
Abstract
Photoaged skin features an appearance of premature aging induced by external factors, mainly ultraviolet (UV) irradiation. Visible aging signs and increased susceptibility to skin-related diseases triggered by UV irradiation have raised widespread concern. As a critical component of human skin, the extracellular matrix (ECM) provides essential structural, mechanical, and functional support to the tissue. Consequently, UV-induced ECM deterioration is a major contributor to photoaging. This review begins by analyzing the structural and functional changes between healthy and photoaged skin in prominent ECM components, including collagens, glycosaminoglycans (GAGs), proteoglycans, basement membrane proteins, and elastic fibers. Furthermore, we explore the key mechanisms driving ECM deterioration in response to UV irradiation, focusing on mitogen-activated protein kinase/matrix metalloproteinase and transforming growth factor-β/Smad signaling pathways, as well as the synthesis and degradation of GAGs. A comprehensive understanding of these changes and underlying mechanisms is crucial for elucidating the biological influence of UV on the ECM, ultimately providing more reliable evidence for the prevention and treatment of skin photoaging.
Impact Statement
The extracellular matrix (ECM), a fundamental component of the skin, experiences substantial changes after exposure to chronic ultraviolet (UV) irradiation, leading to the deterioration of the skin’s structure, function, and appearance. In addition to investigating the structural and functional differences in the ECM between healthy and photoaged skin, including collagen, glycosaminoglycans, proteoglycans, basement membrane proteins, and elastic fiber, this review analyses the mechanisms by which UV irradiation induces these changes. By targeting different molecules implicated in these cellular processes, it becomes feasible to rejuvenate the skin and augment its resistance to diseases.
Introduction
Human skin, the largest organ in the body, serves multiple functions, including acting as a physical barrier against the environment. 1 It is divided into three layers: the epidermis, dermis, and subcutaneous fat. 2 The epidermis comprises densely packed keratinocytes that form a stratified squamous epithelium, providing a protective barrier against environmental assaults. The dermis, a collagen-rich connective tissue, provides mechanical strength, resiliency, nourishment, and an environment that supports the dermal cells. 3 It consists mainly of fibroblasts and various extracellular matrix (ECM) components, including collagens, glycosaminoglycans (GAGs), proteoglycans (PGs), basement membrane (BM) proteins, and elastic fibers. The epidermis and dermis are separated by the BM at the dermal–epidermal junction (DEJ), a widely distributed, specialized, thin, dense, sheet-like structure primarily composed of collagen IV and laminin (LM). 4 Beneath the dermis lies the subcutaneous tissue, composed of adipocytes (fat cells) within a connective tissue framework.
Skin aging is a structural and functional degradation process driven by intrinsic and extrinsic factors. Intrinsic aging, which is genetically determined, occurs inevitably due to natural physiological changes over time. 5 In contrast, extrinsic aging is triggered by environmental factors, with ultraviolet (UV) irradiation being the most predominant.1,6 Skin exposed to UV irradiation gradually exhibits a prematurely aged appearance characterized by deep and coarse wrinkles, mottled pigmentation, sallow color, skin laxity, rough texture, dilated blood vessels, compromised wound healing, and both benign and malignant proliferation.7,8 The premature skin has been ascribed to UV-induced remodeling of the compositional architecture of the ECM, which directly or indirectly correlates with prevalent diseases associated with the aging process.3,9,10
The ECM is a highly organized and dynamic three-dimensional network of macromolecules that not only provides a structural scaffold, supporting the growth, survival, and function of cellular components through anchorage dependence but also acts as a reservoir and presenter of growth factors and cytokines, enabling the intracellular transmission of mechanical and biological signals.11–13 Additionally, the ECM contributes tensile strength, maintaining the structural integrity of the cellular framework. 14 Under normal conditions, skin ECM is in a state of homeostasis, balancing its structural and functional roles. 15 However, UV irradiation disrupts this balance, altering skin cell functions and responses, which leads to evident proteostasis loss.16,17 In particular, certain long-lived ECM macromolecular assemblies in the dermis are considered to progressively accumulate photoaging-induced damage over time, ultimately contributing to the photoaging phenotype. 18 Recent studies employing structural and proteomic analyses have identified novel ECM biomarkers in both the epidermis and dermis that undergo modifications as a result of photoaging. These findings offer fresh insights into the roles of ECM components in skin aging and deepen our understanding of the underlying mechanisms. 18 This review focuses on clarifying the differences in ECM between young and photoaged skin, as well as the mechanisms driving these changes.
ECM in Young and Photoaged Skin
In skin tissue, two distinct types of ECM can be distinguished based on their location and composition: the interstitial dermal matrix and the BM. 19 As shown in Figure 1, the ECM forms an intricate and structurally stable network composed of various multidomain macromolecules arranged in a cell- or tissue-specific manner. 13 However, UV irradiation can compromise this matrix by disrupting the higher-order structure of ECM proteins, damaging macromolecular ultrastructure, and altering overall ECM architecture.20–22 These UV-induced changes affect the quantity, quality, arrangement, and function of ECM constituents, ultimately contributing to skin photoaging.
