
Abstract
Primary cultures of human hepatocytes are a reference cellular model, because they maintain key features of liver cells in vivo, such as expression of drug-metabolizing enzymes, response to enzyme inducers, and generation of hepatic metabolites. However, there is a restricted availability of primary hepatocytes, and they show phenotypic instability in culture. Thus, different alternatives have been developed to overcome the culture limitations and to mimic in vivo tissue material. Herein, culture conditions, such as medium composition, impeller type, and cell inoculum concentration, were optimized in stirred culture vessels and applied to a three-dimensional (3D) bioreactor system. Cultures of rat hepatocytes as 3D structures on bioreactor, better resembling in vivo cellular organization, were compared to traditional monolayer cultures. Liver-specific functions, such as albumin and urea secretion, phase I and phase II enzyme activities, and the capacity to metabolize diphenhydramine and troglitazone, were measured over time. Hepatocyte functions were preserved for longer time in the 3D bioreactor than in the monolayer system. Moreover, rat hepatocytes grown in 3D system maintained the ability to metabolize such compounds, as well as in vivo. Our results indicate that hepatocytes cultured as 3D structures are a qualified model system to study hepatocyte drug metabolism over a long period of time. Moreover, these cultures can be used as feeding systems to obtain cells for other tests in a short time.
Introduction
Because the liver is the main organ for biotransformation and metabolism of numerous endogenous substances and pharmacological agents, various in vitro liver models, including isolated liver slices and perfused livers, primary cultures of hepatocytes, and immortalized hepatic cell lines in different cultivation formats, have been utilized to better understand drug response in humans.1–3 Although cultures of human hepatocytes would be the ideal model to perform such studies, to overcome interspecies differences in all aspects of hepatic function, their source is limited because they can only be obtained from liver biopsies.4,5 Rodent hepatocytes, on the other hand, have been successfully used in pharmacological and toxicological studies.6–9 These resources of hepatocytes have the advantage of being easy to access; the results drawn from these experiments are reproducible and consistent, and therefore optimal when it concerns to the development of new culture methods.
Primary cultures of hepatocytes currently are the system that better mimics the organism. However, hepatocytes grown in culture and maintained under standard in vitro cell culture conditions are known to rapidly lose liver-specific functions, especially in biotransformation involving phase I and phase II enzymes. 10 Therefore, a system where primary hepatocytes maintain in vivo–like hepatic-specific functions in culture for longer periods of time would powerfully contribute for drug metabolism research.
Among many reasons that may account for a deficit of hepatocyte-like functions in vitro, cell adaptation to culture conditions after withdrawal from their organ environment seems to be one of the major problems. Several strategies have been developed to overcome this loss of function, such as protein synthesis, cell membrane integrity, and maintenance of life span limit and of cytochrome P 450 (CYP 450) activity. 11 Cultivation of hepatocytes on different extracellular matrices 12 and in different culture systems, or the supplementation of media with different hormones and growth factors was shown to improve in vitro hepatocyte quality.12–14
Culturing hepatocytes as three-dimensional (3D) structures provide cells with an in vivo–like environment, enabling the retention of important hepatic functions12,15,16 without the need to add extracellular matrix components. Approaches such as gel encapsulating systems and spheroid cultures have dramatically improved our understanding of the role of 3D culture strategies upon hepatocyte functions, but in vitro and in vivo models under well-defined and reproducible conditions are still needed. 17 To fully control a culture environment, one has to resort to bioreactors. Several bioreactors of bioartificial livers have already been applied in therapy.18–22 Further, bioreactors for application in both pharmacological and toxicological studies have been developed.15,23 In our laboratory a system with a fully controlled environment, successfully applied to other cell types, 24 could be adapted to hepatocyte cultures bringing several advantages over the others. Besides providing a better control of cellular environment, it would give a more predictive response of in vitro cultures that would improve the economics of cell-based testing.
Herein, the challenge was to establish a 3D in vitro model for primary rat hepatocytes consisting of a fully controlled minibioreactor that would allow for drug screening. The final result consists of a stirred tank with a controlled environment (temperature, pH, and pO2), which is reproducible, allows access to the cells (sampling), and is amenable to large-scale use. Besides, it could be easily adaptable to other types of cells, such as human hepatocytes or even human embryonic stem cells (hESC)-derived hepatocytes, and to be used as a feeding system for parallel cell tests.
