
Abstract
Mesenchymal stromal cells (MSCs), seeded onto a scaffold and associated with platelet-gel, may represent an innovative treatment to improve bone repair. The preparation of MSCs for clinical use requires the fulfillment of Good Manufacturing Practice indications. The aim of this study was to validate a Good Manufacturing Practice–grade protocol of tissue engineering for bone regeneration, seeding platelet lysate (PL)–cultured MSCs onto an hydroxyapatite clinical-grade scaffold. Six large-scale experiments were performed. MSC expansions were performed comparing fetal bovine serum 10% and PL 5%. We demonstrated that PL lots contain high levels of growth factors possibly responsible of accelerated growth rate, since the number of colony-forming unit–fibroblast and population doublings were always significantly higher in PL cultures. MSCs were characterized for their phenotype and multilineage differentiation capacity, demonstrating appropriate features for both conditions. Gene expression analysis revealed higher expression of typical osteogenic genes of PL-cultured MSCs, when compared to fetal bovine serum MSCs. Cell transformation was excluded by analysis of karyotype, absence of growth without anchorage, and p53/c-myc gene expression. Scaffolds were precoated with retronectin before MSC seeding. MSC adhesion, distribution, and proliferation were demonstrated through the whole surface of the scaffold by scanning electron microscopy analysis or by immunofluorescence and MSC osteogenic differentiation through quantitative reverse transcriptase–polymerase chain reaction of typical osteogenic genes. The present report offers a model of an MSC-based bioengineered device, using an hydroxyapatite clinical-grade scaffold, and supports its potential use in tissue engineering to repair bone defects.
Introduction
One of the great properties of bone is its ability to heal without scarring. However, perturbations of fracture sites may disrupt the repair process, and evidence has been provided that the number and the functional properties of MSCs are reduced in severe bone fractures complicated by nonunions.13,14
Various types of tissue engineering techniques, 15 such as the implantation of bone substitute, 16 the local delivery of growth factors (GFs),17,18 the injection of bone progenitor cells, 19 or the stimulation of the progenitor cells pool 20 have been envisaged to facilitate bone healing.
Cell-based tissue engineering for bone regeneration by autologous MSC transplantation combined with a three-dimensional (3D) scaffold may represent a promising therapeutic approach. Several scaffolds are currently available, of both natural and synthetic origin. In vitro–expanded MSCs seeded onto a hydroxyapatite (HA)/tri-calcium phosphate biomaterial have been used in several experimental animal models,21–25 showing a great potential in promoting bone union. In addition, formation of new vessels is also necessary for osteogenesis, 26 and both processes (new bone deposition and revascularization) are equally important. A variety of molecules are involved in neo-angiogenesis—in particular, the vascular endothelial growth factor (VEGF).27,28 Several researchers have focused their attention on a combined use of MSCs (loaded or not into a scaffold) and platelet-rich plasma (PRP) or platelet-gel 29 for their significant GFs release,30–32 including VEGF, 33 platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), insulin-like growth factors, epidermal growth factor (EGF), and basic fibroblast growth factor (bFGF), all representing powerful angiogenic inducers, and mitogenic stimuli for MSCs. 34 To date, in humans, only few clinical trials have been published using ex vivo–expanded MSCs with porous scaffolds for the repair of critical-sized long bone defects.35–38
Clinical applications of cell therapy represent a complicated challenge, due to the necessity of operating under strict Good Manufacturing Practice (GMP) conditions, 39 to ultimately certify the quality of the cell product. 40 This quality assurance includes not only the determination of the sterility of the cell lot and the compliance with specific clinical needs, but also the identity, purity, safety, and functionality of the MSC-based constructs. To culture MSCs in a clinical-grade medium facilitating the translation into clinical practice, platelet lysate (PL) has been used to replace fetal bovine serum (FBS).41–45 PL medium, besides avoiding the use of animal-derived material, accelerates MSC expansion, reducing the duration of ex vivo manipulation. Another relevant aspect is the possibility that MSCs may undergo malignant transformation, as reported both in humans 46 and in mouse models, 47 with contradictory results. 48 Previous publications have shown that c-myc protein overexpression is linked to transformation in human MSCs. 46 On the contrary, absence or decreased p53 expression is well known to be relevant for the process of neoplastic transformation of various cell types. 49
Here we report a validation of a manufacturing procedure of a tissue-engineered cell product, composed of an HA scaffold (Orthoss®) combined with BM-derived MSCs, comparing FBS versus PL as MSC growth media. PL-cultured MSCs retain their typical characteristics and can adhere, uniformly distribute, proliferate, and differentiate inside an HA-based clinical-grade scaffold. Moreover, manipulated cells do not show any propensity to malignant transformation. These translational data bring from bench to bedside an innovative technology for the treatment of bone defects.
Materials and Methods
Isolation, culture, and growth kinetics of human MSCs
BM cells were collected from the iliac crest of healthy donors (n = 6), who underwent BM harvesting for a related patient. Unfiltered BM collection bags were used, after approval by the local ethics committee. Informed consent was obtained from donors.
All cell expansions were performed in a GMP facility (Laboratory “Stefano Verri,” San Gerardo Hospital, Monza, Italy), only using GMP-grade qualified reagents and following GMP indications. Briefly, mononuclear cells were isolated from BM by density gradient (Ficoll-Paque Plus, GE Healthcare, Milan, Italy), as previously described, 40 and plated at 2 × 105/cm2 in fresh media described below. Cells were cultured until passage 3 (p3), detaching cells with trypsin (Listarfish, Milan, Italy) at subconfluence and seeding at each passage around 3000 cells/cm2.
The colony-forming unit–fibroblast (CFU-F) assay was performed as described by Castro-Malaspina et al. 50 Briefly, total BM cells were seeded at 7.5 × 106 (in triplicate). CFU-F formation was examined after 8- to 12-day culture and counted after staining with crystal violet. Four independent experiments for each of three different donors were performed.
The growth kinetics of MSCs was assessed by plating cells at 103 cells/cm2, in both media in duplicate. Cells were cultured in multiple wells; every 2 days, two wells for both media were trypsinized, the numbers were enumerated, and population doublings (PD) were calculated as PD = [log (X/Xo)]/log 2, where X = harvested cells and Xo = plated cells.
Medium preparation
All media (including PL-lots setup and storage) were prepared under GMP conditions in a class-B room, class-A cabinet. The FBS medium consisted of low-glucose Dulbecco's modified Eagle's medium (LiStarFish; GMP-grade) supplemented with 10% of a selected batch of FBS (Hyclone, Logan, UT) and with 2 mM L-glutamine (LiStarFish; GMP-grade). Antibiotics were not added to the media, since their GMP used was not allowed by regulatory national authorities for this protocol. PL medium consisted of an identical medium, but with 5% freshly thawed PL instead of 10% FBS. Before use, PL medium was filtered with a 0.22 μm filter.
PRP was obtained either from platelet aphaeresis or whole blood units, collected at the Transfusion Center as previously described. 51 The PRP lots have been randomly selected, according to clinical availability at our Transfusion Center. Plasma was added to obtain around 109 platelets/mL. Heparin was used at 200 IU/mL. PRP products were frozen at −80°C and subsequently thawed at 37°C to obtain the release of GFs. After thawing, PL lots were centrifuged three times at 4000 rpm for 15 min, and aliquots were finally stored at −20°C.
