
Abstract
Background:
Circulating autologous CD133+ stem cells have been differentiated into a number of cell types that have the potential for clinical use, including endothelial cells. These cells are infrequently found in peripheral blood specimens, and this limits their use in clinical applications. To address this problem, we have developed an extracorporeal cellular affinity (ECA) column that can recover CD133 expressing progenitor cells with high efficiency.
Methods and Results:
In sheep model, 1800 mL of blood was passed through a Sepharose-based column with affinity for CD133. Unbound cells and plasma were returned to the animal. Our results show that this process has a minimal effect on the hematologic and physiologic parameters of the animal. This recovery approach generated over 600-fold more endothelial colony forming units than conventional buffy leukocytes isolated from a peripheral blood specimen. Ultimately, the increased cell recovery of the ECA column enables the generation of a cell biomass for therapeutic purposes in nearly a third of the time.
Conclusions:
This technology may facilitate the generation of large numbers of progenitor-derived cells for clinical therapies and reduce the time required to attain clinically relevant cell numbers while minimizing loss of other important cell types to the donor.
Introduction
To address these challenges, effective methods to identify and select these cells to expand their clinical utility have been sought.10,11 CD133 (prominin-1) is a surface antigen of the prominin family of pentaspan transmembrane glycoproteins. While the function of CD133 is not yet known, it has been identified as a marker for multipotential progenitor cells12,13 as well as a marker of tumor-initiating cells. 14 Cells expressing CD133 may differentiate into such distinct cell types as hepatocytes, neuronal cells, vascular endothelial cells, renal epithelium, and smooth muscle cells.2–5,15,16 Further, they have demonstrated the ability to reconstitute the entire hematopoietic system.13,17,18 This extensive differentiation potential has led to the use of CD133+ cells in a variety of potential clinical applications.19–26
Notably, CD133+ cells are rare in isolated peripheral blood, ranging from only about 0.01% to 0.0001% of isolated leukocytes.6,7,27,28, The percentage of cells capable of forming endothelial colonies is even lower, approximately 1 colony in 100 million leukocytes. 7 The low numbers of circulating CD133+ endothelial progenitor cells remains a major obstacle to the development of clinical applications of these cells. 9
Currently, CD133+ purification techniques are limited by the volume of blood that can be safely removed from a living donor. As these cells are infrequently found in the peripheral blood, processing larger blood volumes would likely lead to the isolation of increased numbers of CD133+ cells. To this end, we have developed an extracorporeal cellular affinity (ECA) column that can recover CD133 expressing progenitor cells ex vivo with high efficiency. In contrast to conventional in vitro methods of harvest, cells not captured by the column are returned to the body, thus preserving other cell types. Herein, we describe an approach to recover CD133+ progenitor cells from a large volume of peripheral blood in a living donor while minimizing losses of other cell types. ECA columns directed toward CD133 or an isotype control were compared to buffy leukocytes purified from an isolated blood specimen using traditional gradient methods.
Methods and Materials
Animals
Female Dorper cross sheep between the ages of 4 and 12 months were purchased from RSI Farms (Mocksville, NC). All procedures were approved by the Wake Forest Animal Care and Use Committee. Sheep were monitored perioperatively for evidence of neurologic, cardiovascular, allergic, or pulmonary complications. Physiologic parameters included heart rate, blood pressure, end-tidal carbon dioxide, and pulse oximetry before, during, and immediately after cycling of the ECA column or sham-operated animals. Hematological monitoring included complete blood counts (CBC) with differential drawn immediately before column cycling and 24 h postoperatively (n = 8). In control animals blood work was performed immediately after vascular exposure and 24 h postoperatively.
Antibodies
Anti-human CD133 immunoglobulin G (IgG1) monoclonal antibody was prepared from the AC133.1 hybridoma line (ATCC, Manassas, VA) using a hollow fiber unit based on a model that has been previously described 29 with serum-free medium (SFM4MAB; Hyclone, Logan, UT). Antibody was then purified by Hi-Trap Protein G cartridges (GE Healthcare, Piscataway, NJ). Mouse isotype control IgG1 (MOPC-21) was purchased from Sigma (St. Louis, MO). Both antibodies were concentrated into saline pH 7.4 using Ultra-15 centrifugal 50 kDa devices (Millipore, Billerica, MA).