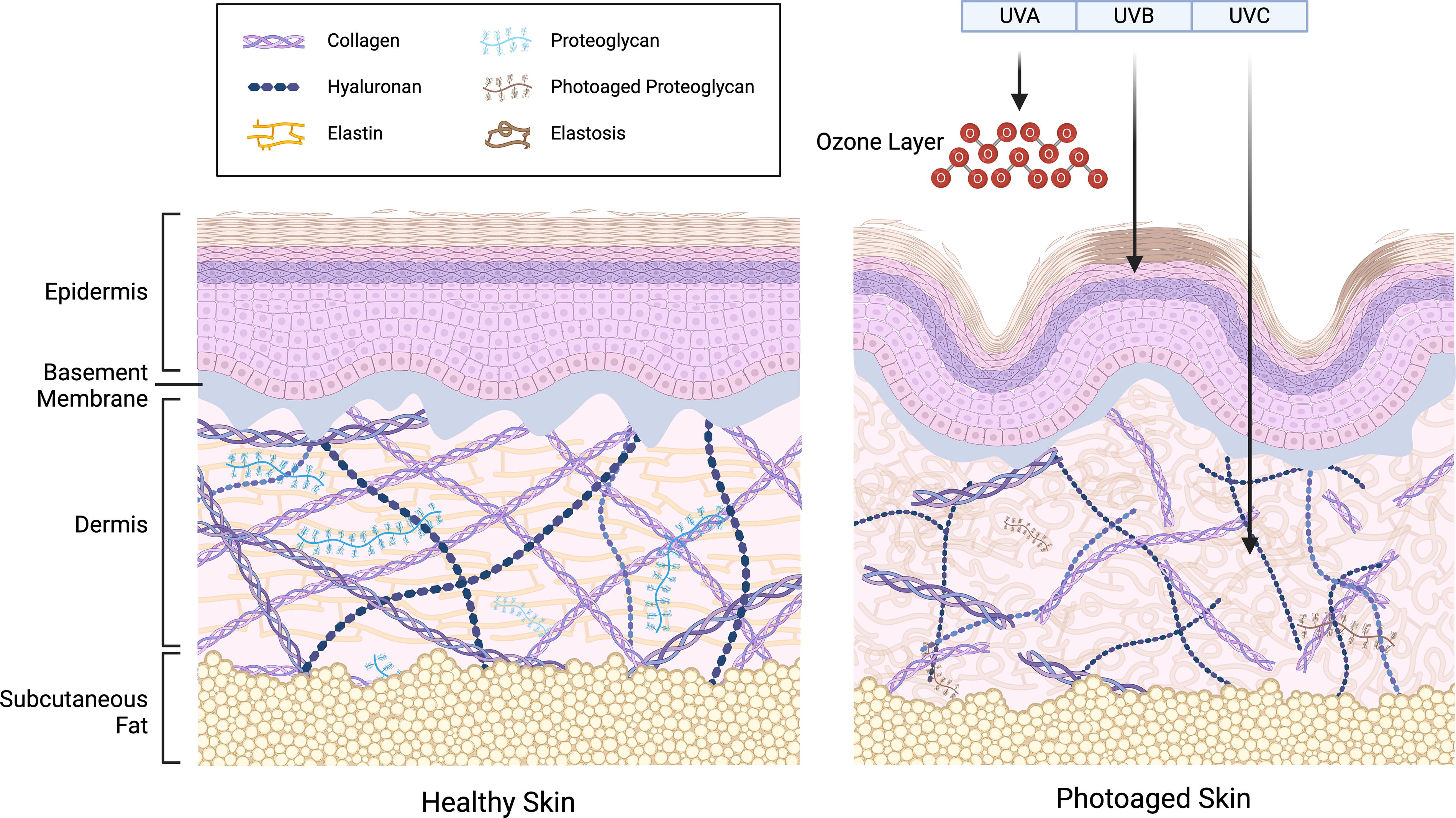
Healthy skin and photoaged skin.