Quantification of PL-derived GFs
Human EGF, TGF-β1, PDGF-αα, PDGF-ββ, VEGF, and bFGF were quantified in the 12 PL lots utilized for MSC culture using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Wiesbaden, Germany), following manufacturer's instructions and as previously described. 31
Cytogenetic analysis
At p3 cells were exposed to colchicines (Sigma Chemicals, St. Louis, MO) for 6 h, detached with trypsin (Lonza, Milano, Italy), washed with phosphate-buffered saline (Lonza, Milano, Italy), subjected to sodium citrate 1% for 15 min at 37°C, and fixed and spread according to standard procedures. Metaphases of cells were Q-banded and karyotyped in accordance with the international System for Human Cytogenetic Nomenclature recommendations. 52 Twenty-five metaphases were analyzed for three expansions.
Clonogenic assay
MSCs (n = 6) were plated at a density of 105/dish in methylcellulose-based medium (MethoCult®; Stem Cells Technologies, Vancouver, British Columbia, Canada) supplemented with 20% FBS. The culture mixture was plated in triplicate in 1 mL in 35-mm Petri dishes (BD Falcon, Milan, Italy). After a 14-day incubation, CFU cells were scored under an inverted microscope. Positive control was represented by hematopoietic CD34+-selected stem cells cultured with 10% 5637-conditioned medium, as a source of appropriate colony-stimulating factors.
Real-time quantitative reverse transcriptase–polymerase chain reaction analysis
c-myc and p53
Quantitative expression of c-myc and p53 was determined by quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR). HL-60 (human promyelocytic leukemia) cell line, known to be p53 knock-out and c-myc overexpressed (German Collection of Microorganisms and Cell Cultures;
MSC-derived p53 and c-myc mRNA levels were compared to healthy donor peripheral blood mononucleated cells, considered as nonmanipulated cells. p53 and c-myc gene expression analysis was performed on 1 × 106 MSCs harvested at p3 from three different expansions in both media, from HL-60 cell line and from peripheral blood mononucleated cells (5 × 106 cells each). One single PL lot was used for all the experiments.
Osteogenic genes
Six osteogenic genes were analyzed (n = 2): alkaline phosphatase (ALP), Collagen type1 (COL1), osteonectin (ON), runt-related transcription factor 2 (RUNX2), osteopontin (OP), and osteocalcin (OC). Cells were seeded at 2 × 104/cm2, and cultured in complete media (10% FBS or 5% PL) supplemented with 100 nM dexamethasone, 0.05 mM ascorbic Acid, and 10 mM sodium glycerol-phosphate (all provided by FARVE, Altavilla Vicentina, Italy), and produced following European Pharmacopoeia indications. Osteogenic medium was freshly prepared and replaced three times weekly for 21 days. mRNA was extracted at 4, 8, 11, 14, 18, and 21 days after osteogenic induction. To contemporarily detect the corresponding mineralized bone matrix deposition, a cell culture plate was kept aside for staining with alizarin red S solution.
RNA extraction was performed using TRIZOL reagent (Invitrogen, S. Giuliano Milanese, Italy), following the manufacturer's protocol. cDNA synthesis and qRT-PCR analysis were performed as described elsewhere. 53 Genes amplifications primers/probe sets were provided from TaqMan Gene Expression Assay (Applied Biosystems, Foster City, CA; assay ID: p53: Hs00153349_m1, c-myc: Hs00153408_m1, GAPDH: Hs99999905_m1, ALP: Hs01029144_m1, COL1: Hs01028970_m1, ON: Hs00234160_m1, RUNX2: Hs00231692_m1, OP: Hs00959010_m1, and OC: Hs00609452_g1). The transcript level of the genes was normalized for the expression of GAPDH constitutive gene. Data were expressed using the comparative Ct method. 54
Flow cytometry analysis of MSCs
The following monoclonal antibodies (mAb) were used, according to the standard protocol: phycoerythrin (PE)–conjugated anti-CD33 mAb, fluoroisothiocyanate (FITC)–conjugated anti-CD34 mAb, FITC anti-CD14 mAb (all from IQP, DL Groningen, The Netherlands); Peridinin–chlorophyll–protein complex–conjugated anti-CD45 mAb, PE anti-CD73mAb, FITC anti human leucocyte antigen clone I (HLA-ABC) mAb (all from Becton Dickinson, Franklin Lakes, NJ); FITC anti human leucocyte antigen clone II (HLA-DR) mAb, PE anti-CD90 mAb; PE anti-CD105 mAb (eBioscience, San Diego, CA); Cyanine 7 (Cy7)–conjugated anti-CD19 mAb (Beckman Coulter, Fullerton, CA). Labeled cells were analyzed by a FACSCalibur flow cytometer (Becton Dickinson), after gating on viable and CD45-negative cells.
MSC multilineage differentiation
The adipogenic, osteogenic, and chondrogenic differentiation capacity of MSCs was determined at p3 for all experiments, utilizing the respective supplements (10% FBS or 5% PL) and the specific conditions, as previously described. 40 To detect the osteogenic, adipogenic, and chondrogenic differentiation, alizarin red S, oil red O, and safranin O (all from Sigma) were utilized, respectively.
Cell loading of Orthoss scaffolds
Orthoss® scaffolds were supplied by (Geistlich Pharma AG, Wolhusen, Switzerland) in cubes of 1 × 1 × 2 cm. Scaffolds were cut into cubes of 125 mm2. The cubes were then coated with fibronectin (Sigma) or retronectin (Takara–Lonza, Milano, Italy), by soaking in a solution containing 50 μg/mL retronectin or fibronectin for 4 h at RT. The cubes were air-dried overnight in a sterile biosafety cabinet. The cubes were placed in a suspension of cells (1 × 106 cells/scaffold) and subjected to vacuum for 60 s to facilitate the flow of the cells into the pores. After a 3-h incubation at 37°C with 5% CO2, scaffolds were cultured with the PL medium for 2 to 4 weeks at 37°C and 5% CO2. The control scaffolds were treated identically, but without cells.
MSC proliferation and osteogenic differentiation in Orthoss scaffold culture
Total cell numbers in 3D scaffold constructs on days 4, 14, and 28 (n = 2) were estimated by counting cells on 10 × fluorescence section images elaborated with the software Adobe Photoshop CS3. Ten serial sections were analyzed for every scaffold at each time point, enumerating the number of cells/mm2 as counted cells on the perimeter multiply by the section thickness (8 μm).
For osteogenic gene expression, cellular scaffold constructs were collected on days 1, 4, 8, 11, 14, and 21 after osteogenic induction. Each scaffold was mechanically broken in a specific lysis cocktail, and mRNA was extracted using RNEasy kit (Qiagen, Hilden, Germany;
Histological analysis of the scaffolds
Scaffolds were processed at 2 and 4 weeks and analyzed with either scanning electron microscopy (SEM) or confocal microscopy.
SEM
Scaffolds were fixed, postfixed, dehydrated, and sputter coated with gold as standard protocols. Samples were examined using a Philips XL-40 scanning electron microscope (FEI, Eindhoven, The Netherlands).
Confocal microscopy analysis
Sections of the frozen scaffolds were treated for standard immunofluorescence and labeled with AlexaFluor 546 phalloidin (Invitrogen) and with TOTO-3 iodide (642/660) (Invitrogen) for nuclei staining. Analysis was carried out on a Radiance 2100 microscope (Biorad Laboratories, Hercules, CA) equipped with a Kripon/Argon laser and a red laser diode.
Statistical analysis
Data are shown as mean ± standard deviation (SD). The statistical differences were analyzed using the paired Student's t-test, and a p-value < 0.05 was considered to be statistically significant.