Antibody conjugation of NHS Sepharose beads
CD133 or isotype control antibodies (5 mg/mL) were conjugated at a ratio of 500 μg antibody/mL N-hydroxy succinimide (NHS) Fastflow Sepharose beads (GE Healthcare, Buckinghamshire, United Kingdom) according to the manufacturer's instructions with one modification. Because starting bead sizes ranged between 45 and 165 μm, it was necessary to remove small beads that would not be retained by the 70 μm pore size mesh dividers in our column. The Sepharose was passed through a 70 μm nylon mesh cell strainer (BD Biosciences, San Jose, CA) before conjugation to remove the small beads. After conjugation and bead washing, beads were stored in 0.1% sodium azide until use.
ECA column
Columns were created using 50 mL Falcon tubes (BD Biosciences) and three 70 μm filter mesh strainers (BD Biosciences) and gas sterilized with ethylene oxide. Columns were assembled with 1 mL of conjugated beads evenly distributed among the three mesh filters of the column. Preoperatively, animals received a single dose of clopidogrel 75 mg and aspirin 375 mg orally the day before surgery. The carotid or superficial femoral artery and the jugular or superficial femoral vein, respectively, of sheep under general anesthesia were exposed. After heparinization with 100 units/kg of sodium heparin as part of accepted routine during vascular manipulation, the vessels were cannulated with the taper cut end of sterile arterial monitoring tubing (Arrow International, Reading, PA). The circuit was completed by connecting the tubing to each end of the column, and blood was cycled for a volume of 1800 mL. To normalize the volume of blood cycled, flow rates were measured by means of a three-way stopcock distal to the column over 10 s into a graduated reservoir and extrapolated into mL per minute. Subsequently, the time of column cycling was adjusted based upon the flow rate to achieve a consistent cycle volume of 1800 mL for each animal.
Cell recovery
After cycling the ECA column, beads were removed from each column and rinsed with sterile saline over 70 μm mesh filters to remove unbound cells. Beads were then treated with 0.05% trypsin-ethylenediaminetetraacetic acid (Gibco, Grand Island, NY) to separate the cells from the beads. Cells were passed through a new mesh strainer to remove the beads, and the cells were pelleted by centrifugation. Cells were resuspended in endothelial cell growth medium (EGM2; Cambrex, East Rutherford, NJ) for endothelial culture or subsequent receptor analysis. Buffy coat leukocytes were prepared from freshly heparinized peripheral blood specimens as previously described using gradient centrifugation. 30
Assessment of CD133 expression
ECA column–derived cells or buffy coat leukocytes were incubated in EGM2 medium for 8 h before analysis to allow for recovery of cell surface receptor expression. Equivalent numbers of buffy coat–derived leukocytes or column-derived cells were incubated with the biotinylated anti-CD133 antibody 293 (Miltenyi, Auburn, CA) for 40 min. Cells were secondarily stained with Fluorescein Isothiocyanate (FITC)-avidin (Vector Laboratories, Burlingame, CA) according to manufacturer's instructions. After being cytospun onto glass slides, cells were mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting medium (Vector) and examined under fluorescent microscopy with dual color imaging.
Endothelial progenitor colony forming unit assay
Cells recovered from 1800 mL of blood using either CD133 ECA or isotype IgG control ECA columns were plated onto fibronectin-coated 24-well plates (Costar, Lowell, MA), at a density of 25,000 cells/well. The entire eluted cell population was plated in this manner. As a control, buffy coat leukocytes collected from the same animal before column cycling were plated at the same density into a matching number of 24-well plates such that the same number of wells was plated for both the ECA column and buffy leukocyte cells. As very few cells were recovered by the IgG isotype control ECA column, it was necessary to plate fewer wells. Medium was changed at 5 and 8 days after the initial plating, and then at day 9 colonies in each well were counted by a blinded observer with experience in identifying endothelial colony morphology. Colony counts were then expressed as the yield from each column (CD133 or isotype control, n = 4 for each) and for the matching number of buffy leukocyte–plated wells (n = 8, four matching sets for each of the two column conditions, CD133 or IgG isotype). In a separate buffy leukocyte colony assay, buffy coat leukocytes purified from freshly heparinized blood were plated in a fibronectin-coated six-well dish containing EGM2 medium. Each well contained the cells derived from a 10 mL sample of blood. Nonadherent cells were serially passaged to new fibronectin-coated plates every 24 h over 3 days. Colonies were then counted as described. Of note, no colonies from either the buffy leukocytes or the ECA-purified cells were observed before 7 days in culture.
