clinical treatment.
Footnotes
Authors' Contributions
Y.B. and N.L. conceptualized and wrote this review article; other authors contributed to the critical revision of article drafts.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Natural Science Foundation of Liaoning Province 2021-YGJC-01 (to N.L.), and the Fundamental Research Funds for the Central Universities DUT22YG211 (to N.L.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.