Results
Analysis of MSC growth rate and transformation propensity
First, we have compared the isolation and culture of MSCs (n = 6) using either FBS or PL medium. Figure 1 shows MSC cultures at p3, grown in FBS or PL medium (Fig. 1a), CFU-F histogram (Fig. 1b), and PD diagram (Fig. 1c). BM cells generated CFU-F at a higher degree in PL medium (n = 3) than in FBS medium (Fig. 1b, p-value = 0.04). The mean of CFU-F per 7.5 × 105 nucleated cells was significantly higher in PL cultures (14.25; range 7.75–21.38) than in FBS cultures (10.22, range 4.92–17.75).
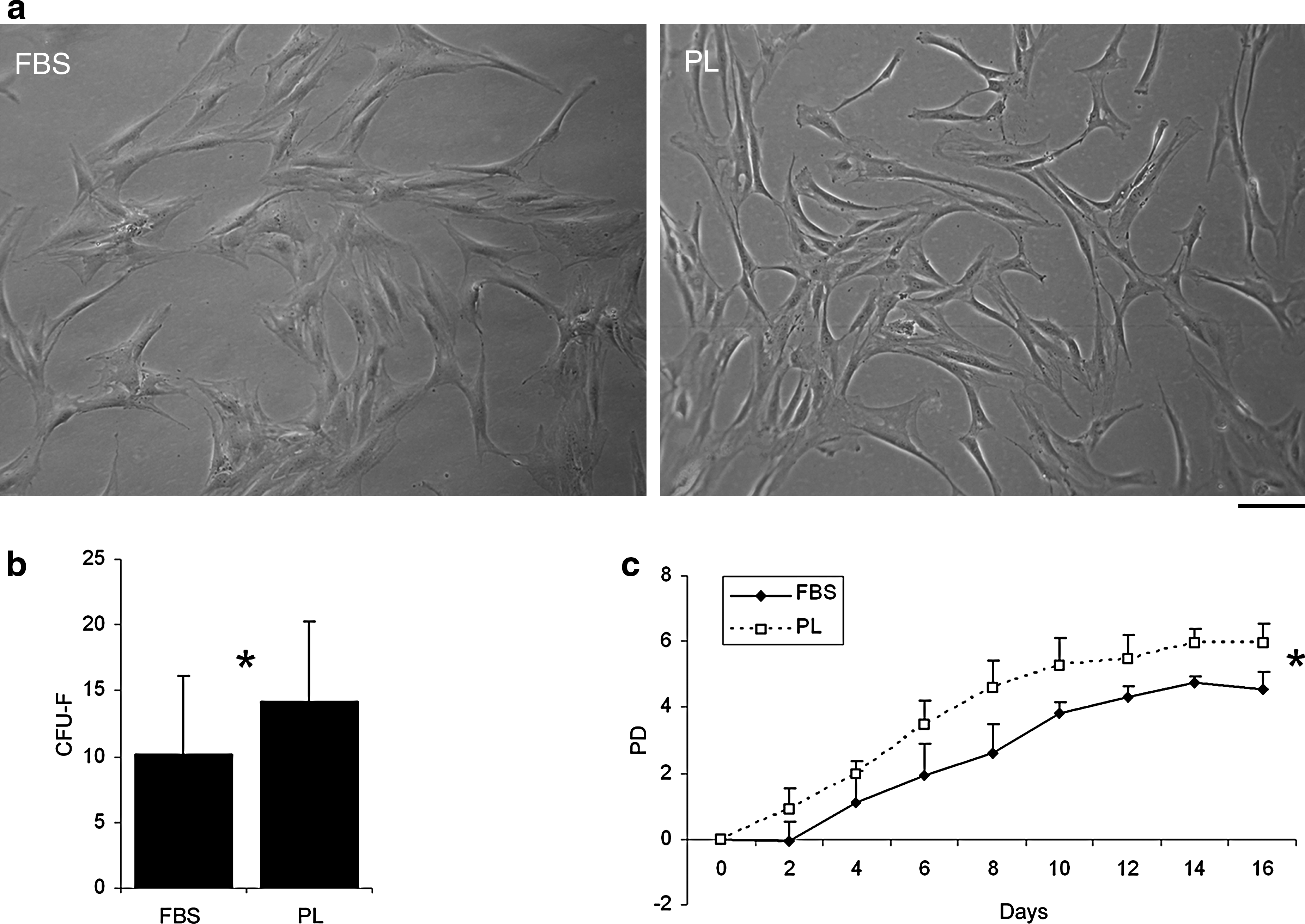
Culture of mesenchymal stromal cells (MSCs) under Good Manufacturing Practice conditions in fetal bovine serum (FBS) versus platelet lysate (PL) medium. MSCs in both media show the typical fibroblastic morphology (
At each passage (until p3) cells produced a subconfluent layer (70–80% confluence) with a fibroblastic shape. The cells showed a more elongated morphology (Fig. 1a) when supplemented with PL medium, as already described in the literature. 42 PL-cultured MSCs proliferated faster than FBS-cultured MSCs, as shown by the PD analysis, resulting in a significant difference in the number of PD (Fig. 1c; the difference acquired a statistically significant value [p < 0.05] at time point 6 till time point 16). Cell viability at p3 (as measured by Trypan blue exclusion) was always, in both conditions, greater than 90%.
To analyze differences in the release of GFs possibly responsible of accelerated growth rate,56–58 ELISA tests were performed to determine the amounts of six human GFs. The 12 PL lots used for MSC cultures were assayed for PDGF-αα, PDGF-ββ, TGF-β1, VEGF, EGF, and bFGF contents. All PL lots contained about 109 platelets/μL. The minimum and maximum values, the average, and the SDs of 12 lots are shown in Table 1. The GF levels analyzed in human plasma were undetectable (data not shown). Analysis on FBS always produced undetectable levels, as expected for a bovine-derived product (data not shown).
The table shows minimum and maximum levels, averages, and SDs for each growth factor tested.
VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor beta; EGF, epidermal growth factor; SD, standard deviation.
Since accelerated growth rate may be responsible for augmented propensity for spontaneous cell transformation, we performed at p3 three different assays (Fig. 2). Classic cytogenetic analysis was performed with conventional Q-bands by fluorescence using quinacrine (QFQ) banding (Fig. 2.1). All donors were characterized by a normal karyotype, both in FBS (Fig. 2.1a) and PL medium (Fig. 2.1b). Moreover, MSCs were seeded for growth in methylcellulose, and no colony was detected in all MSC cultures, analyzed after 4 weeks (data not shown).