Materials and Methods
Rat hepatocyte isolation
Hepatocytes were isolated from male Wistar rats, 6–9 weeks old, with 200–300 g body weight (from Instituto de Higiene e Medicina Tropical, Lisbon, Portugal, Animal House). A two-step collagenase perfusion–based method described by Seglen, 25 with slight modifications, was used. Briefly, the rats were anesthetized with an intraperitoneal injection of ketamine (90 mg/kg body weight) and xylazine (10 mg/kg body weight) solution. The liver was perfused via the vena portae for 10 min with a perfusion buffer I (0.14 M sodium chloride, 6.7 mM potassium chloride, and 10 mM HEPES), adjusted to pH 7.5 with 2.4 M ethylene glycol tetraacetic acid (EGTA) at 39°C. Subsequently, perfusion was continued with a collagenase buffer, consisting of 67 mM sodium chloride, 6.7 mM potassium chloride, 100 mM HEPES, albumin (0.5%) adjusted to pH 7.6, and 4.8 mM CaCl2 · 2H2O, at 39°C for 7 min. The flow rate for the perfusion buffers was 10 mL/min. After perfusion the liver was removed from the animal and dissociated in cold perfusion buffer I with 10 g/L of albumin. The resultant cell suspension was filtered through gauze, centrifuged for 10 min at 50 g, washed once with medium, centrifuged again, and resuspended in medium in a final concentration of not more than 3.5 × 106 cells/mL. For the enrichment of the final hepatocyte population, an additional Percoll-step was included by layering 5 mL of cell suspension over a 25% Percoll solution. After centrifuging at 1300 g at 4°C for 20 min, hepatocytes were obtained as a pellet. The cell pellet was diluted in phosphate-buffered saline (PBS), centrifuged for 10 min at 50 g, and washed twice with PBS for removing the Percoll solution. Finally, the cells were harvested in supplemented Williams' E medium for culturing. The cellular viability of the isolated hepatocytes was assessed by trypan blue exclusion; routinely, values within an 85–95% range were obtained.
Cell culture
Freshly harvested hepatocytes were cultured with Williams' E medium supplemented with 10% fetal bovine serum (FBS) (v/v), 1.4 μM hydrocortisone, 0.032 U/mL insulin, 15 mM HEPES, 1 mM sodium pyruvate, 1 mM of non-essential aminoacids (NEAA), and antibiotics (100 U/mL penicillin/100 μg/mL streptomycin and 40 μg/mL gentamicin) (Williams' E complete medium), or Vito 142 basal medium (Vito medium) (Biochrom AG, Berlin, Germany) with the corresponding supplement supplied by the manufacturer and antibiotics (100 U/mL penicillin/100 μg/mL streptomycin and 40 μg/mL gentamicin), added accordingly.
In static monolayer cultures (two-dimensional (2D) cultures), cells were seeded onto Matrigel®-precoated culture plates at a density of 5 × 104 cells/cm2. Hepatocytes were left untreated for at least 12 h at 37°C in a humidified atmosphere with 5% CO2 in air, allowing cell attachment. The medium was changed the following day to remove unattached cells. The culture medium was renewed every 24 h, and the cells were routinely examined under phase contrast microscopy before every culture medium renewal. The supernatants and cells were collected according to the protocol at the indicated time points and stored at −20°C for further assays.
In stirred tank cultures (3D cultures), single-cell suspensions were seeded in 125 mL spinner vessels or in a 250 mL stirred tank bioreactor. An inoculum of 1.2 × 105 cells/mL or 2.4 × 105 cells/mL was used for a final volume of 70 or 200 mL, in the spinner vessels or in the bioreactor, respectively, with supplemented medium at 15% FBS (to promote cell aggregation). Stirred tanks were placed on a magnetic stirrer, agitated at 60 rpm, and kept at 37°C in a humidified atmosphere of 5% CO2 in air. After 3 h in culture, to avoid settling of the cells, the stirring rate was increased to 80 rpm. After 24 h, 50% culture medium was changed and added to a final culture volume of 125 or 250 mL, in the spinner vessels or in the bioreactor, respectively, and the FBS concentration was adjusted to 10% (v/v). To maintain the aggregates, the operational mode applied was a 50% medium substitution (refeed mode) every 4 days, for nutrient availability and to decrease the accumulation of bioproducts of cellular metabolism that can be toxic to the cells.
Cells were counted using a Neubauer counting chamber, and the cell viability was determined by the trypan blue exclusion method. Cell counting data was presented as percentage of cell survival considering cell number at day 1 as 100%.