Collagens
Collagens dominate the ECM of the skin, accounting for ∼30% of its dry weight, which is primarily synthesized by fibroblasts in the dermis. The collagen superfamily consists of 28 members denoted as I–XXVIII, with detailed information provided in Table 1. Different types of collagens are present in various tissues; specifically, types I, III, IV, V, VI, VII, VIII, XII, XIII, XIV, XV, XVI, XVII, and XIX collagen are found in the skin.23,24
Classification and Molecular Species of Collagens
In healthy and youthful skin, collagen is plentiful, tightly packed, and well-organized, providing tensile strength and resistance to deformation. 25 Type I collagen (COL1) exhibits an antiparallel fibril alignment in the reticular dermis, producing dense fibers. 26 Type III collagen (COL3) is primarily located in the mesh-like papillary dermis, often coexisting with COL1. 26 Nearly 95% of the skin’s structure is composed of COL1 and COL3, which enhance the elasticity, firmness, and resilience of the skin by forming massive extracellular fibers within the dermis. 14 Network-forming type IV collagen (COL4), representing the principal structural components of the BM, constitutes less than 5% of the skin’s collagen and will be further discussed in the subsequent section. 27 Type V collagen (COL5), a minor fibrillary collagen found alongside COL1 and COL3, plays a significant role in tissue formation. 23 Both COL5 and type VI collagen (COL6) are crucial minor collagens that stabilize the dermal matrix, particularly in the papillary dermis, by acting as bridging molecules to bind COL1. 28 Additionally, COL6 and type XXIX collagen (COL29) are involved in microfibril formation. 23 Type VII collagen (COL7) is crucial for maintaining skin integrity by anchoring COL1 in the dermis to the BM at the DEJ as part of the anchoring fibrils.29,30 Type VIII collagen (COL8) plays a role in establishing collagen network systems. 23 Last, type XII collagen (COL12) is a homotrimer with short collagenous domains that bind to COL1-containing fibrils and flexible arms that attach to noncollagenous (NC1) matrix components, crucial for ECM contraction and remodeling. 24
The primary alterations observed in photoaged skin predominantly involve structural and quantitative changes in collagen fibers. In photoaged skin, collagen fibers become fragmented and coarsely disorganized due to aberrant collagen homeostasis, impairing the skin’s structural integrity and mechanical properties and rendering the tissue microenvironment susceptible to skin disorders.25,31 The length and width of collagen fibers both diminish; however, the reduction in width is less pronounced than the decrease in length. 32 Additionally, the surface of collagen fibrils becomes rougher than in healthy skin, and the collagen bundles demonstrate increased stiffness and hardness. 31 Quantitatively, collagen in photoaged skin exhibits decreased biosynthesis and increased fragmentation mediated by proteases, leading to an overall reduction in collagen content, even though dermal fibroblasts continue replenishing collagen at a diminishing rate. 26 This fragmentation is further intensified by collagen denaturation and disruption of its higher-order structure due to direct UV irradiation and reactive oxygen species (ROS) produced photodynamically. 20 Recent studies suggest that two clusters of UV-sensitive proteins involved in collagen fibril assembly in the ECM and protein translation and motor activity may serve as crucial targets in UV-exposed tissues. 33 Moreover, the decline in collagen levels inhibits the mechanical interplay between fibroblasts and the ECM, impairing fibroblast attachment and resulting in smaller fibroblast size, decreased elongation, and collapsed morphology. 34 The deterioration in fibroblast function further contributes to decreased dermal collagen. 34 Clinically, these alterations manifest as wrinkles and diminished elasticity, observable in both intrinsically aged and photoaged skin. 30
