
Abstract
The use of bioengineered cell constructs for the treatment of bone defects has received much attention of late. Often, bone marrow stromal cells (BMSCs) are used that are in vitro–stimulated toward the osteogenic lineage, aiming at intramembranous bone formation. The success of this approach has been disappointing. A major concern with these constructs is core degradation and necrosis caused by lack of vascularization. We hypothesized that stimulation of cells toward the endochondral ossification process would be more successful. In this study, we tested how in vitro priming of human BMSCs (hBMSCs) along osteogenic and chondrogenic lineages influences survival and osteogenesis in vivo. Scaffolds that were pre-cultured on chondrogenic culture medium showed collagen type II and collagen type X production. Moreover, vessel ingrowth was observed. Priming along the osteogenic lineage led to a mineralized matrix of poor quality, with few surviving cells and no vascularization. We further characterized this process in vitro using pellet cultures. In vitro, pellets cultured in chondrogenic medium showed progressive production of collagen type II and collagen type X. In the culture medium of these chondrogenic cultured pellets, vascular endothelial growth factor (VEGF) release was observed at days 14, 21, and 35. When pellets were switched to culture medium containing β-glycerophosphate, independent of the presence or absence of transforming growth factor beta (TGF-β), mineralization was observed with a concomitant reduction in VEGF and matrix metalloproteinase (MMP) release. By showing that VEGF and MMPs are produced in chondrogenically differentiated hBMSCs in vitro, we demonstrated that these cells produce factors that are known to be important for the induction of vascularization of the matrix. Inducing mineralization in this endochondral process does, however, severely diminish these capacities. Taken together, these data suggest that optimizing chondrogenic priming of hBMSCs may further improve vessel invasion in bioengineered constructs, thus leading to an alternative and superior approach to bone repair.
Introduction
One of the alternatives focuses on the use of human bone marrow stromal cells (hBMSCs), which can be harvested with relative ease and can be differentiated toward the chondrogenic and osteogenic lineages in vitro. 4 Additionally, they can be transplanted autologously, which makes them suitable to use in transplantable bioengineered cell constructs. However, a vascularized, mechanically competent, osteoconductive construct that could be used to produce bone in vitro or cause complete osteogenesis in vivo remains to be developed. The issue of core degradation, arising from lack of nutrient delivery to and waste removal from the center of tissue-engineered constructs and caused by insufficient blood supply to the implanted tissue, is of major concern in the field of tissue engineering. 5 First, it takes time for blood vessels to invade a tissue and oxygenate it and provide it with nutrients. This is not an instantaneous process and usually occurs too slowly for cells to survive without nutrients. Second, culture of a scaffold in vitro for several weeks can lead to formation of an extensive extracellular matrix, which can prevent or seriously hamper blood vessel infiltration in vivo by sealing the pores of a scaffold.
The approach used for tissue engineering of bone using hBMSCs is usually similar in different laboratories and involves the induction of direct osteogenic differentiation in a manner akin to intramembranous ossification in vivo. As discussed, the problem with this approach is the long-term survival of these constructs, which is limited because of lack of vascularization. In this study, we hypothesized that it might be preferable to induce bone formation in vivo via an endochondral ossification approach in which hypertrophic chondrocytes, primed in vitro and derived from hBMSCs, would be implanted. This priming of chondrogenic three-dimensional cellular constructs can be performed to different stages of the differentiation and maturation process, namely until a cartilaginous extracellular matrix has been formed or when the extracellular matrix is mineralized. 6 Often this is seen as an obstacle to cartilage tissue engineering and is seen as something to be avoided. 7 However, more recently, the potential benefit of using this approach for bone tissue engineering has been realized. 8 Because chondrocytes can survive in hypoxic environments 9 and because they produce anabolic and catabolic factors important for the conversion of avascular tissue to vascularized tissue, such constructs might stimulate vessel ingrowth and overcome the problem of core degradation. This approach has the benefits of in vitro and in vivo vascularization strategies. 10 In vivo, prevascularization of a construct in an ectopic site before its use has the benefit of creating a material that can be surgically connected to the host vasculature but requires a second surgery to relocate the construct to the implant site. 11 In vitro prevascularization avoids this but still requires some time for anastamosis12,13 with the host, during which time there will be a hypoxic period. The approach presented here combines the ease of in vitro priming with the benefit of the ability to withstand the initial hypoxic conditions while performing in vivo vascularization in the target site. Any undifferentiated hBMSCs at this stage would also stand a better chance of survival than differentiated cells, as has also recently been shown. 14
In in vivo endochondral ossification, vascular endothelial growth factor (VEGF) is known to play a critical role in the establishment of vessel in-growth by effecting angiogenesis, remodeling of extracellular matrix, and ossification. 15 Additionally, it has been shown that chondrocytes and hypertrophic chondrocytes express matrix metalloproteinases (MMPs). These molecules play an important role in the conversion of the non-vascularized cartilaginous matrix to a vascularized mineralized tissue in skeletal development and fracture healing.16–19 Based on these facts and the lack of success of tissue engineering bone, it is more logical to use chondrocytes, which are less oxygen sensitive, to produce bone in vivo via the endochondral ossification route that will then naturally vascularize and mineralize in situ.