Cultures in the bioreactor
To ensure a fully controlled cell culture environment, a glass vessel (Fig. 1) that could be adapted to a commercially available bioreactor control unit (B-DCU; B-Braun Biotech International, Melsungen, Germany) was designed and developed in our laboratory. The internal geometry and the stirrer (paddle impeller) of the vessel are similar to the commercially available spinner vessels. The vessel has multiple (up to eight) upper cap ports for different applications, such as pH and pO2 meters (Mettler-Toledo, Urdorf, Switzerland), that allow online measurement and control of these parameters, as well as easy sampling and the addition or removal of medium or other supplements or solutions. The pH is kept at 7.4 by injection of CO2 and addition of base solution (NaOH, 2M). The dissolved oxygen concentration is maintained at 30% via surface aeration with air. The temperature was kept at 37°C by water recirculation in the vessel jacket controlled by a thermocirculator bath. The bioreactor controller unit was used to monitor and control pH, pO2, and temperature. Data acquisition and process control were performed using Multiple Fermenter Control System for Microsoft Windows (MFCS)/Win Supervisory Control and Data Acquisition (SCADA) software (B-Braun Biotech International).

Minibioreactor apparatus. (
Determination of lactate dehydrogenase activity
The release of intracellular enzymes—in particular, lactate dehydrogenase (LDH)—in the culture supernatant can be correlated with cell viability along the culture. This approach assumes that higher rates of release of enzymatic activity correspond to increased cellular damage and thus a loss in culture viability. 26 Hence, the extent of cell lysis was assessed by determination of LDH released to the medium as previously described by Vassault. 27
Determination of albumin secretion and urea synthesis
The secretion of albumin from hepatocytes was measured by an enzyme-linked immunosorbent assay (ELISA) using NEPHRAT albumin test kit (ref. NR002; Exocell, Philadelphia, PA). The assay was performed according to the manufacturer's description. The results were expressed as μg/day/106 cells at the indicated time point.
The urea synthesis rate was determined using a quantitative colorimetric urea kit (QuantiChrom™ Urea Assay Kit, DIUR-500, ref DIUR-500; BioAssay Systems), according to the manufacturer's instructions. The results were expressed as μg/day/106 cells at the indicated time point.
Testosterone hydroxylation
Testosterone is regio- and stereoselectively metabolized by CYPs to several hydroxylated metabolites that were extracted and analyzed. In this study, 4-androstene-3,17-dione (androstenodione), and 2α, 7α, 6β, and 16β-hydroxytestosterone were analyzed. Briefly, 250 μM testosterone dissolved in culture medium was added to cells and incubated for 2 h at 37°C. Hydroxylated metabolites were extracted with dichloromethane. After centrifugation (2000 g, 5 min), the organic phase was collected to a new clean tube and allowed to evaporate. The pellets were resolubilized in a mixture of methanol and water (50%/50%, v/v) for high-performance liquid chromatography (HPLC) analysis. Separations were performed on a 250 × 4 mm RP18 Lichrocart, 5 μm column using a Merck Hitachi LabChrom Elite chromatograph equipped with an autosampler, column oven, and diode array detector. Data analysis was performed with EZChrom Elite data system.
7-ethoxycoumarin-O-deethylase (ECOD) activity
7-ethoxycoumarin-O-deethylase (ECOD) activity was measured according to Gomez-Lechon et al. 28 with slight modifications. Salicylamide (1.5 mM) was added to the medium to prevent conjugation of 7-hydroxy metabolites (7-HC) of 7-ethoxycoumarin. 29 Therefore, hydrolyzation treatment with β-glucuronidase/arylsulfatase was overcome. The activity is expressed as nmol of 7-hydroxycoumarin formed per hour and per 106 cells.
Uridine diphosphate glucuronoltransferase activity
The uridine diphosphate glucuronoltransferase (UGT) activity was determined by quantification of the substrate, 4-methylumbelliferone (4-MU), before and after cell incubation with the substrate. The procedure was performed according to Gomez-Lechon et al. 28 with slight modifications. Briefly, a 100 μM solution of 4-MU in 0.01 M PBS was incubated with cells for an hour at 37°C. Samples were measured by fluorescence with emission at 450 nm and excitation at 320 nm. The 4-MU remaining concentration was determined based on a standard curve generated in PBS spiked with 0, 1.56, 3.12, 6.25, 12.5, 25, 50, and 100 μM. The activity is expressed as nmol of 4-MU metabolized per hour and per 106 cells.