GAGs and PGs
PGs are composed of core proteins covalently linked to GAG chains. Table 2 shows six types of GAGs: chondroitin sulfate (CS), dermatan sulfate (DS), hyaluronic acid (HA), heparin (HP), keratan sulfate (KS), and heparan sulfate (HS). Except for HA, which is synthesized at the plasma membrane and lacks sulfation, other GAGs are produced in the Golgi apparatus and contain sulfate substituents that covalently attach to core proteins via O-glycosidic bonds.35–38
Molecular Composition of Glycosaminoglycans
A given ECM comprises multiple PGs, each differing in quality, length, and composition. 39 In the dermis, PGs and GAGs form an amorphous network that envelops and integrates with the fibrous and cellular matrix elements of the dermis, constituting ∼0.2% of its dry weight. 34 The most abundant PGs in the skin include versican, decorin, and biglycan, all of which belong to CS/DS-PGs. 35 Heparan sulfate proteoglycans (HSPGs) are commonly found on the surfaces of dermal and epidermal cells and within the ECM, including the BM. Most cutaneous HA is found in the dermis at a concentration of 0.5 mg/kg, and its flexibility and hydrophilic nature allow it to fill gaps within the ECM. 40 Since the sulfate residues on the disaccharide units of GAGs enable PGs to bind various positively charged molecules, GAGs are pivotal in numerous biological processes by interacting with proteins like cytokines, growth factors, and collagen.41,42 Their high negative charge also helps maintain the skin’s water content. 38 Despite representing a minor proportion of the ECM, PGs and GAGs exhibit a remarkable capacity to absorb water—up to 1000 times their volume—and play vital roles in regulating the water-binding capacity and compressibility of the dermis, thus supporting tissue structure and facilitating cell migration.34,37
Previous studies have shown inconsistent trends in the quantity of GAGs and PGs after UV irradiation, mainly due to differences in UV doses and analysis times. 43 Some research indicates an elevation in total sulfated GAGs (tsGAGs), while tsGAGs notably decrease in the dermis during intrinsic aging. 44 These photoaging-dependent elevations of tsGAGs occur primarily in the dermis rather than the epidermis. 38 Regarding HA, photoaged and intrinsically aged skin experience a reduction in epidermal HA, while dermal HA content in photoaged skin significantly increases compared with intrinsically aged skin.35,45 Additionally, the molecular size of HA in photoaged skin is reduced. 35 Mean values of tissue water content increase partly due to the rise in HA, a critical factor in influencing dermal water content. 38
BM Proteins
In the skin, as illustrated in Figure 2, the BM beneath the basal keratinocytes separates the epidermis from the dermis while simultaneously anchoring the two tissues together. The BM framework consists of two distinct networks: polymerized LMs and cross-linked COL4, interconnected and stabilized by the glycoprotein nidogen and HSPGs (mainly perlecan and agrin).46,47 The deposition of the COL4 network below the LM network is crucial for preserving cell–BM interactions and ensuring mechanical stability. 48 The BM functions as a barrier and anchoring substrate for cells, a filter of large molecules, and a reservoir for growth factors, owing to its numerous HP-binding sites. 49 Additionally, the BM determines epidermal polarity and is a barrier against epidermal cell migration. 50
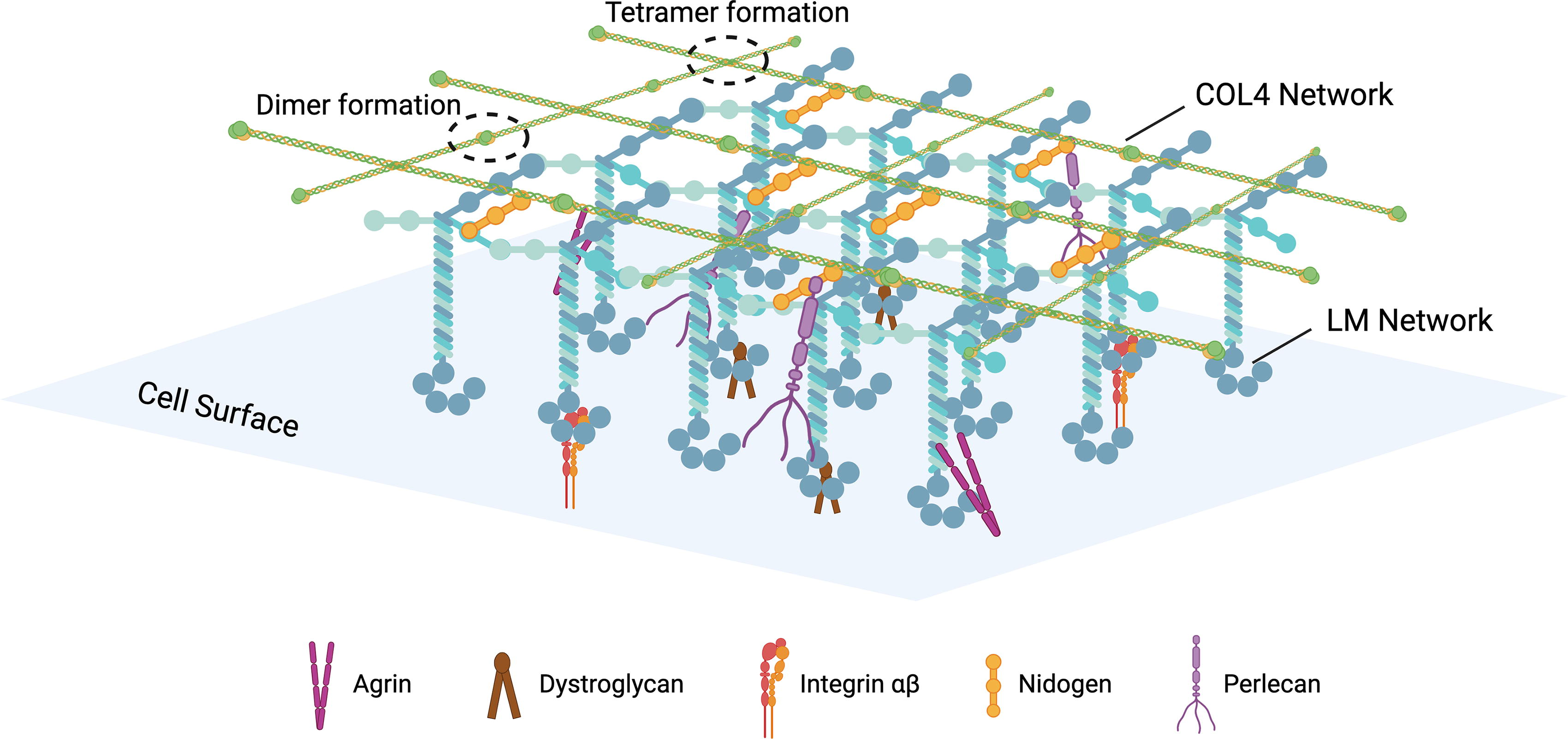
The basement membrane.