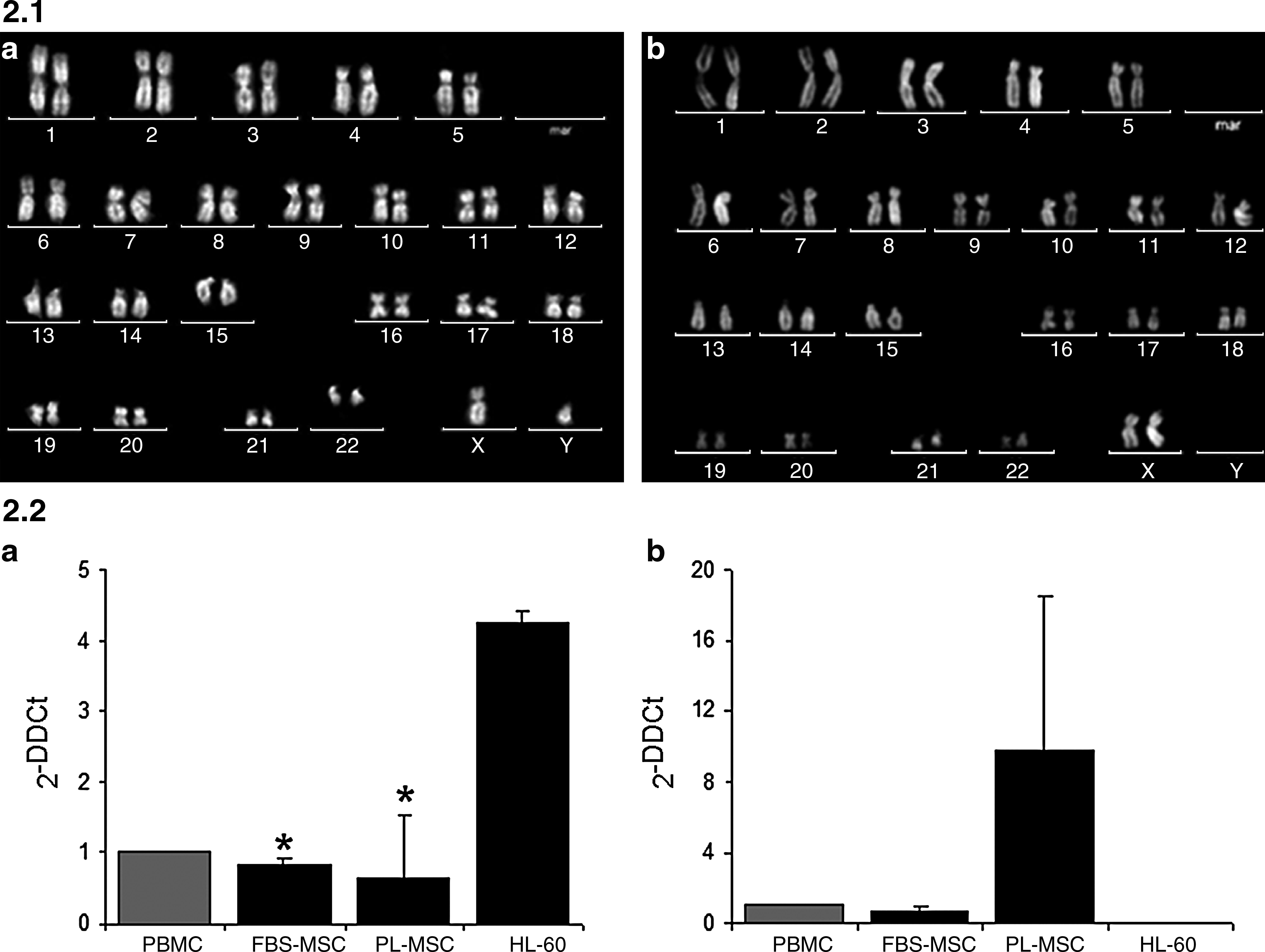
Analysis of MSC transformation propensity. Figure 2.1: Cytogenetic analysis of MSCs cultured with FBS (
As far as concerns p53 and c-myc gene expression analysis, the mean values and the SDs measured at p3 (n = 3) are shown in Figure 2.2. We found a 4.25 mean fold increase of c-myc expression in HL-60 cell line compared to a 0.85 (FBS-cultured MSCs) and 0.65 (PL-cultured MSCs), with a p-value <0.05 for both conditions (Fig. 2.2a), when compared to HL-60. On the contrary, p53 (Fig. 2.2b) was expressed by MSCs in both conditions (FBS or PL), even though at different levels, but it was completely absent in HL-60 cell line, as expected.
MSC phenotypical and functional characterization
PL- and FBS-cultured MSCs were analyzed for their phenotype at p3 (Fig. 3) and for their multilineage differentiation capacity (Fig. 4). PL-MSCs were constantly negative for typical hematopoietic markers and HLA-DR. On the contrary, pL-MSCs showed a high expression of CD73, CD90, CD105, and HLA-ABC (Fig. 3). Similar levels were observed for FBS condition. The table in Fig. 3 shows the complete panel (n = 6) of averages of percentages and SDs for all tested antigens for both conditions.

Immunophenotype of MSCs cultured with FBS or PL medium. A representative cell expansion (out of six performed for each medium) is shown. The antigens CD105, CD90, CD73, human leucocyte antigen clone I (HLA-ABC), CD33, CD34, CD14, CD19, and human leucocyte antigen clone II (HLA-DR) were analyzed by a flow cytometry analysis software (FACS). The histograms show the percentages for each single marker for the selected quadrants, after gating on viable CD45-negative cells. The table represents the averages ± SDs of the nine antigens (n = 6) analyzed for both FBS and PL conditions.
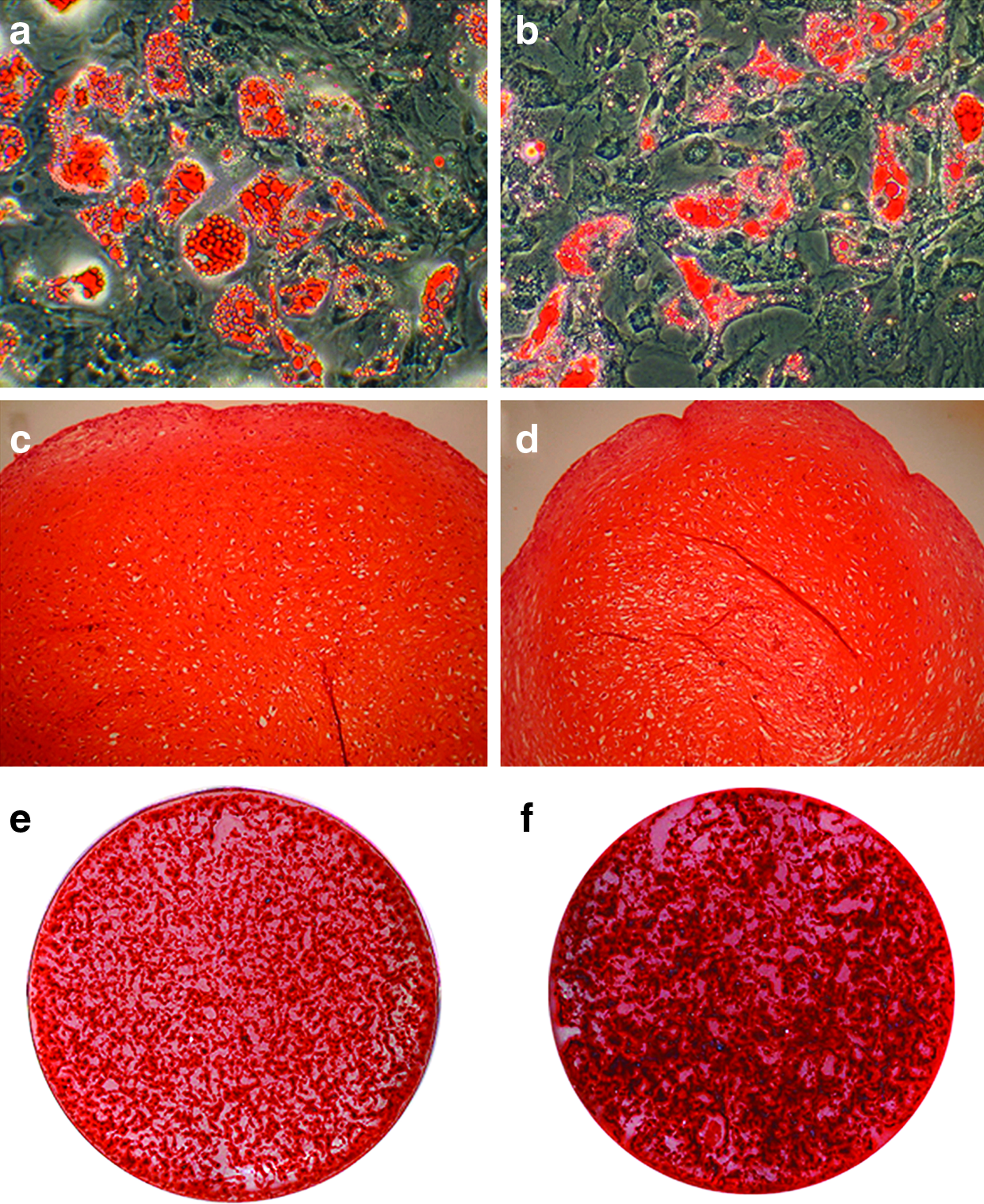
Differentiation into mesodermal lineages of FBS-MSCs (
MSCs were then induced to differentiate into adipogenic, chondrogenic, and osteogenic lineages and analyzed for their differentiation capacity (Fig. 4). Results demonstrated that MSCs cultured in PL medium (Fig. 4b, d, f) were able to differentiate into adipocytes (Fig. 4b), chondrocytes (Fig. 4d), and osteoblasts (Fig. 4f), similarly to FBS-cultured MSCs (Fig. 4a, c, e, respectively). With respect to PL cultures, we observed a more robust and earlier osteogenic differentiation than in FBS-ones, as described in details in the following paragraph.