Preparation of test compounds
All compounds used for metabolic stability assays were purchased from Sigma-Aldrich (Taufenkirchen, Germany). Stock solutions were prepared by dissolving diphenhydramine in acetonitrile (Carl Roth, Karlsruhe, Germany) and troglitazone in dimethyl sulfoxide (Carl Roth) at a final concentration of 10 mM. The highest solvent concentration used was 0.05% (v/v).
Metabolic stability assay
Stock solutions (10 mM) in acetonitrile and dimethyl sulfoxide were diluted in Williams' E complete medium to obtain working solutions. The concentration of final compounds during each assay was 5 μM.
In 2D cultures (24-well plates), the reaction was initiated by the addition of 250 μL of test item working solution at a concentration of 5 μM to the preincubated hepatocytes at 37°C for 5 min. For 3D cultures, 125 μL of the compounds stock solution (10 mM) was added to the 250 mL bioreactor 3D culture resulting in a final concentration of 5 μM. Cell concentration in each culture system was as described in the Cell Culture section. Controls consisting of compounds dissolved in supplemented medium, but not in the presence of cells, incubated at 37°C, were always performed in parallel. To stop the reaction, 250 μL of the test item samples was removed from the incubations at the respective time points and processed for acetonitrile precipitation. Isolation of the test items was performed by addition of 107 μL acetonitrile containing the internal standard griseofulvin (ISTD, 1 μM) to 250 μL sample and calibration standard, respectively. After vigorously shaking (10 s), sonification (10 s), and centrifugation (5000 g, 10 min), an aliquot of the particle-free supernatant was subsequently subjected to liquid chromatography-mass spectrometry (LC-MS/MS) for analysis. The HPLC system consisted of a MS Plus pump (Surveyor, Waltham, MA) and an auto sampler (Surveyor). Mass spectrometry was performed on a TSQ Quantum Discovery Max triple quadruple mass spectrometer equipped with an electrospray (ESI) interface (Thermo-Finnigan, Waltham, MA) and connected to a PC running the standard software Xcalibur 1.4.
Statistics
The data presented for the different culture parameters represent mean values for three independent experiments with two determinations each. Comparison of mean values between 2D monolayer, 3D spinner, and 3D bioreactor cultures, as well as the effect of inoculum concentration study, was performed using an appropriate one-way analysis of variance (ANOVA) test. Data evaluation between 2D and 3D spinner cultures maintained with Williams' E and Vito medium, on the other hand, was performed using a two-way ANOVA test. Multiple comparisons between groups in each case were performed using a Tukey's honestly significant difference test. Level of confidence was set at α = 0.05 for all tests, with p-values lower than 0.05 considered statistically significant.
Results
3D culture optimization
Evaluation of culture conditions that better granted specific hepatocyte cellular functions, using previously established biochemical assays, was examined. Albumin secretion and urea secretion by hepatocytes are indicators for long-term functional performance of hepatic cultures, whereas the activity of phase I (such as CYP 450) and phase II enzymes mirrors the capacity of hepatocytes to metabolize xenobiotics. On the other hand, general culture parameters, such as LDH, glucose, and lactate levels, have also been considered. The spheroid performance as 3D structures cultured in spinner vessels (3D cultures) was evaluated against conventional cultures in monolayer static conditions (2D cultures).
Immediately after isolation, cells were inoculated in 3D and 2D conditions and allowed to stabilize overnight. Cultures were maintained for 2 weeks. Stirring rate in suspension cultures was kept at 60 rpm overnight. On day 1, cell adhesion to the plastic surface of the culture dish was observed in static conditions, whereas in suspension cultures small tightly packed groups of cells, of approximately 100 μm, were detected. During the remaining culture time, stirring rate was increased to 80 rpm in 3D cultures; spheroids became larger, reaching sizes of 100–300 μm in diameter (Fig. 2). Such size range was not critical regarding the formation of necrotic centers as assessed by LDH (data not shown).

Phase contrast microscopy, with an amplification of 100 ×, of a 3D hepatocyte spheroid, cultured in stirred tanks and sample collected at day 3 of culture.
In both monolayer (2D) and spheroid (3D) cultures, no cell proliferation was observed, as expected for hepatocyte primary cultures. However, in general, approximately 2.5-fold higher viability for a longer period of time (as shown in Fig. 5D) could be achieved when hepatocytes were cultured as 3D structures.