In vitro, pellet cultures of chondrogenic differentiated hBMSCs show a similarity to endochondral ossification; they progressively produce collagen type II and collagen type X, proteins specifically produced by hypertrophic chondrocytes.20,21 When a phosphate donor is added to the culture medium, mineralization can occur. 22 Therefore, this culture system offers a simple model for the development of in vitro tissue engineering approaches for the creation of vascularized bone in vivo. This study aimed to assess the possible advantages of using an endochondral ossification model of bone formation for use in tissue engineering approaches. To that end, hBMSCs were seeded into highly biocompatible collagen–glycosaminoglycan (GAG)scaffolds23–25 and primed in vitro to progress along the osteogenic or chondrogenic routes before subcutaneous implantation into nude mice. In vitro, a simple model of endochondral ossification using pellet cultures enabled better understanding of the process.
Materials and Methods
Isolation and expansion of human bone marrow cells
hBMSCs of three donors, all female aged 33 to 60, were obtained during total hip arthroplasty after informed consent with approval of the local medical ethical committee (METC 2004-142). Bone marrow aspirates were taken from the greater trochanter. Heparinized aspirates were seeded at a density of 30 to 90 × 106 nucleated cells per T175 flask. After 24 h, non-adherent cells and cell debris were washed out. hBMSCs were further expanded in low-glucose Dulbecco's modified Eagle medium (DMEM) with 10% fetal calf serum from a pre-selected batch to maintain the multipotential capacities of the cells, 50 μg/mL of gentamicin, and 1.5 μg/mL of Fungizone (all Invitrogen, Carlsbad, CA) and 0.1 mM of L-ascorbic acid 2-phosphate and 1 ng/mL of fibroblast growth factor (Instruchemie B.V.Delfzijl, Delfzijl, the Netherlands). Cells were cultured at 37ºC under humidified conditions and 5% carbon dioxide (CO2). Medium was changed twice a week. When cultures neared confluence, they were trypsinized using 0.05% trypsin and replated at a density of 2000 cells/cm2. Cells from the second to the fourth passage were used for the pellet cultures. hBMSCs purchased from Cambrex (now Lonza, Verviers, Belgium) were used in the scaffold study.
hBMSC seeding onto collagen–GAG scaffold and subcutaneous implantation
Cells were rinsed with phosphate buffered saline (PBS, 100 mM sodium chloride, 80 mM sodium phosphate, 20 mM dibasic anhydrous sodium phosphate ) and detached with trypsin-ethylenediaminetetraacetic acid (Invitrogen). The resulting suspension was centrifuged (650 g, 5 min at 20°C), re-suspended in 2 mL of control medium, and aspirated through a 20-gauge needle to obtain a single cell suspension of 1.5 × 106 cells/mL. Collagen–GAG scaffolds (8 mm2, generously donated by Massachusetts Institute of Technology, Cambridge, MA), prepared using a freeze-drying method previously described, 24 were seeded with 300,000 cells per scaffold, as previously described, with the exception that a smaller volume of 100 μL of cell suspension per side was used. 23 Seeding efficiency was estimated to be 50% according to recent experimental data using the same approach. 26 These scaffolds were prepared from bovine collagen type I and chondroitin sulphate in a freeze-drying process using dehydrothermal crosslinking. After seeding, 3 mL of supplemented DMEM was added to each well. After 2 days, medium was replaced with chondrogenic (25 μg/mL of L-ascorbic acid 2-phosphate, 100 mM of sodium pyruvate (Invitrogen), 1:100 insulin-transferrin-selenium (ITS; BD Biosciences, Bedford, MA), 10 ng/mL of transforming growth factor beta-2 (TGF-β2), (R&D Systems, Abingdon, United Kingdom) and 100 nM of dexamethasone (Sigma, St. Louis, MO)) or osteogenic (25 μg/mL of L-ascorbic acid 2-phosphate, 10 mM of β glycerophosphate (Sigma), and 10−8 M of dexamethasone) medium, as previously described.27,28 Half of the medium was replaced every 3 days. Scaffolds were treated for 21 days and were maintained in a humidified atmosphere of 95% air/5% CO2 at 37°C. At day 21, five scaffolds of each condition were harvested for histology, and five scaffolds were implanted subcutaneously into the backs of nude mice. Two incisions were made along the central line of the spine, one at the shoulders and one at the hips. Two scaffolds were implanted at each point, one to the left and one to the right at random. Animals were allowed to recover and were sacrificed 4 weeks later. This procedure was carried out with approval from the local animal ethical committee (EUR334).
Chondrogenic differentiation in pellet cultures
After detachment of the cells with 0.05% trypsin, 0.5 mL of medium, containing 200,000 cells, was put in polypropylene tubes. Tubes were then centrifuged for 8 min at 120 g. To induce chondrogenic differentiation, pellets were cultured for 21 days in high-glucose DMEM containing 50 μg/mL of gentamicin and 1.5 μg/mL of Fungizone (Invitrogen), l-ascorbic acid 2-phosphate, 100 mM of sodium pyruvate (Sigma), 1:100 ITS (BD Biosciences), 10 ng/mL of TGF-β (R&D Systems), and 100 nM of dexamethasone (Sigma).
After 21 days of culture, three groups were made that were cultured for another 2 weeks in different media:
Chondrogenic differentiation medium: This group was kept on the same medium. Phosphate-containing chondrogenic medium: In this group, 10 mM of β glycerol-phosphate (BGP) was added to the chondrogenic medium. Osteogenic medium: This group was switched to medium with 10 mM of BGP and 10−8 M of dexamethasone, without TGF-β2.