The principal collagen component of the BM is COL4, with additional contributions from type XV and XVIII collagen. 51 COL4 family comprises six genetically distinct yet highly homologous α-chains, which assemble into three types of triple helical structures through various chain combinations, with the α1α1α2 COL4 being predominantly identified in DEJ BMs.23,52 Each COL4 molecule features an N-terminal 7S domain, a Gly-X-Y repeat sequence capable of forming a collagenous triple helix, and a C-terminal NC1 domain. 52 COL4 protomers pair at their C-terminal NC1 domains and group into hexametric networks at their N-terminal 7S domains.27,46 COL4 binds to soluble glycoproteins, PGs, and growth factors in a tissue-specific manner. Consequently, the COL4 network provides a scaffold for attaching other proteins within the BM and is chiefly accountable for its mechanical properties.27,46
LMs, consisting of α, β, and γ chains, are heterotrimeric glycoproteins. At the distal end of the α chain, five laminin G-like domains are present, which contain the major cell-adhesive sites of LM. The human genome encodes 11 LM chains, with α3, α5, β1, β3, γ1, and γ2 present in the BM, where they assemble into three primary isoforms—LM311 (α3β1γ1), LM332 (α3β3γ2), and LM511 (α5β1γ1)—with LM332 and LM511 serving as major components in the DEJ BM.49,53–55 Each LM chain features N-terminal globular domains, and interactions between these domains are essential for forming the LM network, a key initiator in BM assembly at the basolateral surface of cells, crucial for BM formation.46,56 Specifically, LM511 forms an extensive polymer network interconnected by nidogen and perlecan, which attaches to the COL4 scaffold. 49 LM332 uniquely binds to COL7 and COL17, creating a complex structure in the BM, which physically extends from basal keratinocyte cytoskeleton to papillary dermal collagen fibrils. 46 The integration of LM into the BM also involves cross-linking between LM332 and LM311. 57
Ubiquitous in the BM, nidogens are glycoproteins characterized by three globular-like domains (G1–G3) connected by link-like and rod-like segments, which exist in two isoforms: nidogen-1 and nidogen-2. 58 Nidogen-1, along with the less prevalent nidogen-2, binds to LM via the γ-subunit short arm, serving as a bridge between the LM network to COL4 network.59,60 Moreover, both isoforms interact with perlecan, COL1, and COL4, while only nidogen-1 binds to fibulins. 58 Additionally, integrin binding requires the LG1-3 region in LM, with a3β1, a6β1, a7β1, and a6β4 identified as the primary LM-binding integrins. 61 Core components of the BM—LM, COL4, perlecan, and nidogen—regulate cell adhesion, migration, polarization, proliferation, and differentiation by directly or indirectly binding to cell surface receptors, including dystroglycan, integrins, receptor tyrosine kinases (RTKs), and discoidin domain receptor 1. 58
The BM undergoes repetitive damage in sun-exposed skin, exhibiting multilayering and partial disruption, which results in both epidermal and dermal deterioration, accelerating skin aging.50,62 In youthful skin, the undulating BM at the DEJ forms rete ridges, which extend into the dermis to project dermal papillae, increasing surface area and enhancing adhesion between the epidermis and dermis. 63 However, in photoaged skin, this corrugated structure becomes flattened. 50 Additionally, compared with young skin, all BM proteins are less abundant in photoaged skin. 32 The decrease of COL17, a hemidesmosome component, potentially results in hemidesmosome instability and competition among epidermal stem cells. 64 Additionally, the decrease in LM511 reduces the abundance of MCSP-positive and K15-positive epidermal stem/progenitor cells within the epidermis. 65
Elastic Fiber
Elastic fibers correspond to only 2% of total dermal protein, consisting of an insoluble inner core of elastin surrounded by a mantle of fibrillin-rich microfibrils.66,67 Elastin consists of monomeric precursor tropoelastin units that are cross-linked at lysine residues to form a stable and insoluble biopolymer.66,68 Because of its extensive cross-linking, elastin is highly durable and serves as a metabolically stable component throughout the human lifespan. 69 Fibrillin, an ECM glycoprotein, includes three fibrillin isoforms in humans—fibrillin-1, -2, and -3—that assemble into microfibrils and provide structural support and elasticity to connective tissues.68,70 In addition to representing the core scaffolds for elastic fiber formation, fibrillin microfibrils also decorate the surface of elastic fibers and form an independent network. 71 Three members of the fibulin family (fibulin-1, -2, and -5) are also located at the interfaces of elastic fibers. 67 Elastic fibers are essential for skin compliance (the ability to deform easily) and resilience (the ability to recoil), collectively known as skin elasticity, allowing the skin to undergo extensive deformation and passive recoil without energy input.34,67
In healthy and youthful skin, elastic fibers intersect the BM at the DEJ, exhibiting a distinctive and highly ordered architecture, featuring microfibrils arranged perpendicularly in the papillary dermis and large-diameter elastic fibers in the reticular dermis.34,71 In mature elastic fibers, GAGs are located alongside the amorphous elastin core, where they play multiple roles in maintaining the structural and functional integrity of the fibers. 69
UV irradiation directly contributes to the loss of cutaneous elasticity, inducing structural alterations within elastic fibers. 66 Photoaged skin exhibits an accumulation of abnormal elastic fibers, a condition known as solar elastosis. 30 Solar elastosis occurs through a cycle of elastic fiber degradation, subsequent ECM production, and reconstitution into an organization distinct from the original structure. 66 The extent of solar elastosis reflects cumulative sun exposure, reflecting the degree of ECM degradation. 72 In terms of the mechanism of ECM degradation caused by UV irradiation, skin photoaging has been demonstrated to increase the enzymatic susceptibility of elastin. 68 Photoaged skin also exhibits remodeling and depletion of fibrillin microfibrils in the papillary dermis, as UV irradiation increases the proteolytic susceptibility of specific regions within the fibrillin-1 protein structure.71,73,74 Moreover, UV irradiation alters the colocalization of fibulin-2 and fibulin-5 on elastic fibers and affects the structural integrity of fibulin-1. 18
Mechanisms
The MAPK/MMP signaling pathway
Mitogen-activated protein kinases (MAPKs) cascades are pivotal signaling pathways downstream of receptors/sensors that detect endogenous or exogenous stimuli. In Figure 3, UV irradiation influences the expression of various components of the MAPK pathway.