Effect of inoculum concentration, impeller type and medium composition on cultures performance
Two different inoculum concentrations (1.2 × 105 and 2.4 × 105 cells/mL) and two impeller types (ball and paddle) were compared when optimizing the 3D culture conditions. As for culture performance, the albumin secretion levels and ECOD activity did not vary much with different impeller (data not shown) or inoculum concentrations within 3D cultures, although they were consistently higher in 3D than in 2D conditions (Fig. 3) (p < 0.05). Albumin production decreased dramatically in 2D cultures, reaching residual levels after 2–3 days in culture, whereas 3D cultures allowed albumin secretion to continue for more than 8 days in culture (Fig. 3A). ECOD activity, on the other hand, was generally five times higher in 3D cultures than in 2D cultures (Fig. 3B) (p < 0.05). No significant difference could be observed between cultures with an inoculum of 1.2 × 105 and 2.4 × 105 cells/mL at days 2 and 7 of culture (p > 0.05). Urea synthesis was significantly higher in spheroid cultures with an inoculum of 1.2 × 105 cells/mL (p < 0.05). Moreover, overall synthesis of testosterone metabolites was also higher in the later culture conditions, detectable up to day 12. In summary, spheroid cultures, with an inoculum of 1.2 × 105 cells/mL using a paddle impeller, resulted in enhanced hepatocyte functionality.

Effect of culture time in (
Regarding medium composition, Williams' E medium and Vito medium were selected. Our results showed that although spheroids cultured in Vito medium allowed 20% higher survival rate maintenance, 3D cultures in Williams' E resulted in significantly higher albumin secretion (Fig. 4A) (p < 0.05), for longer periods of time, never reaching residual levels before day 9, whereas with Vito medium residual levels were observed after 3 days in culture. Overall, CYP 450 activity, determined by measuring ECOD (Fig. 4B) and testosterone metabolites (data not shown), was significantly higher in 3D when Williams' E medium was used (p < 0.05). Therefore, Williams' E medium was adopted in the following cultures.

Effect of culture time on (
However, in cell cultures a compromise between cell viability and cell functionality must be achieved; in hepatocyte cultures for pharmacological purposes, it is critical to keep hepatocytes functionality for as long as possible; therefore, Williams' E medium overall results showed greater advantage over Vito medium results.
Metabolic performance of bioreactor cultures
Because the optimized 3D cultures in spinner vessel improved liver-specific functions, we went forward to apply the optimized parameters to a bioreactor (fully controlled environment) (Fig. 5A–C) where an equivalent or better culture performance could be expected. Cultures were maintained for a total of 21 days in the bioreactor, as well as in spinner vessels and in monolayer, herein used as control cultures. Higher survival rates could be achieved when hepatocytes were cultured as 3D structures (Fig. 5D), confirming the results obtained in spinner vessels described above.

Profiles of (
In both 3D cultures, after an adaptation phase that lasted about 3–4 days, cells showed relatively stable metabolic activity over at least 10 more days of culture. LDH leakage rates were high at day 1 but declined during the culture time, followed by a slow increase from day 14 in 3D cultures. Biochemical data for days 4–14 showed no significant difference among 3D cultures, whereas 2D cultures reached residual levels after 2–3 days in culture without recovering. In 3D bioreactor cultures, urea secretion (Fig. 6A) values were maintained at a stable level, showing only a slight decrease from day 14 onward. Still, urea production was significantly higher in 3D bioreactor than in the other two culture systems (p < 0.05). Albumin production (Fig. 6B) in 3D bioreactor culture after day 1 was maintained at nearly constant levels throughout culture time, decreasing approximately 75% only on day 21. Significantly higher albumin production values were obtained in 3D culture systems (p < 0.05), although even higher in the 3D bioreactor cultures (p < 0.05).