To induce mineralization of the extracellular matrix, pellets were switched to a phosphate-containing culture medium, as in medium 2 above. Because it has been shown that the presence of TGF-β in the culture medium might negatively influence hypertrophy, 29 the induction of mineralization was also evaluated in pellet cultures switched to a culture medium more resembling an osteogenic differentiation medium (medium 3).
A pure osteogenic condition was not included in the pellet culture experiment based on the lack of sufficient bone formation in the in vivo condition that we see as a non-viable option for bone repair in these settings.
Gene expression analysis
At harvesting, pellet cultures were suspended in 300 μL of RNA-Bee (TEL-TEST, Friendswood, TX). To allow optimal suspension of all cells, pellets were disintegrated using a pestle and were subsequently flushed through a 28-gauge needle (Monoject, Sherwood Davis & Geck, St. Louis, MO). For RNA precipitation, a commercially available kit was used (Qiagen, Hilden, Germany). Reverse transcription in complementary DNA (cDNA) was performed on 200 ng of total RNA using a RevertAid First Strand cDNA Synthesis Kit (MBI Fermentas, St. Leon-Rot, Germany). Primers were designed using PrimerExpress 2.0 software (Applied Biosystems, Foster City, CA) to meet TaqMan or SYBR Green requirements and were designed to bind to separate exons to avoid co-amplification of genomic DNA. BLASTN ensured gene specificity of all primers listed in Table 1. The following genes were analyzed: Sox-9, collagen type II, collagen type X, runt-related transcription factor 2/core-binding factor alpha (runx2/cbfα-1), and alkaline phosphatase (ALP).
FW, forward; RV, reverse; FAM, FAMlabelled Taqman Probe.
Amplifications were performed as 25-μL reactions using TaqMan Universal PCR MasterMix (ABI, Branchburg, NJ) or qPCR Mastermix Plus for SYBR Green I (Eurogentec, Nederland B.V., Maastricht, the Netherlands) according to the manufacturers' guidelines. Real-time reverse transcriptase polymerase chain reaction (PCR) was done using an ABI PRISM 7000 with SDS software version 1.7. (Applied Biosystems, Nieuwekerk a/d Ijssel, Netherlands). Data were normalized to glyceraldehyde-3-phosphate dehydrogenase that was stably expressed across sample conditions (not shown). Relative expression was calculated according to the 2-ΔCT formula using averages of triplicate samples for each donor. Because of insufficient RNA extracted from Donor C, PCR analysis could be performed for RNA only from pellets of Donors A and B.
Immunohistochemical and histochemical staining
For immunohistochemistry, pellets and scaffolds were fixed overnight in 4% phosphate buffered formalin and embedded in paraffin. Six-μm sections were made, deparaffinized in xylene, and rehydrated through graded ethanol. Three pellets of each condition were analyzed per donor. Five chondrogenically primed scaffolds and four osteogenically primed scaffolds were analyzed and sectioned at 10 μm. One osteogenic scaffold could not be located and retrieved. As a control, scaffolds were also cultured in unconditioned medium and analyzed in the same manner (data not shown).
Immunohistochemistry for collagen type II and collagen type X
To analyze collagen type II expression, sections were incubated with 0.1% pronase for antigen retrieval and 1% hyaluronidase (both Sigma). Sections were incubated for 2 h at room temperature with mouse monoclonal antibody against collagen type II (II-II6B3 antibody, 1:100; Developmental Studies Hybridoma Bank, Iowa City, IA, under contract N01-HD-6-2915 from the National Institute of Child Health and Human Development).
For collagen type X, sections were incubated with 1% pepsin in 0.5 M of acetic acid at pH 2.0 for 2 h at 37°C (Sigma) and 30 min in 1% hyaluronidase (Sigma). The sections were incubated overnight at 7°C with mouse monoclonal antibody against collagen type X (1:30, X53, Quartett, Berlin, Germany).
A biotin-labeled secondary antibody (Biogenex HK-325, 1:100) was used for both staining procedures, followed by ALP-conjugated streptavidin (Biogenex HK-321-UK, 1:100). This ALP activity was demonstrated by incubation with a new fuchsin substrate (Chroma, Kongen, Germany). Control for all antibodies was performed using an isotype immunoglobulin G1 monoclonal antibody. Counterstaining was performed with Gill's hematoxylin (Sigma).
Quantification was determined by multiplying the intensity of the staining (ranging from 1 to 3, where 1 is light positive staining and 3 intense) by the fraction of stained area over the pellet area, resulting in a maximum score of 300 (3 × 100%). Scores of three sections were averaged. Two independent observers performed these scorings blind.
With regard to choice of markers, collagen type II is the definitive chondrogenic marker, and collagen type X is the most widely accepted marker of chondrocyte hypertrophy and terminal differentiation. For gene expression analyses discussed below, we also examined classical markers of chondrogenesis, hypertrophy, and osteogenesis, namely the chondrogenesis transcription factor, Sox 9, the osteogenic marker ALP, and the early osteogenic, hypertrophic transcription factor cbfα-1/runx2. 30
Von Kossa and thionine staining
For evaluation of mineralization, slides were immersed in 5% silver nitrate solution (Sigma) for 10 min, rinsed in ultra pure water and exposed to light for 10 min. Excess silver nitrate was removed with 5% sodium thiosulphate (Sigma), and cells were rinsed in distilled water. For evaluation of proteoglycan content, paraffin sections were counterstained with 0.4% thionine in 0.01 M of aqueous sodium acetate, pH 4.5, for 5 min. Thionine stains GAGs, as can be seen in the chondrogenic conditions. 31
Positive calcium phosphate surface over the pellet surface resulted in the fraction of mineralized area. Averaged results of three sections per pellet were used for further analysis.