The MAPK/MMP signaling pathway. Mammalian MAPK family are serine-threonine protein kinases, comprising of ERK1/2, ERK5 (also known as BMK1 or MAPK7), JNK1/2/3, and p38 (including p38α, p38β, p38γ, and p38δ). Each of these kinases functions as a central module in one of the four distinct MAPK signaling cascades, transmitting extracellular signals including UV to intracellular targets. 75 Typical MAPK cascades consist of three layers of protein kinases, the topmost kinases, MAPKKKs, respond to the stimulus and then mediate the phosphorylation and activation of MAPKKs, which in turn phosphorylate and activate MAPKs to regulate various substrate proteins. MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase.
UV-induced MAPK synthesis
G-proteins, categorized into heterotrimeric G-proteins and small GTPases (Ras, Rho, Rab, Arf, and Ran), serve as molecular switches in signal transduction by modulating signals through the GTP/GDP cycle.76,77 UV irradiation activates G protein-coupled receptors (GPCRs), leading to the dissociation of active Gβγ subunits from Gα subunit, which sequentially activates the epidermal growth factor receptor (EGFR). 78 The primary mechanism of EGFR transactivation involves GPCR-mediated pathways that engage intermediaries such as Sarcoma kinases, Ca2+, PKC, PKA, Pyk2, and protein tyrosine kinases.79,80 These pathways stimulate members of the A Disintegrin and Metalloproteinase (ADAM) family of metalloproteinases to cleave pro-HP-binding EGF (pro-HB-EGF), producing the mature ligand that activates EGFR. 81 RTKs, including EGFR, are cell surface proteins characterized by an N-terminal extracellular ligand-binding domain and a C-terminal intracellular tyrosine kinase domain. Typically, RTKs exist as inactive monomers and dimerize upon ligand binding. 82 At the plasma membrane, activated RTKs induce Ras activation by recruiting GRB2/SOS complexes, with Kinase Suppressor of Ras (KSR) serving as an Extracellular Signal-Regulated Kinase (ERK) scaffold to enhance Ras-dependent activation of the ERK signaling cascade. 82 Beyond the plasma membrane, activated RTKs can also recruit these complexes at focal adhesions, endosomes, and the Golgi apparatus. 82 Subsequently, Ras recruits Raf to the membrane, facilitating Raf dimerization and subsequent phosphorylation and activation of MEK1/2, ultimately leading to the activation of ERK1/2.83,84
Low ROS concentrations are constantly produced in vivo, participating in various physiological processes. As ROS level increases, oxidative stress occurs, which is believed to be a significant driver of the aging process. UV irradiation of varying wavelengths generates different types of ROS: Ultraviolet A (UVA) mainly produces singlet oxygen (1O2) through photosensitive reactions with endogenous chromophores, such as porphyrin and riboflavin, which also create superoxide anion radicals (•O2−) via Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase activation and photosensitization of advanced glycation end products. Ultraviolet B (UVB) stimulates the production of •O2− by activating NADPH oxidase and respiratory chain reactions. 85 ROS-dependent inactivation of protein tyrosine phosphatase leads to the continuous phosphorylation of EGFR. 85 Additionally, apoptosis signal-regulating kinase 1, an upstream activator of p38 and JNK, is activated. 86 ROS signaling orchestrates the assembly of a scaffolding complex dependent on mitophagy and p62, amplifying the MEKK/MEK5/ERK5 signaling pathway. 87 Moreover, ROS triggers tumor necrosis factor receptor-associated factor 6 (TRAF6), a ubiquitin E3 ligase, autoubiquitination that facilitates recruitment of TAB2 and its binding to TAK1, which, in turn, enables the direct TRAF6–TAK1 interaction and promotes TAK1 ubiquitination. 88
UV irradiation exposure to chromophore tryptophan leads to the formation of 6-formylindolo[3,2-b] carbazole (FICZ), a high-affinity aryl hydrocarbon receptor (AhR) ligand, which is translocated to the nucleus and the cell membrane by activating the cytoplasmatic AhR. 89 DNA damage caused by UV irradiation is enhanced by AhR via suppression of NER, which is sensed by ATM kinase then subsequently activates p38 and JNK. 90 The FICZ–AhR signaling pathway activates the MEK/ERK signaling cascade and is involved in the EGFR and ERK1/2 pathways.91,92
Increased MMP initiated by MAPK
Matrix metalloproteinases (MMPs) belong to a family of ubiquitous endopeptidases capable of degrading ECM proteins. 93 MMPs can be classified into five primary subgroups, as detailed in Table 3. There is a high degree of similarity among the elements in the promoters of some MMPs, including AP-1 and SP-1.90,96
Classification and Function of Human MMPs in Photoaging
MMPs can be classified into five primary subgroups owing to their linear sequence similarity, domain organization, and substrate specificity.94,95
MMP, matrix metalloproteinase.