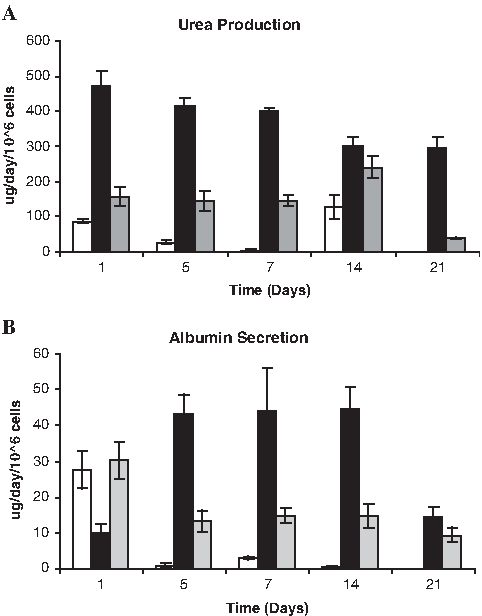
Functional capacity of hepatocytes over long-term cultivation periods was assessed by determining (
The testosterone metabolism and ECOD activity are critical routine assays to determine the activity of the CYP 450 (CYP) isozyme system. In Figure 7A, it is shown that ECOD activity was significantly improved in 3D cultures when compared to 2D cultures from day 1 onward (p < 0.05), with even higher values being obtained in the bioreactor (p < 0.05). Concerning the testosterone metabolism, metabolites such as androstenodione and 2α, 7α, 6β, and 16β-hydroxytestosterone were considered. The formation of those metabolites by isolated hepatocytes cultured in 2D and 3D conditions (spheroids cultured in spinners vessels or in the bioreactor) during 21 days was quantified. The quantity of hydroxytestosterone metabolites produced by freshly isolated cells was variable depending on the metabolite and on the culture system. However, clearly testosterone hydroxylation profiles were always higher in both 3D systems. In monolayer cultures, results showed that the testosterone metabolism dropped dramatically after day 4 in culture, where no more activity could be detected. In contrast, in 3D bioreactor cultures, activity increased with culture time to approximately 4.5-fold that of fresh cells after 3–4 days in culture. Testosterone metabolites were detected up to day 16 in the bioreactor culture and up to day 12 in the spinner vessels. Metabolism of testosterone to 6β and 2α-hydroxytestosterone was similar in both 3D culture systems, showing average values of 4.4 pmol/min/106 cells and 4.6 pmol/min/106 cells, respectively. On the other hand, metabolism of testosterone by combined CYP 2B1, 2C11, and 2B2 activity (resulting in the formation of androstenodione) in the bioreactor was 5.2-fold greater than in the spinner vessel. Metabolism of 7α-hydroxytestosterone and 16β-hydroxytestosterone in the bioreactor was 3.2- and 2.7-fold greater than in the spinner vessel, respectively.

Hepatic metabolic function was assessed by determining the activities of (
Among phase II enzymes, UGT is involved in conjugation processes of substrates. Figure 7B shows results for UGT activity. A significant improvement of UGT activity in the bioreactor system could be observed (p < 0.05) reaching a maximum activity at day 14 when compared to the other two culture systems. Although a considerably lower activity was found, the hepatocytes cultured on 3D structures in the spinner vessel still exhibited a UGT activity significantly higher than those cultured in monolayer (p < 0.05).
Metabolic stability assays
To further validate the newly developed bioreactor-based 3D primary hepatocytes culture system, metabolic stability of a couple of compounds (troglitazone and diphenhydramine) was examined both in the 3D bioreactor and in the 2D culture system. The incubations were always performed in supplemented medium because this was mandatory for the bioreactor and data had to be comparable. No influence of the incubation medium in the compounds metabolism was observed. In Figure 8A and B, it is shown that, at day 2 of culture, cells in both culture types were able to metabolize the compounds equally. Moreover, at day 21 bioreactor cells in 3D cultures were still able to metabolize the compounds, whereas in 2D cultures, because cell viability was very low (Fig. 5D), the same could no longer be performed.
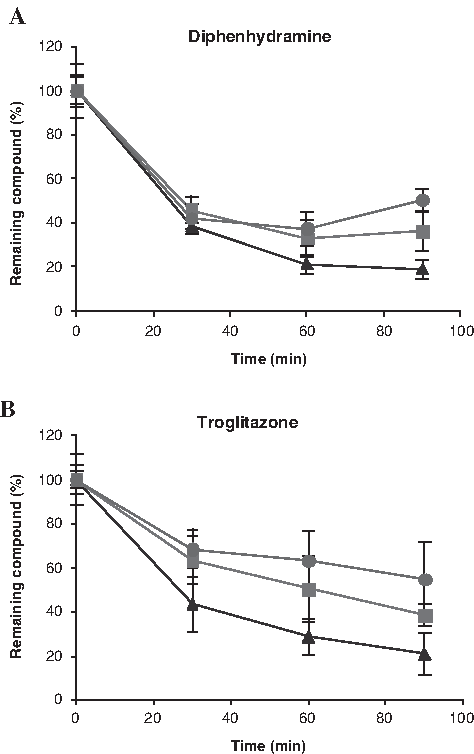
Metabolic stability of (
Discussion
An important scientific achievement in this study was the development of an alternative in vitro hepatocyte culture system that can be used for both toxicological and pharmacological studies.