VEGF expression and MMP release
Culture medium of days 14, 21, and 35 were collected from each condition. Immediately after collection, the medium was centrifuged at 1000 g for 10 min at 4ºC and stored at −80ºC until further use.
Vascular endothelial growth factor
Expression of VEGF was analyzed on 24-h conditioned culture medium of days 14, 21, and 35. For each condition, medium of three pellets was analyzed in triplicate using a commercially available sandwich enzyme-linked immunosorbent assay kit (R&D Systems, Abingdon, United Kingdom) according to the manufacturer's protocol.
MMP release
Release of MMPs was determined by concentrating 100 μL of the conditioned culture medium using ultra-filtration (YM-30, Millipore, Billerica, MA), leaving a sample volume of approximately 15 μL. Sample loading buffer (Biorad zymogram buffer, BioRad, Hercules, CA) was added to the samples (2:1), which were incubated for 10 min at 37°C. Electrophoresis in a gelatin gel (BioRad) was performed, at 100 V and 25 mA together with a marker (BioRad). Thereafter, gels were incubated for 2 h in a renaturing buffer containing 2.5% Triton X-100 (Sigma) in 25 mM Tris–hydrochloric acid (HCl, pH 7.4) followed by incubation for 72 h in an activation buffer (2 mM of calcium chloride, 5μm of zinc chloride, 25 mM of Tris-HCl, pH 7.5, Fluka, Zwijndrecht, Netherlands). Gels were stained using Coomassie brilliant blue and destained with Roti destain (both Carl Roth, Karlsruhe, Germany). Areas of proteolytic activity appeared as translucent halos against a blue background of stained, non-degraded gelatin. Gels were scanned using a Kodak IS scanner (La Hulpe, Belgium).
Statistics
Analyses of immunohistochemistry and VEGF expression were performed on three pellets of each of the three donors for all conditions. An analysis of variance test and post hoc Bonferroni test of the means between culture days and between different culture media on day 35 were performed. P < 0.05 was considered to indicate statistically significant differences. Statistical analysis was performed on averages of triplicate samples per pellet using Graph Pad Prism 5.00 for Windows (GraphPad Software, San Diego, CA).
Results
Priming of hBMSCs in vitro and subsequent in vivo implantation
After 3 weeks in vitro priming, chondrogenic scaffolds were positive for GAGs and negative for mineralization (Fig. 1A), whereas osteogenically primed scaffolds were mineralized throughout (Fig. 1B). After implantation, the osteogenic scaffolds continued to differentiate along the osteogenic route however, producing an inferior matrix with questionable levels of cell survival 4 weeks after implantation, as evidenced by fewer cells within the mineralized portions (Fig. 1D). In the chondrogenic condition, despite cell survival and formation of a cartilage-like matrix with some hypertrophic cells visible, no osteogenesis was observed (Fig. 1C). No appreciable matrix formation of either kind was observed in unprimed control conditions (cultured on expansion medium, data not shown), showing the promise and importance of the initial in vitro culture period and the possible benefits of well-timed implantation.

von Kossa staining with thionine of primed scaffolds. Glycosaminoglycan production (thionine staining, white arrows) in chondrogenically primed scaffolds after 3 weeks in vitro, (
Collagen type X immunostaining of chondrogenically primed implanted scaffolds showed positive staining in all five of the retrieved scaffolds (Fig. 2A). Osteogenically primed scaffolds showed only weak positive staining in one of four scaffolds (Fig. 2B).
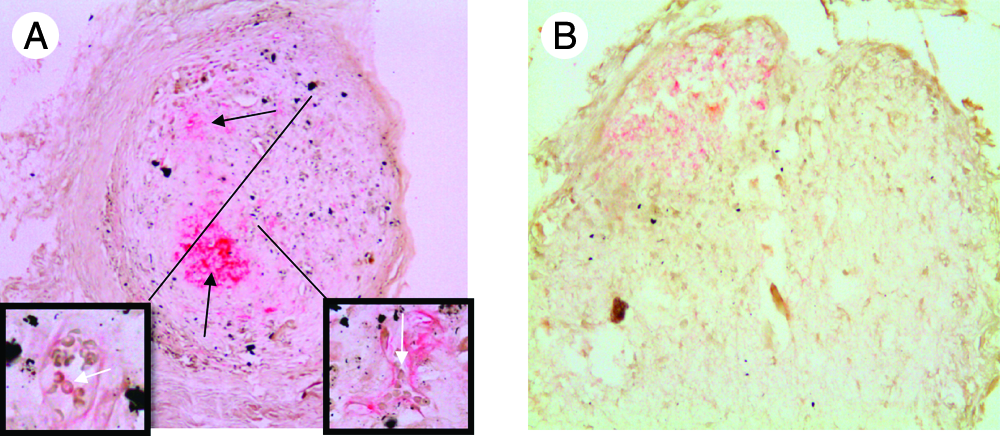
Collagen type X staining of implanted scaffolds. After implantation of primed scaffolds, immunostaining for collagen type X showed that all chondrogenically primed scaffolds were positive for collagen type X (
In the chondrogenic scaffolds, mature blood vessels were visible in all implanted scaffolds (Fig. 2). In three of five of the scaffolds, blood vessels contained erythrocytes; in the two other scaffolds, we found what appeared to be blood vessels without red blood cells. This shows not only good cell survival of chondrogenically primed cells but also good host integration and suggests progression along the endochondral ossification route. Conversely, no vascularization was visible in the osteogenically treated scaffolds, possibly because of too much matrix deposition or a lack of release of inductive factors. This lack of vascularization in the osteogenically primed constructs might explain the lack of cells in the scaffolds due to cell death.