MSC osteogenic differentiation: Mineral matrix deposition and gene expression analysis
Both PL- and FBS-MSCs were cultured in osteogenic medium, and at 4, 8, 11, 14, 18, and 21 days, gene expression analysis (n = 2) and alizarin red staining (n = 6) were performed (Fig. 5). When exposed to osteogenic medium over 21 days, PL-cultured MSCs underwent more robust and earlier osteogenic differentiation (only 8 days in PL cultures compared to 14 days in FBS cultures, in the case of the experiment shown in Fig. 5.2), generating more extracellular mineralization than FBS-cultured MSCs, as shown by alizarin red S staining.
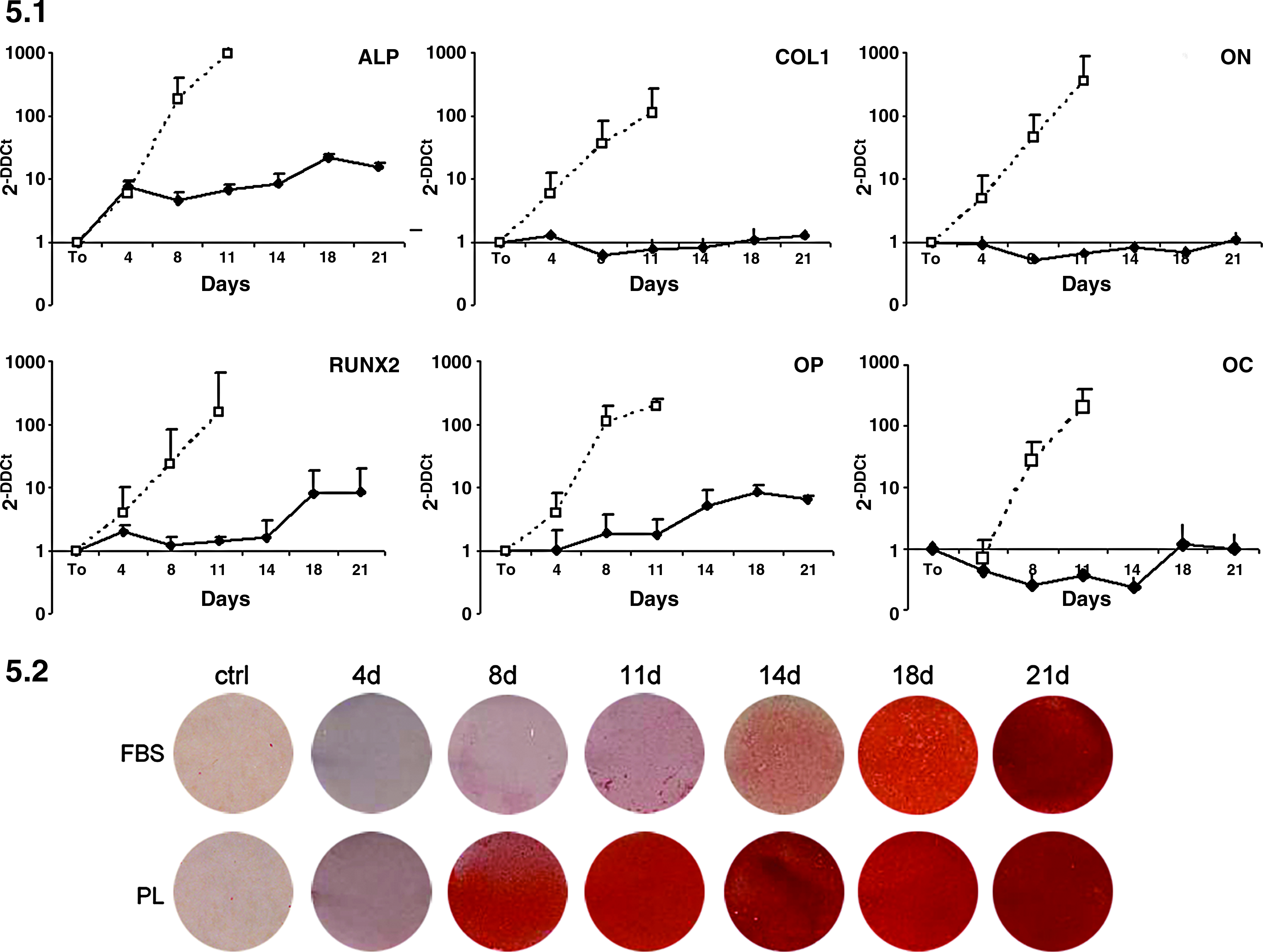
MSC osteogenic differentiation on monolayer culture. FBS- and PL-MSCs were cultured in osteogenic induction medium at 4, 8, 11, 14, 18, and 21 days. Figure 5.1 shows mRNA expression levels of alkaline phosphatase (ALP), collagen type1 (COL1), osteonectin (ON), runt-related transcription factor 2 (RUNX2), osteopontin (OP), and osteocalcin (OC) on differentiated cells under FBS (continuous line) and PL (dashed line). Averages and SDs of two independent experiments are shown. Figure 5.2 shows alizarin red S staining of mineralized matrix of cells cultured under the indicated conditions. The shown experiment is one of the two analyzed by qRT-PCR in Figure 5.1.
The osteogenic differentiation was confirmed by qRT-PCR, under osteogenic permissive condition, during a 21-day in vitro time-course study. The expression of the six typical osteogenic genes has been observed both in FBS and PL cultures. Interestingly, the expression of all the factors ALP, COL1, ON, RUNX2, OP and OC is significantly higher in PL-cultured cells. Of note, in PL medium the analysis was stopped at 11 days because of the inadequate quality of RNA likely due to the low number of viable cells observed and to an abundant quantity of mineral matrix.
MSC culture onto HA scaffolds
Further experiments were performed only using PL-cultured MSCs, to manufacture a GMP-grade construct. Cells were seeded onto the Orthoss scaffolds and cultured in PL medium. Several culture conditions and quality controls were assessed.