The choice of a 3D system was based on the fact that hepatocytes retain both high viability and differentiated liver functions when cultured as 3D aggregates. This was shown by Li et al. 30 who successfully cultured viable spheroids on nontissue culture plastic plates for 1 month. Li et al. and Yoffe et al. 31 were also among the first authors to report hepatocyte aggregates cultured in rotating wall vessels. Aggregates could be cultured for a prolonged period with differentiated cell morphology and functions.
The advantage of our system over previously described in vitro models is that it conjugates several characteristics that are important for functional hepatocytes culture and for culture practicability in general. The cells were cultured in a fully controlled environment bioreactor where sampling throughout the whole culture time was made possible. It is a setup based on commercially available equipment and therefore accessible to any laboratory or company. Such system decreases the biological cell-to-cell and culture-to-culture variability that has thus far limited the size of cell-based experiments. Further, not only this minibioreactor did allow for oxygen control in suspended cultures,32,33 but also the stirred tank environment enabled a better medium oxygenation further resembling the physiological blood flow. 34 It has been shown that the absence of oxygen in culture can lead to a cellular metabolic state of sensitive or even rapid hypoxic injury, due to the depletion of dissolved oxygen; regular culture conditions do not allow this control as a bioreactor would. 33
In this study, before the application of the 3D hepatocyte cultures to the bioreactor system, some culture parameters were tested in spinner vessels to establish a culture system for hepatocytes in vitro maintenance. The maintenance of a viable cell culture in suspension conditions requires optimization of several cultivation parameters. In particular, for spheroid cultures, it is mandatory to control their size hydrodynamically, to avoid necrotic centers resulting from oxygen and nutrient transport limitations. Experimental parameters such as stirring rate and impeller design can be optimized to circumvent such drawbacks without the decrease in culture viability. 35 Moreover, medium composition and inoculum concentration are also crucial parameters to be considered when developing a new culture system. Therefore, parameters such as inoculum concentration, medium formulation, impeller type, and stirring rate were studied herein. Concerning the media, higher survival rates, but lower hepatocyte-specific activities were observed with Vito medium than with Williams' E medium. Probably, it was due to the fact that Vito medium was designed for culturing human rather than rat hepatocytes, although it was specially developed for culturing hepatocytes in bioreactors. 36 On the other hand, both the lower inoculum concentration and paddle impeller type showed better results (Fig. 3), the lower inoculum being an advantage to minimize the low availability of hepatocytes.
In this context, the optimized conditions (1.2 × 105 cells/mL inoculum, cultured in supplemented Williams' E medium with a paddle impeller) were used in the bioreactor. Hepatocyte-specific functions such as albumin and urea secretion are important indicators when evaluating hepatocyte cultures. Albumin secretion is a general marker of protein synthesis in hepatocytes as it is synthesized almost entirely by the liver. It is a source of amino acids for various tissues. Decreased albumin level can serve as an index of impaired synthetic protein capacity by the liver. 37 Likewise, the conversion of ammonia to urea is a vital liver function. The ammonia produced through amino acid deamination can be detoxified by the combination of ammonia with CO2 to form urea in the liver. 38 In this study, albumin secretion rapidly decreased in monolayer cultures but not in 3D cultures. This is not an unexpected finding because studies of the albumin metabolism have shown that the synthesis and albumin secretion by isolated hepatocytes occur only in early culture phases. 37 However, in our culture system this effect was circumvented because albumin secretion could be detected throughout a significant period of time, decreasing only after day 14. Although higher values were observed in both 3D culture systems, in contrast to the 2D system, the bioreactor clearly showed higher values than the spinner vessels. Accordingly, in both 3D cultures urea production levels were clearly higher than those produced by hepatocytes cultured in monolayer. Still, higher levels were observed in the bioreactor than in the spinner vessel, suggesting that not only does growth in 3D structures enhance liver-specific functions, but also a fully controlled environment, resembling the in vivo conditions, is an issue in this type of cultures. One parameter further controlled in the bioreactor is the dissolved oxygen, a key factor on hepatocyte cell long-term cultures. 34 Therefore, an oxygenated environment may be the reason why better results are observed in the bioreactor.