Immunohistochemistry
Confluent cells plated in chamber slides were fixed with formalin, rinsed in PBS, blocked, and then incubated with primary antibodies toward von Willebrand factor (vWF; DAKO, Carpinteria, CA), vascular endothelial growth factor receptor (VEGFR2; Pharmingen, San Diego, CA), and endothelial nitric oxide synthase (eNOS; #610297; Transduction Laboratories, San Jose, CA) using 1:25 and 1:50 dilutions. For staining of lectin, cells were incubated with biotinylated UEA Lectin (Vector). Primary antibodies or lectin were then localized with FITC anti-rabbit, FITC anti-mouse, or FITC-avidin (Vector) at 1:100 dilution.
Growth curves
Growth curves were determined for cells isolated using a CD133 ECA column and cells isolated using the traditional leukocyte buffy coat method. The cells were plated and counted using a standard hemacytometer at each passage. To normalize cell plating density between column and buffy leukocyte–derived cells, cells were seeded at a consistent density of 10,000 cells/square cm. Cells were progressively passaged until a total cell number of 10 million cells was reached in each group. This number was chosen as it is routinely used to seed bioengineered vascular grafts in our laboratory. Because of a low expected frequency of colony appearance in the buffy leukocyte group, a larger number of specimens were studied for this group (n = 18).
Statistics
Intraoperative physiologic data and changes in cell counts (pre- vs. postoperative) were compared between column and control animals with Wilcoxon rank sum testing using exact methods. Endothelial cell growth curves for column-isolated versus buffy leukocyte–derived cultures were compared using survival analysis, with the survival endpoint defined as number of days to reach ≥ 10 million cells. Data for the growth curve analysis are expressed as a median time to achieve this target cell number with confidence intervals as shown.
Results
Hematologic and physiologic stability during column use
The ECA column consists of a Sepharose matrix with antibody-based affinity toward the progenitor antigen CD133 (Fig. 1). The extracorporeal circuit consists of an arterial cannula carrying blood to the column and a venous cannula that returns the blood to the anesthetized sheep. From a physiologic aspect, sheep vitals including heart rate, blood pressure, end-tidal carbon dioxide, and pulse oximetry were not significantly different between column-cycled versus sham-operated control animals (p > 0.05 for all variables) (Table 1). Given the expected turbulent environment of a device cycling high volumes of blood and the theoretical potential for allergic reaction or disturbed hematologic parameters, perioperative hematologic and physiologic parameters were assessed in sham-operated control and column-cycled animals. CBC with differential were assessed before and after cycling of the ECA column as well as in control animals. A comparison CBC, drawn 24 h after the surgery, showed a postoperative elevation of neutrophils and eosinophils that was small but statistically significant from control animals. No significant neutropenia, depletion of platelets, or decrements in hematocrit of column-cycled animals were observed relative to control animals (Table 2).

Schematic of ECA column. An arterial cannula carries peripheral blood to the column, which contains three tiers of mesh filters retaining antibody-coated Sepharose beads (70–165 μm diameter, inset). In this study, CD133+ cells are retained in the column, while unbound cells and plasma are returned to the donor by a venous cannula. Color images available online at
Sham control includes vascular exposure and vessel cannulation without column cycling. ECA, CD133 extracorporeal cell affinity; BP, blood pressure; CO2, carbon dioxide; O2, oxygen.
Sham control includes vascular exposure and vessel cannulation without column cycling. Units for pre and post values are cells/mm3 except hematocrit, which is expressed as percentage. ECA, CD133 extracorporeal cell affinity; Pre, cell count measured intraoperatively; Post, cell count drawn 24 h postoperatively; WBC, white blood cell.