First of all, a clinical-grade precoating material (retronectin) was compared with standard fibronectin, for supporting MSC adhesion on HA. After 2 weeks of culture, we could observe a detectable cell adhesion in both conditions (Fig. 6.1), with no differences between fibronectin-precoated (Fig. 6.1a) and retronectin-precoated (Fig. 6.1b) scaffolds. Following experiments were performed only with retronectin-precoated scaffolds.
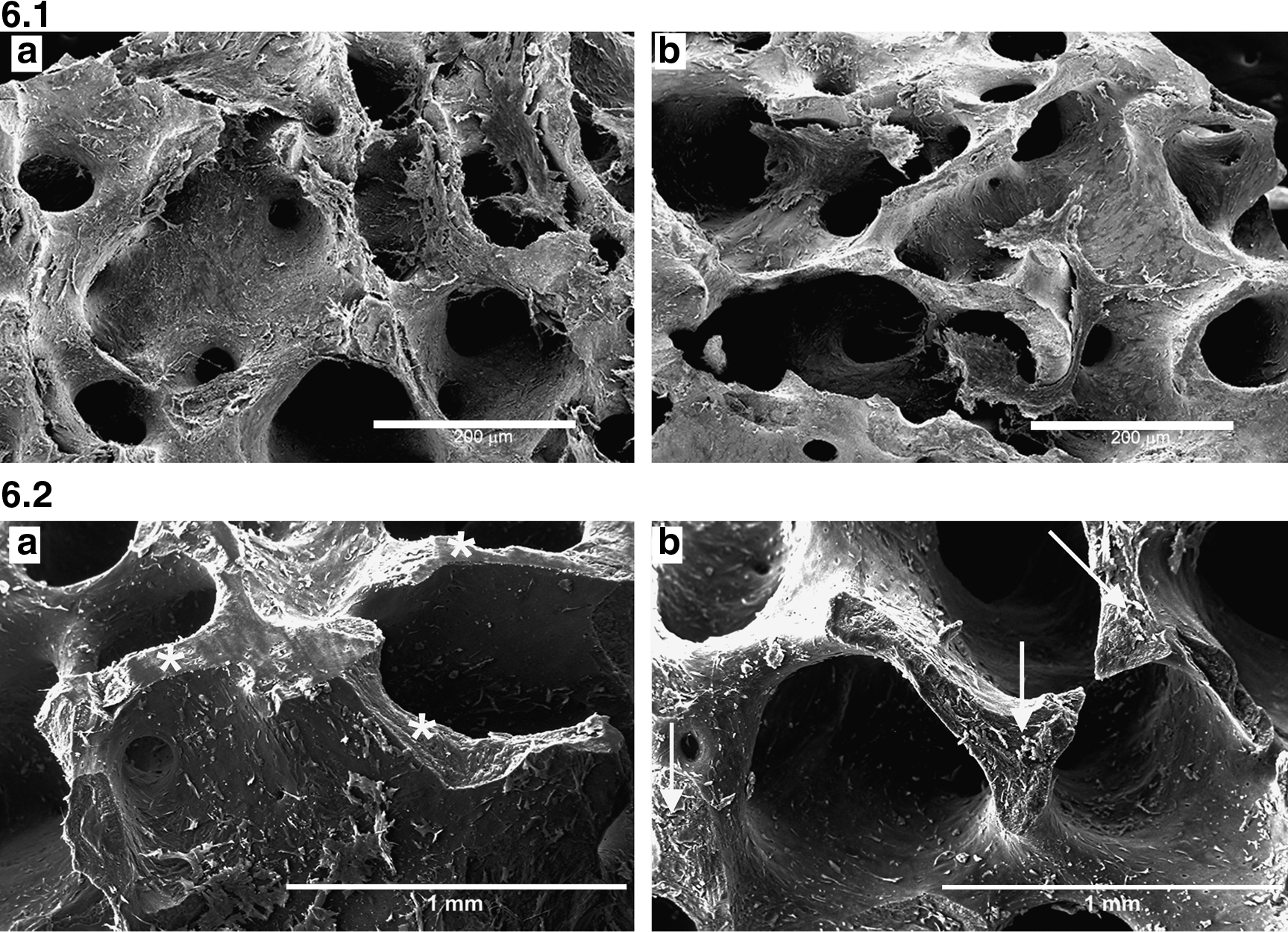
Scanning electron microscopy analysis. Figure 6.1: PL-cultured MSC adhesion on the fibronectin-precoated (
Cell distribution on the 3D matrix was therefore determined, to observe if our method could facilitate the flow of the cells into the most internal pores of the scaffolds (Fig. 6.2). Scaffolds cultured for 2 weeks with MSCs were processed for SEM analysis and fractured in the middle. Figure 6.2 demonstrates an equal cell distribution both in the internal (white asterisks, Fig. 6.2a) and in the external (white arrows, Fig. 6.2b) surface. To observe more optical plans of the scaffold and confirm the previous results, immunofluorescence experiments were then performed (Fig. 7). Serial sections of the scaffold, cultured with MSCs for 2 weeks, have been treated by specific staining to observe cytoplasm (with phalloidine, which stains for actin cytoskeleton filaments) and nuclei (with TOTO3). Figure 7 demonstrates adherent cells along the entire perimeter of a scaffold hole.

Confocal microscopy image of a scaffold cryosection. Cells were immunolocalized with phalloidin (red), and nuclei were stained with TOTO3 (blue). The boxes show high magnification of a selected area.
Subsequently, we have evaluated the viability of PL-cultured MSCs inside the scaffold and their ability to proliferate among the HA meshes and to colonize the entire area (Fig. 8). At 4 weeks of culture we could observe several layers of MSCs adherent to the scaffold (Fig. 8.1b), demonstrating MSC proliferation inside the scaffold. An identical monolayer was not observed everywhere along the scaffold, and it is likely that variability in cell concentration is maintained (Fig. 8.1c, single cells vs. 8.1 b confluent layer). Cell proliferation was further demonstrated by counting cells on 10 × fluorescence sections images elaborated with the software Adobe Photoshop CS3, as shown in Figure 8.2 (n = 2).