Biotransformation of xenobiotics by cultured hepatocytes can be assessed in terms of activity using specific substrates. Synthetic substrates metabolism by primary cultures of hepatocytes cultured in plates and in stirred tanks (spinner vessels and bioreactor) was examined by measuring phase I and phase II enzyme activities. The functionality of CYP 450 enzymes in the culture systems was assayed via the regiospecific hydroxylation of testosterone and the 7-ethoxycoumarin O-deethylation (ECOD activity), whereas phase II enzymes were assayed by UGT. Metabolism of several endo- and exogenous substances is carried out by a set of phase I and phase II enzymes. Their activities determine the overall therapeutic and toxic profiles of a drug. 39
The 6β-hydroxylation of testosterone is predominantly mediated by CYP 3A, the major isoform being 3A4 in humans 40 and 3A1 in rats. 41 Oxidation of testosterone to 4-androstene-3,17-dione in rats is predominantly catalyzed by CYP 2B1 with minor contributions from CYP 2C11 and CYP 2B2. 42 In human hepatocytes, CYP 2C19 and to a lesser extent CYP 2C9, are responsible for production of 4-androstene-3,17-dione, with potential contribution from CYP 2B6. 43 2α-Hydroxylation is predominantly mediated by CYP 2C11 in rat. 42 A CYP 2C11 orthologous enzyme has not yet been identified in humans, but 2α-hydroxylation is presumably mediated by an enzyme belonging to the 2C subfamily.44,45 ECOD activity in rat hepatocytes is mostly mediated by CYP 1A and 2A families. 46
Research in the area of drugs biotransformation in static 2D primary human and rat hepatic sandwich cultures has revealed a preservation of the major forms of phase I and phase II enzymes that respond to rifampicin by increasing their activities. 12 However, the long-term maintenance of the phase I enzymes Ethoxyresorufin-O-deethylase (EROD) (CYP1A1/2) and ECOD (CYP2B1/2), as well as of the phase II enzyme UGT, was achieved in sandwiched primary porcine hepatocyte cultures in a dynamic flat-sheet bioreactor with a gas-permeable membrane 47 and in a small-scale bioreactor with an hepatic sandwich model and a gas-permeable membrane. 15 Similarly, our work using a fully controlled bioreactor showed that these activities were also well preserved in rat hepatocytes. Here, the importance of an adequate oxygenation for the phase I, but mostly phase II metabolism, is emphasized by the lower activities in spinner vessels and by the lack of an effective phase I and phase II enzymatic activities in the conventional 2D monolayer culture dishes after 2–3 days in culture.
The metabolic stability of a drug candidate is an important consideration in determining its potential for human use. Those studies are usually conducted during earlier stages of drug development to allow the selection of structures with the most appropriate stability for further development. Because of the presence of all hepatic drug metabolizing enzymes and cofactors at physiological levels, intact hepatocytes represent a more relevant experimental system than liver microsomes for general metabolic stability screening. An interesting achievement in the application of intact hepatocytes for the evaluation of metabolic stability is the incubation of the test compound and hepatocytes in 100% human serum, therefore providing an experimental condition similar to humans in vivo. For instance, it has been reported that hepatic clearance in vivo can be predicted more accurately from data obtained with hepatocytes incubated in serum than from data obtained in the absence of serum, using either rat or human hepatocytes. 48 Thus, the use of a compound whose behavior in metabolic stability assays is known can be considered as reference for the evaluation of a culture system under development. In this work, metabolic stability of reference compounds (diphenhydramine and troglitazone) was evaluated with the new in vitro culture system using serum-supplemented medium. In fact, the results clearly showed that compound clearance was as in vivo, thus highlighting its potential for pharmacological studies.
This work shows that it is possible to culture primary rat hepatocytes as 3D structures using a fully controlled bioreactor and that this culture strategy improves hepatocyte functionality. By more closely resembling the in vivo environment, due to its three-dimensionality and cell–cell interaction, this model supported long-term viability and the maintenance of several differentiated hepatocytes properties, including xenobiotic metabolism, for up to 21 days. In contrast, traditional monolayer cultures lost these properties after 3–4 days in culture. Therefore, this system offers a much improved alternative culture system for predictive in vitro studies, to study mechanisms of CYP isoforms and to perform metabolic stability studies. It is also a new method for testing drug interaction under 3D conditions, because it has the advantage of offering a better in vitro–in vivo correlation.
This culture system can be implemented for primary human hepatocytes avoiding interspecies differences. Another promising application is for human stem cell–derived hepatocytes culture, which is currently being assessed in our laboratory.
Footnotes
Acknowledgments
This work was supported in part by the European Comission Project Vitrocellomics (LSHB-CT-2006-018940) and the Fundação para a Ciência e Tecnologia (BPD-26623). This work was performed at Animal Cell Technology Laboratory, IBET/ITQB-UNL, Portugal.
Disclosure Statement
No competing financial interests exist.