In vitro pellet culture system
Gene expression
All genes were observed to increase in expression over time upon addition of chondrogenic factors. At day 35, comparison of expressions in the mineralizing conditions that had been switched 14 days before was unchanged for Sox 9, cbfα-1/runx2, and ALP. There was a decrease in expression of collagen type II and X upon the addition of phosphate, although this decrease was not observed in the osteogenic condition, and in no instances was it significantly different from the chondrogenic condition (Fig. 3).
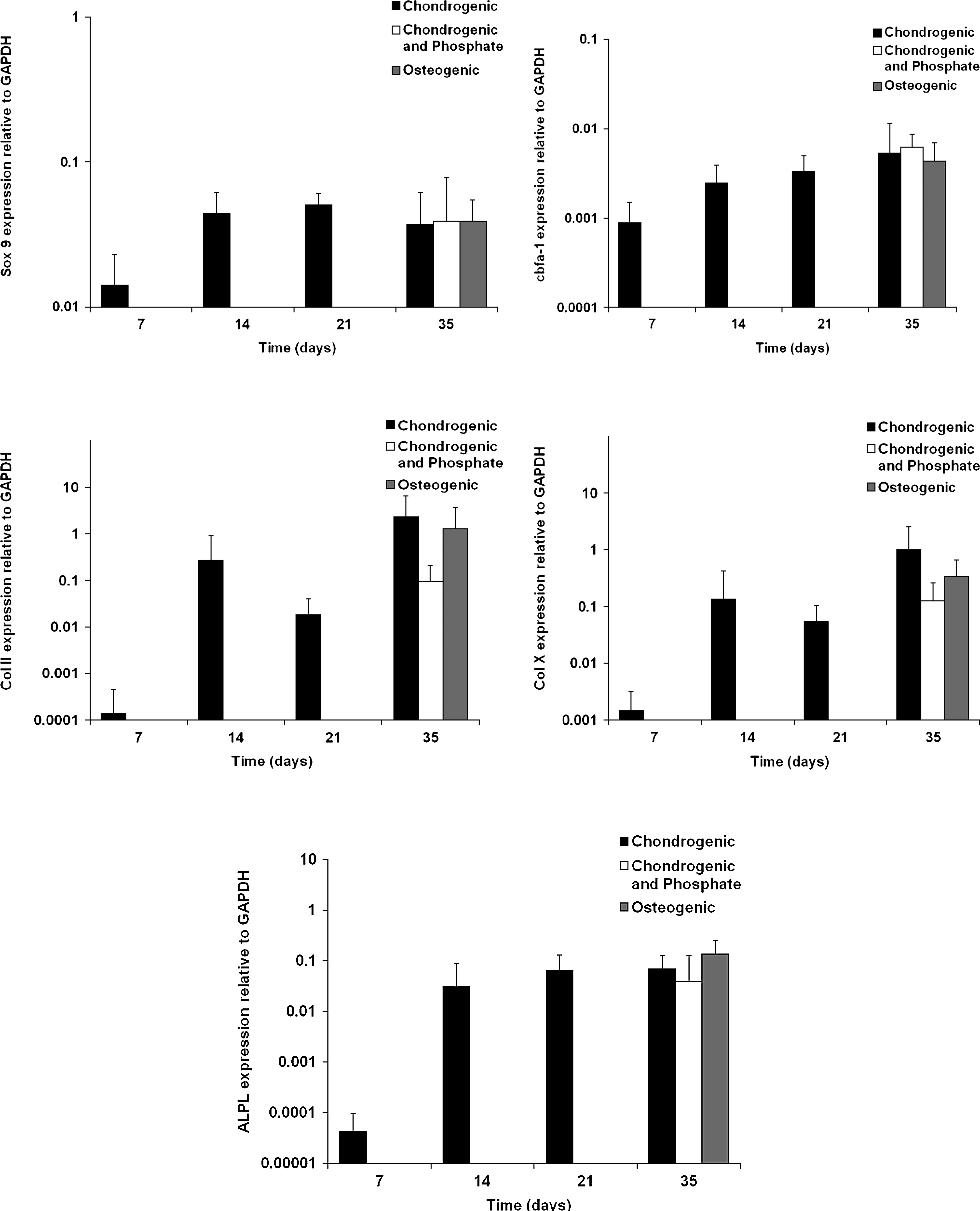
Gene expression analysis of pellets. Gene expression analysis of markers of chondrogenesis, hypertrophy, and osteogenesis showed a steady increase over time in expression of all markers cultured in chondrogenic medium. Switching to either of the two mineralizing media for 14 days did not lead to significant differences in gene expression of any of the markers. Data represent three pellets per donor for Donors A and B only.