Characterization of cells recovered by the ECA column
To demonstrate that cells eluted from the column were enriched for CD133+ cells, eluted cells were compared to buffy leukocytes for expression of the marker CD133 by fluorescent immunohistochemistry. Cells were cultured for 8 h to allow for recovery of CD133 antigen expression. Cells were then stained using FITC-labeled anti-CD133 antibody and counterstained with DAPI. Comparable cell densities are illustrated with the DAPI nuclear counterstain (Fig. 2A, B), and while buffy coat–derived cells exhibited only a rare positive staining cell (Fig. 2A), ECA column–recovered cells demonstrated a predominance of CD133+ cells (Fig. 2B). On average, 44% of cells were noted to be CD133+ among eluted cells.
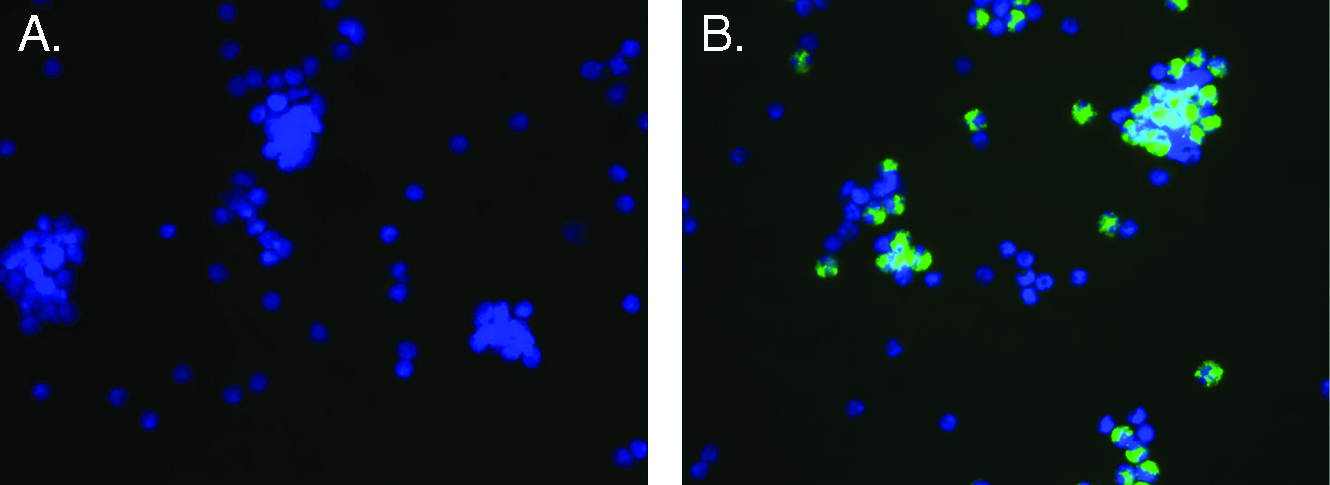
Comparison of CD133 expression on ECA column–eluted cells compared to buffy coat leukocytes. Buffy coat leukocytes (
Increased endothelial colony forming units from column-purified cells
To determine if the increased yield of column-purified CD133 cells would translate into an increased number of colony forming units compared to either IgG isotype control ECA column or buffy leukocytes, cells from each group were plated at a similar density with an equivalent number of cells. Twenty-five thousand cells eluted from CD133 or mouse isotype IgG control ECA columns were compared to buffy coat leukocytes from the same animal. Cells were plated under identical conditions and assessed for colony forming units at 9 days. Cells recovered from the CD133 ECA columns generated an average of 444 colonies/column, while no colonies were generated from either the mouse isotype control recovered cells or buffy leukocytes at the same cell density (Fig. 3). To define the volume of blood from which buffy leukocytes would generate an endothelial colony, a separate assay with a larger number of cells was conducted using the equivalent of 10 mL peripheral blood worth of buffy-derived leukocytes. The average initial leukocyte count recovered from this volume of blood averaged at 13.5 million cells. When cultured for 4 weeks, the average yield was only 0.72 colonies from each 10 mL of peripheral blood (n = 18). Further, only 44% of wells plated with leukocytes from this volume of blood formed any colonies. When compared to a 10 mL blood specimen processed as buffy coat leukocytes, a single run of the ECA column generates over 600-fold more endothelial colonies. For a blood volume normalized comparison, the number of buffy leukocyte–derived colonies extrapolated to 1800 mL would be 130 (= 0.72 × 180), while the number of colonies actually recovered by the ECA column for this volume of blood was over three times as high at 444 colonies. Based upon this data, even when adjusted for blood volume, the ECA column still conferred an advantage in terms of colony generation when compared to buffy mononuclear cultures.