UV irradiation-induced MAPK activation plays a crucial role in upregulating the expression of MMP-1, MMP-2, MMP-3, MMP-9, MMP-11, and MMP-12.90,94 AP-1 protein, composed of Jun family members (c-Jun, JunB, and JunD) and Fos family members (cFos, FosB, Fra-1, and Fra-2), elevates due to UV irradiation since upregulation of p38 and ERK enhances the expression of cFos, whereas increased p38 and JNK expression promotes c-Jun expression. 97 JNK and p38 can phosphorylate SP1, which binds to the promoter regions of MMP-2, MMP-9, and MMP-11 to induce their transcription. 90 Moreover, UV irradiation induces the activation of nuclear factor-kappaB (NF-κB) via the p38/CK2/NF-κB axis, during which CK2 phosphorylates IκBα at multiple C-terminal sites, as well as JNK/NF-κB axis, which translocate to the nucleus upon activation.98,99 NF-κB is responsible for the elevation of MMPs, including MMP-1 and MMP-3. 100
MMPs are regulated by a family of specific endogenous tissue inhibitors of metalloproteinases (TIMPs), consisting of four members: TIMP-1, TIMP-2, TIMP-3, and TIMP-4. 101 These TIMPs inhibit MMP activity by binding to the active sites of MMPs, making the balance between MMPs and TIMPs crucial for the proper remodeling of the ECM.101,102 However, in aged skin, the levels of TIMPs do not increase proportionally to the elevated levels of MMPs. 103 The quantity of TIMP-1 in both photoaged and intrinsically aged skin also decreases. 104 Interestingly, low-dose UV can protect skin by elevating TIMP-2 to withstand MMP-2/9.102,105 This imbalance hastens collagens’ progressive fragmentation, thereby accelerating skin aging.
The TGF-β/Smad signaling pathway
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that regulates fibroblasts, connective tissue, and epithelial cells to produce and remodel ECM as a potent fibrogenic signal. 106 It also suppresses the activity of MMPs while upregulating TIMPs and plasminogen activator inhibitor-1, both of which inhibit MMP activation.25,107
In Figure 4, UV irradiation inhibits the TGF-β signaling pathway by reducing TβRII mRNA expression. This reduction is partially mediated by enhanced protein binding to the 38-bp sequence in the TβRII proximal promoter.109,115 UV irradiation may also lead to the internalization of TβRII, accelerating its degradation. 110 UV irradiation inhibits the phosphorylation and nuclear translocation of Smad2 and Smad3. 110 It also causes gene mutations in crucial elements of the TGF-β pathway, including TGFβRI, TGFβRII, Smad2, and Smad4. 114 Furthermore, as a consequence of progressive degradation of ECM, the adherence of fibroblasts is compromised, leading to diminishment in fibroblast dimensions, reduced elongation, and a collapsed structural form.34,112 Diminished fibroblast dimensions and mechanical forces further downregulate the TGF-β type II receptor. 116 UV irradiation reduces the activity of the transcription factor Smad-binding element, inhibiting TGF-β signaling. 117

TGF-β signaling pathway. TGF-β signal transduction is mediated by the TGF-β receptor (TβR), a complex of three transmembrane receptors, consisting of TβRI, TβRII, and TβRIII. Smads occur as three distinct subclasses: receptor-regulated (e.g., Smad2, Smad3), common partner (e.g., Smad4), and inhibitory Smads (e.g., Smad7). 108 The binding between TGF-β and TβRII activates TβRI, leading to the formation of the TβRI/TβRII complex. Subsequently, TβRI triggers the phosphorylation of transcription factors Smad2 and Smad3. After that, Smad4 binds to it and forms a heterometric complex then translocates to and accumulates in the nucleus, where it acts as a transcription factor to upregulate ECM-related genes, including those encoding collagens, fibronectin, decorin, and versican, via Smad-binding elements (SBEs).34,109–111 Interestingly, UV irradiation rapidly induces Smad7 expression partially due to increased gene transcription since Smad7 gene promoter contains a methylation-free CpG island which can bind Smads, AP-1, and Sp-1, the latter two of which are upregulated in photoaged skin.108,112 Smad7 antagonizes TGF-β signaling by interacting with E3 ubiquitin ligases such as Arkadia, Smurf1, or Smurf2, recruiting them to TβRI and promoting its degradation. 113 Smad7 also functions as a TGF-β inhibitor through the prevention of Smad2/3/4 complex formation. 114 TGF-β, transforming growth factor-β.