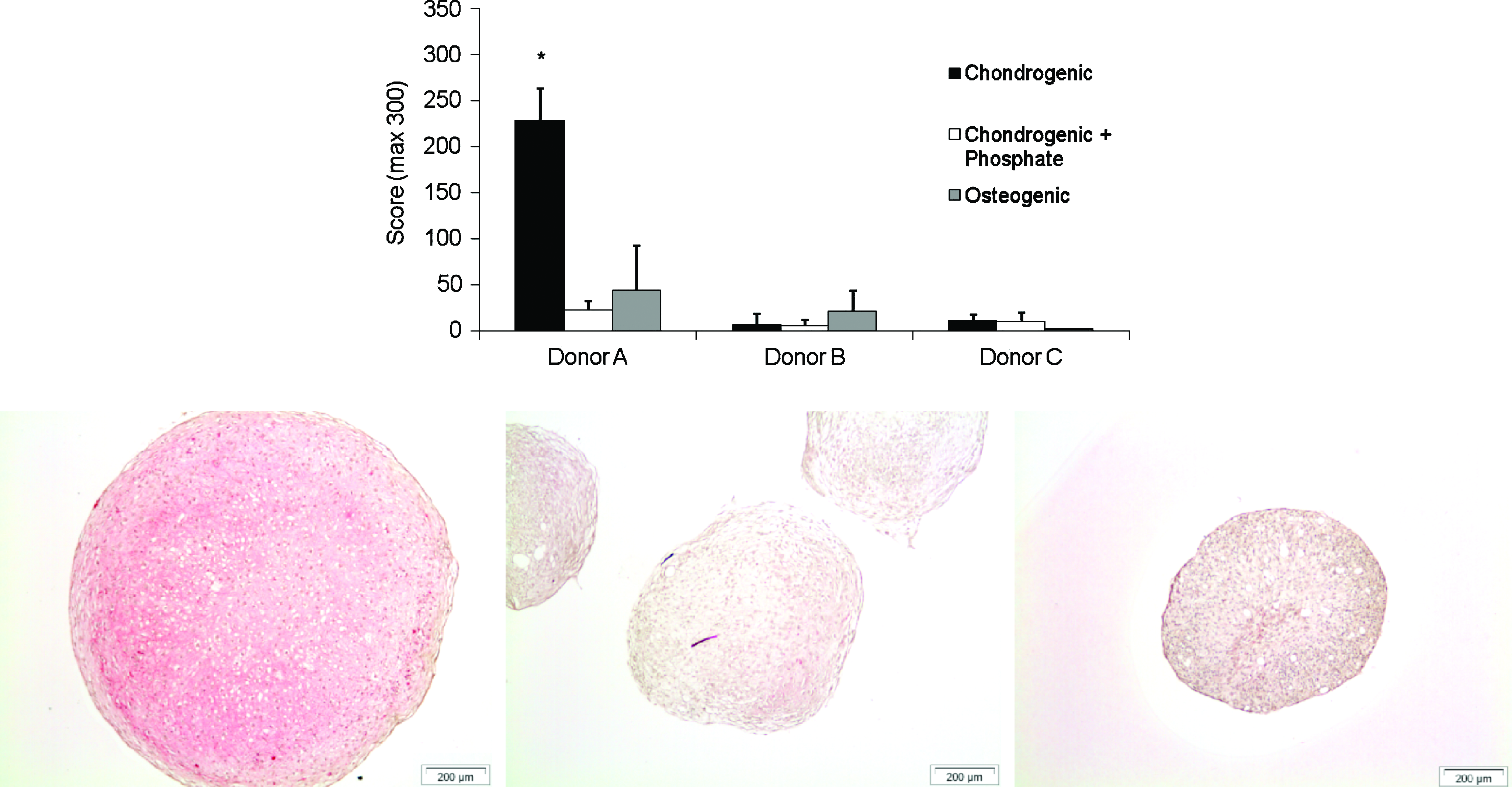
In vitro, collagen type II positive staining was found in chondrogenically differentiated pellets of all donors at day 35 (Fig. 4A). Although they all showed positive staining, variation in stained area and intensity was high, ranging from a score of 57 to the maximum score of 300. This variation was donor dependent. Collagen II expression was mainly seen in the center of the pellets (Fig. 4B).

Collagen type II immunohistochemistry and scoring of pellets. To some extent, positive collagen type II staining (red) was observed in all pellets in a donor-dependent manner, as is often seen. Upon addition of a phosphate donor for 14 days, collagen type II expression was vastly reduced in all three donors. Three sections per pellet with three pellets per donor were analyzed to score the amount of staining (1-way analysis of variance; *p < 0.05). Representative images of the chondrogenic condition are displayed for each donor respectively. Color images available online at
Collagen type X expression was found in all patients at day 35. This expression was again donor dependent and only highly expressed in Donor A, who also produced more collagen type II. Collagen type X and II expressions were reduced in both conditions switched to mineralizing media (Fig. 5).
Collagen type X immunostaining of in vitro–cultured pellets. Staining of pellets for collagen type X showed a similar pattern of expression to collagen type II, with donor-related variation in expression levels with decreased expression in the presence of a phosphate donor. Sections were scored in triplicate with three pellets per donor (1-way analysis of variance; *Significant difference from chondrogenic plus phosphate and osteogenically switched conditions; p < 0.05). Representative images show collagen type X expression in the chondrogenic condition after 35 days of culture. Color images available online at
Von Kossa staining showed calcium phosphate depositions in pellets that were switched to phosphate-containing medium after being cultured in chondrogenic differentiation medium for 21 days. Mineralization was found in two of three donors (A and B) in the chondrogenic plus phosphate switched condition and in the condition switched to osteogenic medium, with no mineralization observed in the chondrogenic condition. The mineralized area was found in the core of the pellet cultures and covered approximately half of the pellet area. Few viable cells were visible in the mineralized portions. This lack of cells was not seen in non-mineralized tissue. No difference was found between the chondrogenic condition containing BGP (culture medium 2) and the osteogenic condition (culture medium 3). (Fig. 6).