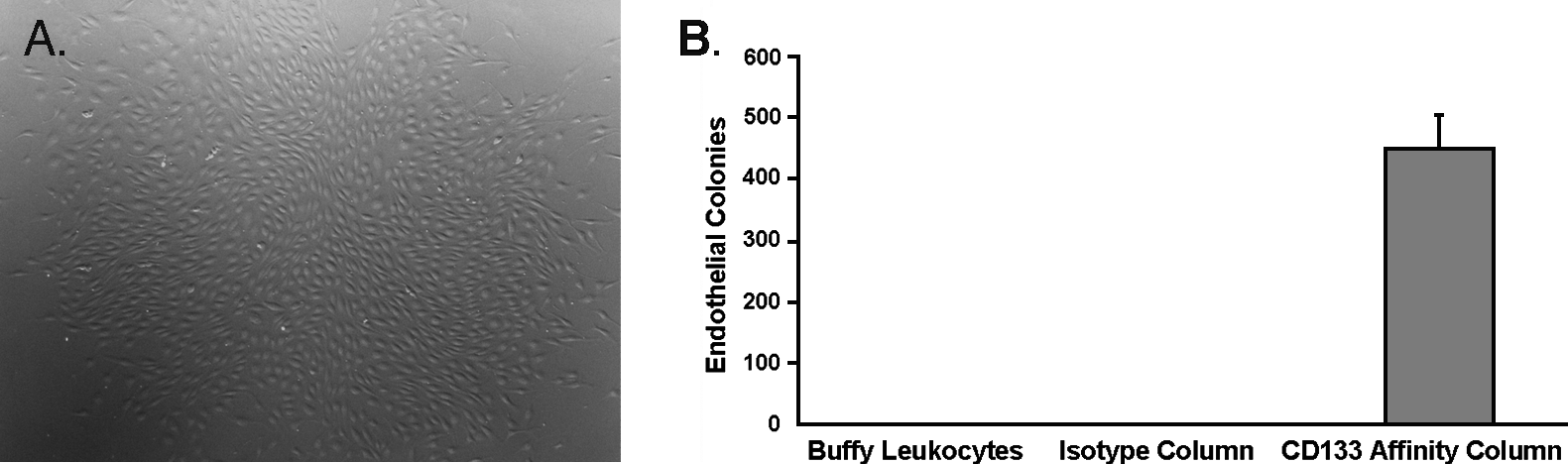
Endothelial colony assay of column-eluted cells versus controls. An image of a representative endothelial colony (40 ×) from the CD133 ECA column is shown (
Characterization of column-derived endothelial cells
To establish that endothelial cells differentiated from buffy leukocyte–derived progenitors were phenotypically identical to those derived from CD133 ECA–purified cells, immunohistochemistry was performed for four endothelial cell antigens. Both buffy leukocyte and ECA column–derived cells were found to express the endothelial markers eNOS (Fig. 4A, B), europeus 1 (UEA lectin) (Fig. 4C, D), VEGFR2 (Fig. 4E, F), and vWF (Fig. 4G, H).