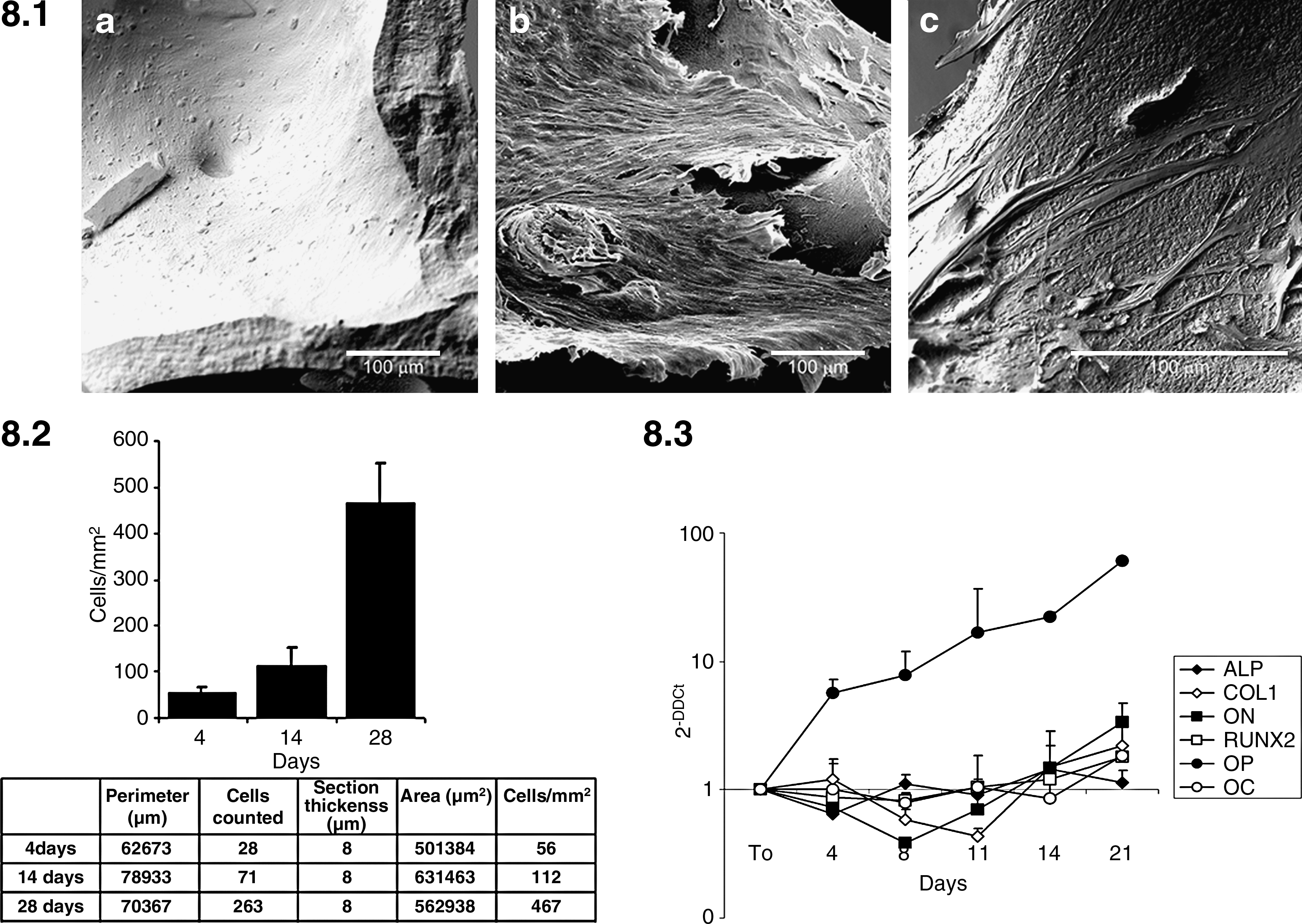
Cell proliferation and differentiation on the scaffold. Figure 8.1: SEM analysis of seeded MSC onto the scaffold at 4 weeks. Cells form confluent layers (
Finally, we have demonstrated that PL-cultured MSCs were able to differentiate inside the scaffold into osteogenic lineage, even if slower than in a monolayer culture on plastic dishes (Fig. 8.3). In particular, at 14 and 21 days, increased expression of all the evaluated genes was detected. OP mRNA was upregulated from the very beginning of the culture. Figure 8.3 shows the transcription profile of ALP, COL1, ON, RUNX2, OP and OC of PL-cultured MSCs inside the scaffold (n = 2).
Discussion
Our article describes a complete GMP-grade validation of a process for the manufacturing of a tissue-engineered cell product, composed of BM-derived PL-cultured MSCs combined with an HA scaffold (Orthoss®) to be used for the treatment of bone defects.
Because of the multiple limitations associated with the use of bone autograft 59 or allograft, 60 tissue engineering has gained renewed interest. Recent studies have investigated the biological causes of bone degenerative processes (such as nonunions), providing evidence that the number, the proliferation capacity, the physical properties, and the repair ability of osteoblasts are reduced at sites proximal to nonunions.13,14 For these reasons, novel cell–based tissue-engineered approaches, which can overcome these cell abnormalities, are strongly warranted. To date, in humans, only few clinical trials have been published using ex vivo–expanded MSCs in conjunction with porous scaffolds for the repair of critical-sized long bone defects.35–38 This difficulty is definitely due to the necessity of operating under GMP conditions, 39 which also implies the strict definition of parameters allowing the use of cells by ensuring that they are in a biologic condition able to guarantee their function, and avoiding any potentially dangerous cell transformation. If the product is composed of cells and biomaterials, both components have to be characterized in their identity, purity, safety, and functionality to finally determine their compliance with specific release criteria for a clinical use, as we recently demonstrated for a MSC-based clinical device using collagen scaffolds. 40
The first aspect that has to be determined for a GMP-grade validation is the assessment of straightforward conditions to obtain sufficient cell numbers for further manipulations. Therefore, PL has been proposed to replace FBS.41,42,43,44,45 In the present study, we have tested 12 random batches of PL, and compared them with FBS. In line with these previous reports, we demonstrated that 5% PL is superior to 10% FBS in terms of cell proliferative capacity. An accelerated cell growth (and therefore a shortening of the culture) can be also further achieved by the use of different culture conditions and cell concentrations at each passage, as we have recently demonstrated. 61 In a protocol of GMP-grade MSC manipulation, PL medium is to be recommended, since besides avoiding the use of animal-derived materials, an accelerated growth rate diminishes the period of culture. PL lots have to be standardized in their platelet concentration, which is the most relevant element for the release of GFs.56,57,58 To analyze differences in the release of GFs possibly responsible of accelerated growth rate, ELISA tests were performed to quantify EGF, TGF-β1, PDGF-αα, PDGF-ββ, VEGF, and bFGF, well known in the literature to induce MSC proliferation and, at least for TGF-β1, MSCs further differentiation. We have observed that all PL lots contain a great quantity of the above-mentioned GFs, particularly if compared to human plasma, whose GF levels were undetectable. In line with our findings, previous reports have demonstrated that the use of allogeneic human serum causes MSC growth arrest and ultimately cell death. 62 Even though it has been reported that the use of autologous serum may determine an adequate growth of autologous MSCs, 63 the amount of autologous serum necessary for sufficient expansion would exceed the amount a donor could provide. The formulation of PL-release criteria in clinical-grade cell expansions would be desirable, but this objective was not obtained, at least for the GFs tested. The amount of GFs in PL lots is extremely variable and donor dependent, and we could never establish a minimum value for any of the GF tested. The actual contribution of the above-mentioned GF for accelerated growth rate is still a controversial topic, and although we observed a wide difference in GF contents among the 12 PL batches, there was no relevant change in the rate of MSC proliferation from batch to batch, included the ones with lower GF values. These observations are in line with what previously described by Weibrich et al., 30 who analyzed 213 different PRP lots. Certainly, such a great amount of GFs is somehow responsible for accelerated MSC proliferation, but further studies are necessary to determine which ones are truly relevant. Our article is not conclusive at this regard. We actually know that PL will have variable potency for different patients, and this is expected in any cell biology process. For cell expansions we actually use randomly available PL lots from our Transfusion Centre, since GF analysis was not informative in any case and every single lot (included the ones with lower GF values) was excellent for expanding MSCs and preserving their characteristics.
Another relevant aspect that has to be taken into account for clinical application is the possibility that MSCs may undergo malignant transformation, as reported both in humans 46 and in mouse models, 47 with contradictory results. 48 The propensity to transformation has been investigated through several methods. The method most commonly used is the analysis of MSC metaphases. In fact, Rubio et al. 46 demonstrated that transformed MSCs always showed chromosome alterations such as trisonomy, tetraploidy, and/or chromosome rearrangements. All our MSC cultures were characterized by a normal karyotype, both in FBS and PL medium. Moreover, MSCs were seeded for growth in methylcellulose and no colony was detected, while transformed MSCs described by Rubio et al. lost contact inhibition and grew in semisolid agar. The same authors also showed that c-myc protein overexpression was linked to transformation in human MSCs. On the contrary, absence or decreased p53 expression is well known to be relevant for the process of neoplastic transformation of various cell types. 49 We did not observe any downregulation of p53 or upregulation of c-myc in our expanded cells. The large variability of p53 expression is not due to different lots of PL, because a single lot was used; therefore, we do not predict a correlation of this variability with some of the GFs released by PL.