Von Kossa staining of in vitro cultured pellets. Culture of pellets for 35 days on chondrogenic medium resulted in no mineralization in any pellet from any donor, as evidenced by the 0 scores for the chondrogenic group. When a source of phosphate was added, mineralization was observed in all pellets in two of three donors, with no difference between the chondrogenic plus phosphate and osteogenic conditions. Despite the presence of cells within the pellets, Donor C demonstrated no mineralization capability. Representative images of stained pellets show mineralization when phosphate was added to the chondrogenic medium (black stain). Color images available online at
When pellets were cultured in chondrogenic differentiation medium for 49 days, collagen type II and X increased further (data not shown). When cultures were switched after 35 days in chondrogenic differentiation medium, the incidence and amount of mineralization was not different from in those conditions switched after 21 days. Mineralization again occurred in two of three donors.
VEGF expression
VEGF was present in medium of pellets in chondrogenic medium on days 14, 21, and 35 except for with one donor, which showed VEGF release at day 14 but little thereafter. This donor, Donor C also showed little collagen type II or X expression, and insufficient RNA was available for gene expression analysis. When pellets were switched to the culture medium containing BGP, independent of the presence of TGF-β2 or the concentration of dexamethasone, VEGF expression was considerably lower than in the chondrogenic condition (Fig. 7). Also, the conditions that were switched after 35 days of culture showed no reduced VEGF expression (data not shown). Although some variation between donors was found, the overall effect on VEGF production was the same.

Release of factors related to endochondral ossification. Release of vascular endothelial growth factor (VEGF) from pellets was measured over time. As was seen with collagen type II and collagen type X, release of VEGF from pellets stopped after addition of a phosphate donor for 14 days after 21 days in chondrogenic medium (
MMP release
Gelatin zymography demonstrated that MMPs were present in the culture medium collected at day 35 from all cultured pellets. A steady increase in release from day 14 was also observed in the chondrogenic condition (data not shown). The two bands with high activity showed up at approximately 62 kDa and 59 kDa. A third activity band was seen at approximately 50 kDa. Analysis of culture medium collected from pellet cultures that were switched to phosphate-containing culture medium demonstrated far less activity at 62 kDa and no activity at 59 and 48 kDa at day 35 and day 49 (not shown) of culture (Fig. 7B).
Discussion
In this article, we assessed the possibility for in vitro chondrogenically primed hBMSCs to offer a better repair option for bone tissue engineering approaches. In vivo, we demonstrated better cell survival after chondrogenic priming of hBMSCs in vitro, with significant blood vessel invasion after 4 weeks in vivo, although no bone formation was observed, possibly because of insufficient time allowed. We hypothesized that in vitro priming of hBMSCs along the chondrogenic lineage would prompt the release of factors leading to tissue vascularization and bone formation in vivo. We demonstrated that mineralization of cartilage tissue can be induced in vitro but that this inhibits the angiogenic and remodeling properties of chondrogenically differentiated pellet cultures. Crucially, in a model of endochondral ossification, we showed that pellet cultures of chondrogenically differentiated hBMSCs express collagen type II and collagen type X and produce VEGF and greater amounts of MMPs and that all of these qualities are quickly reduced after mineralization in a model of endochondral ossification, as was initially hypothesized. This might suggest that mineralized pellet cultures do not possess the same angiogenic and remodeling properties that can stimulate graft–host integration in vivo and that the likelihood of obtaining a bony union in bone defects might therefore be less than in chondrogenically differentiated pellet cultures.
Progressive production of collagen type II and subsequent collagen type X suggests that differentiated hBMSCs in pellet culture resemble chondrogenic differentiation and hypertrophy, as seen in the growth plate during longitudinal growth. Lack of phosphate in the chondrogenic culture medium does, however, prevent further resemblance to endochondral ossification because the formation of calcium phosphate depositions is inhibited. To examine whether pellet cultures need the presence of phosphate in the culture medium only to mineralize the extracellular matrix or whether they must be cultured in a more osteogenic medium with a lower concentration of dexamethasone and in the absence of TGF β, we evaluated the mineralization of chondrogenic differentiated pellet cultures that were switched after 21 days to these two phosphate-containing culture media. Fourteen days after switching, abundant amounts of mineralization were seen in the core of the pellets at the same area as the cartilaginous tissue production was found, suggesting that this is possible also in vitro, given the correct signals and timing. Patterns of VEGF release, collagen expression, and MMP release were similar between this condition and a standard osteogenic condition switched from chondrogenic culture at the same timepoint. The spatial production of collagen type II, collagen type X, and mineralization, as seen in subsequent sections, suggests that endochondral ossification had occurred. This is line with published data by Mueller and Tuan, although they observed the requirement for the removal of TGF-β for mineralization to occur. 32 Gene expression of markers of chondrogenesis, hypertrophy, and osteogenesis was also seen to increase over time, as has been demonstrated previously.7,20,32
VEGF, which hypertrophic chondrocytes produce, is known to have distinctive roles in endochondral ossification. First, it is an important mediator in angiogenesis. Blocking the activity of VEGF inhibits blood vessel formation in the hypertrophic zone. 15 Second, it has been shown that VEGF has chemoattractant effects on mesenchymal progenitor cells and osteoblasts and influences osteoblast proliferation and differentiation.33,34 Cells of the monocyte–macrophage lineage are also affected; chondroclasts are less present when VEGF activity is blocked.15,35,36 Analysis of VEGF on the culture medium of chondrogenically differentiated pellet cultures showed considerable production of VEGF at 14 days of culture. In two donors, the expression remained stable during the 5-week culture period. This suggests that these pellet cultures have potential to stimulate incorporation in vivo after transplantation. Most importantly, amounts of VEGF release appeared to be related to collagen type II and X expression and were almost completely inhibited by matrix mineralization. This is very much in line with the in vivo findings showing no vascularization of the osteogenically primed scaffolds and some vasculature connected to the host within the chondrogenically primed scaffolds. It will be important to elucidate the timing of these events in the future along with cell death events to understand the role of cell death in this process. The type of cell death undergone by hypertrophic chondrocytes is a continuing debate37–39 and warrants investigation, although it is beyond scope of this article.