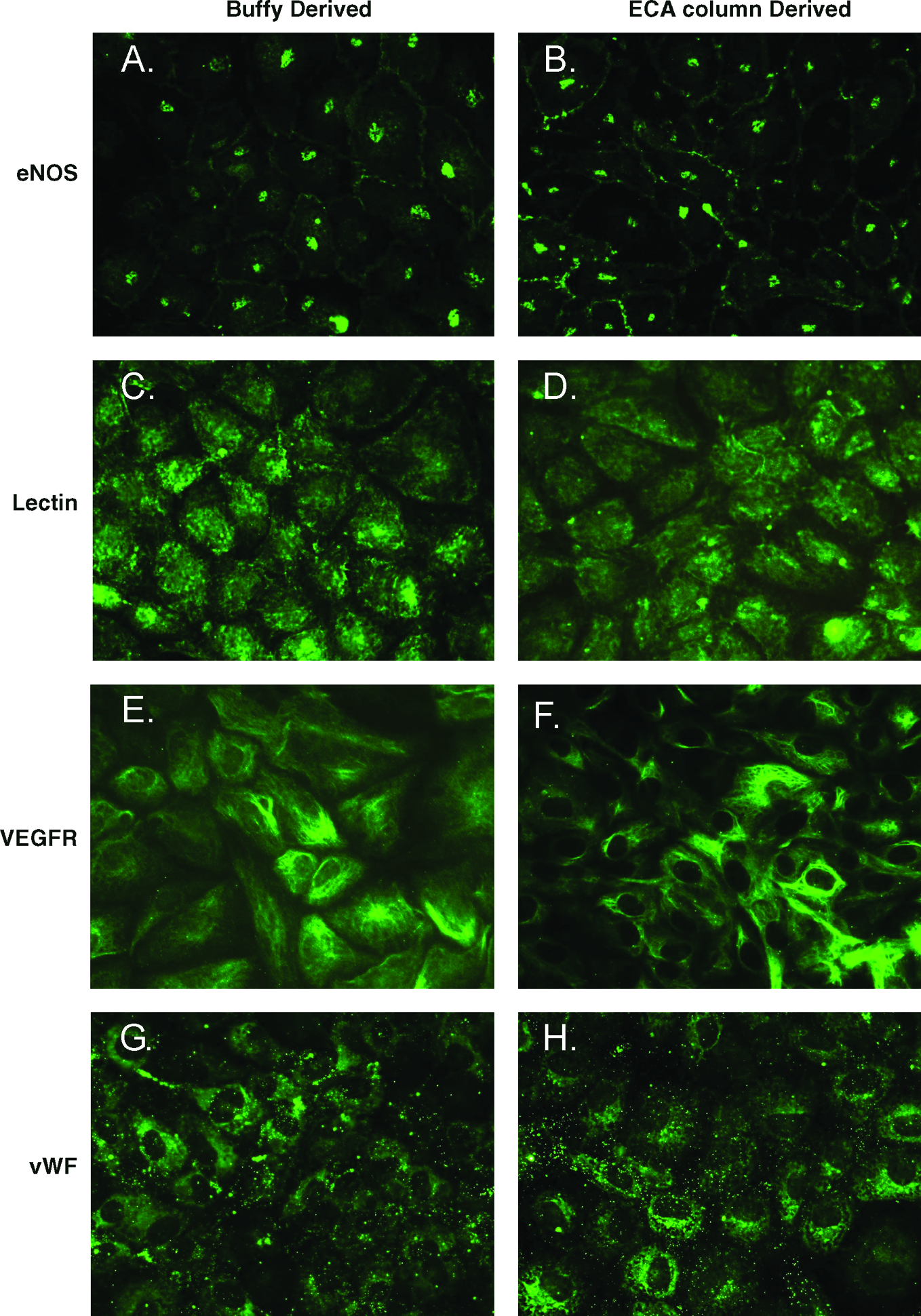
Comparison of immunohistochemical staining of endothelial markers between buffy leukocyte–derived endothelial cells and column-derived endothelial cells. Immunohistochemical staining for eNOS (
Improved growth kinetics of column-derived endothelial cells
Because this novel column approach for isolating CD133+ cells can isolate more progenitor cells, we expected that this would reduce the time needed to produce an adequate number of cells for use in clinical applications. Thus, we quantified the time advantage of this approach by comparing the growth kinetics of cells isolated from a 10 mL blood specimen isolated with standard methods to the kinetics of cells obtained from a single column cycling. To quantitatively compare the endothelial expansion of a standard ECA column run to the yield from buffy leukocytes isolated from the 10 mL peripheral blood specimen, resultant colonies were expanded to an endpoint of 10 million cells with normalization of cell density with each passage. As illustrated in Table 3, the increased starting number of progenitor cells from the ECA column conferred an expected expansion advantage, attaining the endpoint nearly three times earlier (p < 0.0001) than the buffy leukocyte–derived endothelial cells. Notably, despite maintenance at a similar cell density as their column-derived counterparts, 25% of buffy-derived colony sets exhausted their growth potential before attaining the 10-million-cell target.