Besides accelerating growth rate with no trace of tumoral transformation, we also proved that PL medium did not alter the phenotype of expanded MSCs. In accordance with previous reports,41,43,45 MSCs retained their ability to differentiate into osteogenic and adipogenic lineages, demonstrating that PL does not obstacle the multipotency of these cells. An analysis of gene profile was performed to ensure the quality of the cell product. Since our interest was the osteogenic differentiation of MSCs, several genes implied in this process were analyzed, to exclude the possibility of a dystrophic mineralization. 64 The best conditions, the best cell sources, and environment for bone regeneration have been a matter of study for a long time. Differentiation is usually characterized by increased AP activity, RUNX2 expression, and formation of mineralized bone–like nodules containing COL1, OC, ON, and OP. In line with this, Zhang et al. 55 recently demonstrated the influence of the ontological and anatomical origin of MSCs, being human fetal MSCs the most promising candidate for bone tissue engineering. Frank et al. demonstrated the heterogeneity across different subpopulations within the same primary culture. 12 Our results clearly show a very potent effect of PL, combined with a clinical-grade osteogenic induction cocktail, in inducing an accelerated and greater expression of the mentioned genes. Our results in qRT-PCR analysis of osteogenic genes in FBS culture were comparable to literatures,12,55 while in PL cultures data really overpassed any expected values. Concerning the already reported decrease in osteoblastic potency after culture in the presence of PL, 58 these results are not confirmed in our hands, since all the genes were strongly upregulated, as measured by qRT-PCR. We may suppose that different methods of platelet activation (such as thrombin vs. thermic shock) or different platelet final concentration in serum-free medium or different methods of cell culturing could, at least partially, explain the observed differences. Moreover, when using PL, we observed a very high release of TGF-β, which belongs to the TGF-β superfamily, a group of morphogenetic proteins important during the healing of bone. 65 Nevertheless, we are well aware that this remains a controversial topic.
When envisaging the use of a GMP-grade combination of MSCs and an osteoconductive support, the final step is ultimately represented by the establishment of a correct 3D system to create a suitable environment to promote cell adhesion and proliferation, and secondly tissue growth and differentiation. HA has been often employed as bone substitute for human clinical application. In vitro–expanded MSCs seeded onto a HA and/or tricalcium phosphate ceramic biomaterials have been used in several experimental animal models,21–25 but a formal validation of such biological composed devices under GMP conditions has never been provided. The Orthoss® scaffold is a safe and highly purified deorganified bone mineral, very similar to human cancellous bone. Incorporation and remodeling of the bone substitute material was observed in the absence of any toxic effect.66,67 With the goal of promoting MSC adhesion to the HA scaffold, we first studied the adequacy of a clinical-grade precoating material (retronectin). Retronectin showed an excellent capacity in permitting cell adhesion. Immunofluorescence studies further demonstrated cell adhesion around the whole perimeter of the scaffold holes. SEM analysis showed that the seeded MSCs could colonize the most internal parts of the scaffold, receiving nutrient from the medium. Even though we do not provide a direct measurement of viable cells, a certain degree of cell viability was indirectly demonstrated by SEM analysis (by the observation of proliferating cells forming confluent cell layers, as shown in Fig. 6), by immunofluorescence (demonstrating integrity of cells by coloration of cytoplasm and nuclei, as shown in Fig. 7), and by the cell count estimation, as shown in Figure 8. Analysis of MSC osteogenic gene expression demonstrated the expression of each single gene tested, even though at lower levels than in monolayers conditions. This could be explained by several factors, including technical difficulties in extracting mRNA from MSCs after mechanical destruction of the scaffold. In addition, since the cells have to reach a suboptimal confluence to differentiate, we can suppose that the variability we found in our scaffolds can somehow affect the success of the assay. It is also likely that our static culture method needs to be further improved to possibly obtain a confluent layer ubiquitously through the whole scaffold. In fact, various investigators68,69 demonstrated that spinner flask culture methods give beneficial effect on cell growth and differentiation inside the scaffold. Concerning the demonstration of osteogenic potential of the proposed device, we are aware that this is an in vitro characterization and that only an in vivo model could finally prove it. Our data give anyway, in our opinion, a sufficient proof of the tendency of these cells to undergo osteogenic differentiation. The only critical point is represented by OP expression, whose high levels have been described in such a context by our group for the first time. An unexpected single-gene profile does not necessarily mean that the osteogenic process is surely impaired or that this is accompanied by a propensity for a different differentiation process. Only a demonstration in vivo would finally prove if this phenomenon has a significant impact on the process of osteogenesis or it is only due to our peculiar ex vivo culture conditions.
Our results by SEM analysis and confocal microscopy show that Orthoss® can provide a suitable 3D environment for cell adhesion, permitting adequate distribution, and growth of MSCs and allowing cell differentiation. Nevertheless, we are fully aware that additional issues have to be considered when envisaging the in vivo implant of such a device in sites of bone degeneration. One of this issue is if our proposed 3D device can permit cell migration both inside and outside the product itself. We were not able to measure if any migratory activity in vitro was present in our construct. This issue may be relevant for clinical application, since migration toward surrounding damaged tissues would be desirable. Nevertheless, we can hypothesize that such a phenomenon will most likely occur, possibly induced by the presence of chemokines released by various inflammatory cell types present in the implantation area. The second (probably the most important) issue to be considered is represented by induction of new vessel formation. Different studies have indeed demonstrated that formation of new vessels is necessary for osteogenesis. 27 For such a purpose, we envisage the use of our PL-cultured MSCs on the HA scaffold mashes combined with platelet-gel, at the moment of the surgical treatment of nonunions, since it has been widely described as an optimal source of angiogenic factors. 29
In conclusion, the present report offers a model of an MSC-based bioengineered device, suggesting that Orthoss scaffold loaded with MSCs has excellent characteristics in terms of safety and osteogenic potential, and support its potential use, combined with platelet-gel, in tissue engineering to repair bone defects. We initially envisage the use of such a product in degenerative bone diseases, since they represent so far the main field of application of MSC-based cell therapy in the area of bone local defects. Clinical trials are planned for nonunions and avascular osteonecrosis. Nevertheless, this product could be used for other kinds of nondegenerative bone defects. When such a project will be transferred in a phase I clinical trial, initial information concerning its feasibility, safety profile, and biological functionality will be provided.
Footnotes
Acknowledgments
The authors are sincerely grateful to Prof. Edoardo Marinoni, who strongly, initially wanted and supported this research, but passed away in January 2007, while the first preliminary promising results were observed.
This work was supported by grants from the “Progetto Regione Lombardia 2006–Cellule Staminali” and “Progetto Sangue”-DGR VIII/3376-Regione Lombardia-2007.
The authors would like to thank the “Fondazione Matilde Tettamanti,” “Comitato Maria Letizia Verga,” and “Comitato Stefano Verri” for their generous and continuous support; Geistlich Pharma AG (Wolhusen, Switzerland) for their strict collaboration; and EON Medica s.r.l. (Monza, I) for providing Orthoss scaffolds.
The authors also thank Prof. Mahesh A. Choolani, Dr. Jerry Chan, and Dr. Zhi-Yong Zhang for sharing some of their protocols.
Disclosure Statement
No competing financial interests exist.