To further evaluate the angiogenic and remodeling activities of the cell constructs, we performed gelatin zymography to analyze the relative amounts of total MMPs released by the various pellets. In the conditioned medium of chondrogenically differentiated pellet cultures, three bands were found, one at approximately 62 kDa, one at approximately 59 kDa, and a third at approximately 50 kDa, the approximate weights of several MMPs including but not limited to MMP-2, pro-MMP-13, and MMP-13, respectively. Many of these MMPs play an important role in the remodeling of the cartilaginous extracellular matrix during endochondral ossification by breaking down collagen type II and non-collagenous matrix proteins such as aggrecan to allow vessel in-growth.16–19,40,41 Lower levels of MMPs in the mineralizing samples are in agreement with the in vivo findings, in which no blood vessel invasion was observed in the osteogenic condition.
There was large inter-donor variability in this study. We show relatively little variation within pellets from the same donors. Often in this research, field donors are pre-screened, and those that do not “perform” well are not included in experiments, but we believe that it is just these donors who are most likely to require treatment in the future, and it is therefore relevant to examine how they respond to this type of stimulation and to present this data. Donor C produced collagen type II and no collagen type X and subsequently no VEGF. Having performed all experiments on multiple pellets of each donor, we are confident of our results. From this data, we conclude that chondrogenic priming of MSCs will lead to greater VEGF release with concomitantly greater blood vessel invasion in vivo. Induction of mineralization in pellets or scaffolds prevents this release of VEGF and leads to inferior mineralized matrix production in vivo, with no blood supply and non-viable cells. The poor performance of Donor C in all respects confirms that this is not simply a standard response of hBMSCs in pellet culture and that this approach requires further investigation and optimization. This is not the first article to illustrate large variation between donors. Scharstuhl et al. 42 demonstrated as much as 1000-fold differences in expression of chondrogenic markers between patients, and it is important not to overlook this variability and to use it as a means to develop better treatments and approaches to regenerative medicine.
Also of interest is the better control of this process that could be garnered through further understanding of how it works. A host of intra- and extracellular signaling molecules including but not limited to, insulin-like growth factors, members of the fibroblast growth factor family, and the Wnt signaling pathway, tightly control endochondral ossification. 43 It is conceivable that tailored addition of growth factors or agonists at temporally distinct periods in the differentiation process could vastly improve the efficacy of such an approach, particularly for donors who do not respond well to the standard cocktail of growth factors. This also would apply to attraction of vessels in vivo, determining the correct combination to lead to optimum vessel–osteoblast–osteoclast attraction upon implantation.
This study contributes to the view that chondrogenically differentiated pellet cultures of hBMSCs resemble endochondral ossification, the more so because we demonstrated that mineralization of the extracellular matrix can occur when a phosphate donor is present. More importantly, we demonstrated for the first time the critical relationship between VEGF release, MMP release, and matrix mineralization, which can have huge effects on the success of implanted tissue-engineered constructs. Although we are aware of the limitations of using an immunocompromised animal in this model, we felt it was important to assess the potential of human cells, and this was the best model for doing so. We demonstratee that induction of mineralization before transplantation might have severe effects on the angiogenic and remodeling properties, suggesting that transplantation of chondrogenically primed tissue at the correct stage of differentiation, and before mineralization, might prove much more successful than fully differentiated tissue. Finally, we showed that chondrogenically primed hBMSCs are capable of expressing the hypertrophic marker collagen type X, attracting the host vasculature, and surviving long term in vivo. Although this pattern was observed in all samples, this was performed on commercially provided cells from a single donor. Our data suggest that determination of the optimal time of implantation of these primed cells, combined with a longer in vivo period, will lead to successful bone formation in vivo via the more common route of endochondral ossification.
Footnotes
Acknowledgments
The authors would like to acknowledge the assistance of Corinna de Ridder for implantation of scaffolds and funding assistance from the Dutch Programme for Tissue Engineering, Marie Curie Intra European Fellowship, Science Foundation Ireland, and Programme for Research in Third Level Institutions
Disclosure Statement
No competing financial interests exist.