Discussion
Stem and progenitor cells have been recognized as cell sources for various cell-based applications in the field of regenerative medicine due to their ability to differentiate into multiple cell types that could lead to functional tissue restoration.31–35 Progenitor cells are known to reside in multiple tissues in our body system as well as in tissues from all stages of development. While some stem and progenitor cells can be reliably obtained and induced into specific cell lineages for various cell-based applications, many may require establishment of reproducible cell isolation and culture systems. Further, efficient and consistent yield of these cell populations is necessary for clinical use.
Among these are a number of applications specifically by progenitors described to express the marker CD133.2–5,19–26,30 Cells expressing CD133 constitute a population of multipotential progenitor cells and can be found in circulation.6,27,28 These cells are reported to generate populations of endothelial cells, smooth muscle, hepatocytes, neural cells, and cells of the hematopoietic system.2–5,23 Current in vitro techniques for isolation of these cells are limited by the finite amount of blood that can be collected from an autologous donor and, in turn, the number of cells available for downstream expansion. In fact, patients most likely to benefit from progenitor cell therapies, such as patients with coronary artery disease, end-stage renal disease, and diabetes, are also the least likely to tolerate large volume blood donation for in vitro processing. We envisioned a system that would allow selective recovery of these circulating progenitor cells from the entire blood volume of the donor without affecting other blood cell types.
A number of systems have proven useful for cell sorting and isolation in vitro.36,37 These include a magnetic bead system using antibody-coated beads to isolate CD133 cells specifically from isolated leukocyte specimens. 36 These beads might, at first glance, seem applicable to extracorporeal recovery as well; however, the subcellular bead size would likely be difficult to retain in a high flow vascular circuit. In contrast, the ECA column system described in this study employs large, antibody-conjugated Sepharose beads. These beads are easily retained by an appropriately sized mesh strainer and readily allow unbound cells to pass back to the donor. Our animal model indicates that, because the majority of blood components are returned to the donor, this process does not significantly disrupt hemodynamic and other parameters even when a substantial blood volume of the animal is cycled through the column. A potential concern with an ECA column–based approach for progenitor recovery is the possibility of deleterious effects on cell counts, reflected as neutropenia, anemia, or thrombocytopenia. However, comparison of CBC before and after ECA column use revealed only a small but statistically significant rise in neutrophil and eosinophil counts. These changes are rather expected because a murine anti-CD133 antibody was used on this column, and the sheep immune system will naturally recognize the murine epitopes as foreign. Importantly, there were no statistically significant decrements in platelet count when compared to sham-operated controls. No animals exhibited acute or delayed toxicity, and there were no deaths among the study animals as observed over at least 2 months.
We have used buffy leukocyte cultures extensively and have found that only a small number of progenitors can be recovered from the small blood volumes that can be safely recovered in a single setting. As is shown in the data presented in this study, only 0.72 colonies, on average, were generated from 10 mL of peripheral ovine blood using traditional methods. This is similar to published data for human colony assays. In one of these studies, only a single colony was isolated from a comparable 20 mL of blood. 28 As a result of these low yields, a prolonged duration of culture is required to obtain the large numbers of cells that are required for clinical applications. Minimizing this culture duration would reduce the risks of senescence and culture-related infection before use in therapeutic applications. This study suggests that use of a column-based system allows isolation of a much larger population of progenitor cells, which significantly minimizes the culture time needed to attain the required number of progenitor-derived cells. It is interesting to note that ECA column–derived cells generated a larger number of colony forming units than would be expected by extrapolation from our buffy leukocyte colony assay for an equivalent amount of blood. Explanations for this phenomenon might include a paracrine effect from a higher density of column isolated CD133+ cells or, alternatively, an absence of negative regulation by contaminating cell types that would otherwise be present in buffy leukocyte populations. In support of the latter concept, we have been unable to enhance the number of colony forming units by simply increasing the density of buffy coat leukocytes in culture, and, in fact, the appearance of colonies actually decreased substantially when buffy leukocytes were plated at higher density (Tillman, 2007, unpublished data). Our results suggest a high degree of enrichment for CD133+ cells, but purity of only 44%. We suspect that many of the remaining cells may not yet have recovered expression of CD133 after trypsin elution.