Besides being inhibited, the protective effects of TGF-β can be compromised by prolonged or excessive UV exposure, thereby accelerating the photoaging process. UV-induced increases in TGF-β ligand levels trigger the release of MMP-2 and MMP-9 into the dermis and recruit neutrophils, further contributing to the damage and disorganization of collagens and solar elastosis. 114 Activation of TGFβ/Smad3 signaling in the dermis may induce a fibrotic response, potentially linked to premature collagen production in photoaged skin. 114 TGF-β additionally extends the infiltration duration of neutrophils, leading to incremental collagen degradation and the generation of abnormal elastic fibers that contribute to ECM destruction. 118
The synthesis and degradation of GAGs
As previously discussed, the levels of GAGs in photoaged skin exhibit significant variability. UV irradiation affects GAGs by influencing the enzymes responsible for their synthesis and degradation, altering their content, structure, and functionality. HA synthesis relies on several enzymes, with the HA synthases (HAS) family being pivotal in catalyzing the polymerization of UDP-GlcUA and UDP-GlcNAc precursors. 119 The production of other tsGAGs and additional glycosylation processes is facilitated by their respective GAG type-specific glycosyltransferases and sulfating enzymes. 38 Additionally, the degradation of GAGs also requires a variety of enzymes. The equilibrium between GAG synthesis and degradation has been altered by UV irradiation, thereby contributing to the process of cutaneous photoaging. This article will further explore these changes, focusing on HA and HS.
The HAS family mainly mediates HA synthesis on the plasma membrane, which directly secretes into the ECM. 38 The HAS family, including HAS-1, -2, and -3, is upregulated by UV irradiation, leading to increased HA production. 43 HA degradation is mainly catalyzed by hyaluronidases (HYALs) and cell migration-inducing proteins (CEMIPs, alias HYBID, and KIAA1199). UV exposure differentially regulates HYAL1, HYAL2, and HYAL3 mRNA expression, depending on the time, dose, and specific HYAL involved. 120 HYALs are a family of endolytic glycoside hydrolases responsible for cleaving the β1–4 linkage between N-acetylglucosamine and glucuronic acid. 120 Moreover, HYBID expression is notably elevated in the UV-exposed skin. 121 CD44, a receptor for HA, also plays a role in UV-induced HA degradation. CD44 facilitates the uptake of HA degradation products via HYAL-2, transporting them to lysosomes, where HYAL-1 further degrades them. 97 UV irradiation causes an accumulation of CD44, which is associated with the epidermal hyperplasia. 122 Consequently, disrupting the balance between HA synthesis and degradation may increase HA catabolism in photoaged skin.
Mammalian HS-degrading enzymes, known as heparanases, include heparanase-1 and -2. Heparanase-1 is the primary enzyme responsible for cleaving the glycosidic bond with a hydrolase mechanism, while heparanase-2 has regulatory functions, sometimes inhibiting heparanase-1 activity. 123 UV irradiation induces the upregulation of heparanase-1 expression and downregulation of heparanase-2 expression in the epidermis, resulting in increased degradation of HS.47,120 Moreover, exostosin-1 and exostosin-2, working together in the synthesis process of HS, are differentially regulated by UV irradiation—exostosin-1 is upregulated, while exostosin-2 is downregulated. 124 These mechanisms collectively contribute to the degradation and functional damage of HS. Furthermore, the disruption of HSPG function leads to the release of cytokines such as FGF-2, FGF-7, and Vascular Endothelial Growth Factor (VEGF), which enhance growth factor interactions between the epidermis and dermis. 125 As a result, these growth factors participate in epidermal hyperplasia, changes of blood vessels, lymphatic vessels, keratinocytes, and fibroblasts.37,125 Degradation of HS also results in the loss of LM in the BM. The globular domain of LM binds to cellular receptors, including HS; meanwhile, the PGs perlecan and agrin provide collateral anchorage of the LM network. 61
Conclusion
Skin photoaging is a widespread issue largely driven by environmental factors, particularly UV irradiation. After UV exposure, visible signs of premature aging, such as deep wrinkles, mottled pigmentation, sallow complexion, skin laxity, and rough texture, appear, all of which are closely associated with the ECM of the skin. UV-induced ECM deterioration results in structural and functional alterations of the skin, further precipitating the onset of skin cancer, vascular lesions, immune suppression, and other diseases. Clarifying the mechanism by which UV irradiation damages the ECM is essential for the prevention and treatment of skin photoaging.
While recent studies have advanced our understanding of the mechanisms underlying UV-induced ECM damage, significant gaps remain. Future research should focus on identifying novel ECM biomarkers of photoaging to enable earlier detection and targeted interventions. Further investigation is also needed to elucidate the complex interplay between ECM components and cellular responses to UV exposure. Additionally, developing advanced models, such as three-dimensional skin cultures and in vivo imaging techniques, could help map the ECM’s dynamic responses to photoaging in more physiological conditions. Addressing these gaps will be crucial for establishing comprehensive prevention and treatment strategies for skin photoaging.
Footnotes
Authors’ Contributions
E.L.: Conceptualization, data curation, formal analysis, writing—original draft, and review and editing. Z.X.: Validation, resources, and writing—review and editing. Yunjun L.: Supervision and writing—review and editing. Ye L.: Project administration and writing—review and editing.
Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could result in a conflict of interest.
Funding Information
This work was supported by the