In a clinical setting, treatment with granulocyte colony stimulating factor (G-CSF) increases the numbers of circulating CD133+ endothelial progenitor cells by a factor of 8- to 10-fold.38,39 Capitalizing on this increase in progenitors by G-CSF still requires a means to harvest cells safely from the blood. Approaches used clinically include phlebotomy and apheresis, but these are associated with loss of other cell types and procedural complications, 40 respectively. In contrast, the ECA column described here targets specific leukocyte subsets—in this case, CD133+ cells—with no decrease in other leukocyte populations and no significant decrements in other cell types. For these reasons, even in the context of G-CSF mobilization of peripheral progenitors, a column-based approach may improve progenitor yields.
For this study, we used the marker CD133 as a marker of endothelial progenitors although CD34 has similarly been used for this purpose. 41 CD133 is believed to be a more primitive marker than CD34, and perhaps for this reason, cells expressing CD133 have been shown to have a self-replicating potential and exhibit a higher degree of engraftment in bone marrow transplant when compared to CD34+ cells that do not express CD133.12,13,42–44 Thus, CD133 expressing cells may have more plasticity clinically than their CD34+/CD133− counterparts. Notably, cells expressing CD133 are known to be less prevalent in older humans patients. 45 While this study examined relatively young sheep, future studies will compare yields between young and older animals. The latter group, in particular, represents a population where this system is expected to be most useful for progenitor recovery as compared to conventional approaches.
The arteriovenous column circuit described here is advantageous because it is perfused passively by arterial pressure at a rate averaging 120 mL/min. This approach would be directly applicable in the majority of renal failure patients who undergo arteriovenous dialysis, wherein the column might be used in line with a dialysis cartridge. A venovenous approach using a syringe, roller, or valve pump system for blood propulsion has also been modeled (data not shown), although we have not yet compared this approach to the arteriovenous method described here. This latter system could be applied percutaneously in a manner similar to a venovenous dialysis catheter, thereby avoiding surgical procedures altogether. The current system uses antibody crosslinked to beads; however, the beads could be replaced with matrix channels crosslinked to a binding moiety. One option could be a cardiopulmonary bypass blood filter covalently crosslinked to an antibody or ligand. These improvements would likely lead to a device that is more simple to apply in a clinical setting.
In this present study, we describe differentiation of recovered progenitors into endothelial cells. However, CD133+ cells are reportedly capable of differentiating into a much broader range of cell types. Thus, the ECA column system may prove useful in the isolation, differentiation, and expansion of other lineages.3,5,13 Additionally, by substituting the antibody used in the column, this system could be used to isolate other cell types that express different cell surface markers, such as alternate peripheral progenitor markers or even nonprogenitor cell types, such as cancer markers. In the former setting, this approach may have utility in recovery of receptor-specific peripheral mobilized stem cells for use in bone marrow autografts and in the latter setting, for palliative cytoreduction of circulating tumor cells.
In conclusion, the ECA column system allows for substantially higher yields of circulating CD133 progenitor cells with increased growth potential that can, in turn, reduce the time necessary to reach clinically useful numbers of endothelial cells. Given reports of decreased absolute numbers of endothelial progenitor cells in patient populations with advanced age, diabetes, renal failure, hypercholesterolemia, hypertension, and smoking,39,45–47 this technology may prove particularly useful in these patients who stand to benefit most from progenitor-based therapies.
Footnotes
Acknowledgments
The authors thank Dr. Jennifer Olson for editorial assistance. The authors also thank Mandy Lockard and Cindy Andrews for technical assistance.
Disclosure Statement
The authors have no conflicts of interest to disclose for this manuscript.