
Abstract
Since the derivation of human embryonic stem (hES) cells, their translation to clinical therapies has been met with several challenges, including the need for large-scale expansion and controlled differentiation processes. Suspension bioreactors are an effective alternative to static culture flasks as they enable the generation of clinically relevant cell numbers with greater efficacy in a controlled culture system. We, along with other groups, have developed bioreactor protocols for the expansion of pluripotent murine ES cells. Here we present a novel bioreactor protocol that yields a 25-fold expansion of hES cells over 6 days. Using immunofluorescence, flow cytometry, and teratoma formation assays, we demonstrated that these bioreactor cultures retained high levels of pluripotency and a normal karyotype. Importantly, the use of bioreactors enables the expansion of hES cells in the absence of feeder layers or matrices, which will facilitate the adaptation of good manufacturing process (GMP) standards to the development of hES cell therapies.
Introduction
Currently, hES cells are cultured in static tissue culture flasks, which are disadvantageous as they result in culture heterogeneity, low cell yield, and low reproducibility because of the lack of control of the culture conditions. Also, hES cells are particularly sensitive to dissociation, which is required for passaging, cryopreservation, and other applications, both in static and bioreactor culture systems. 8 Recently, it has been discovered that an inhibitor of Rho Kinase (ROCKi; Y-27632) increases the survival rate of dissociated, single hES cells. 9
Although rho kinase (ROCK) proteins have been viewed as being functionally redundant, it has recently been demonstrated that ROCK-1 has higher specific activity than ROCK-2 and that ROCK-1 is essential for focal adhesions and stress fiber formation. 10 β2-Integrins signal through Rho, the canonical upstream activator of ROCK, to facilitate cytoskeletal changes involved in integrin segregation and clustering, resulting in increased adherence to substrates.11,12 Previously, we have showed that ROCKi facilitates the formation of ES cell aggregates, in static suspension culture. 13 This observation facilitated the development of a novel bioreactor expansion process to generate clinically relevant numbers of pluripotent hES cells. However, the use of this molecule was only successful for the bioreactor expansion of hES cells when it was combined with rapamycin treatment. Rapamycin is a macrolide antibiotic and immunosuppressant of the phosphoinositide kinase family, and its downstream target is mammalian target of rapamycin (mTOR). mTOR activity mediates AKT-activated cell proliferation and survival. The TOR protein is essential for cell growth and development and is involved in regulating cell cycle progression, cell size, cell migration, and survival.14–16 Disruption of the gene encoding TOR is lethal in all species. 14 Rapamycin affects the expression of growth factors, such as vascular endothelial growth factor, insulin, and insulin growth factor receptor,14,17 the latter being necessary for the correct functioning of the hES cell niche. 18
Using ROCKi for 24 h and continuous treatment with 0.1 nM rapamycin, we have been able to transition hES cells from static culture into stirred-suspension bioreactors. This system maintained cultures with high expression levels of Nanog, Oct-4, and other markers of pluripotency, a normal karyotype and the ability to form teratomas in vivo.
Materials and Methods
Maintenance culture of hES cells
The H9 hES cell line was used in accordance with the Canadian hES cell research guidelines. Undifferentiated hES cells were maintained as described previously. 19 Cells were cultured in static tissue culture flasks on a feeder layer of human foreskin fibroblast (HFF) cells inactivated with 10 μg/mL Mitomycin C (Sigma, St. Louis, MO), or coated with Matrigel™ in 35-mm gelatin-coated tissue culture dishes (Nunc, Rochester, NY), with mTeSR (StemCell Technologies, Vancouver, Canada) under standard conditions (37°C, 5% CO2, saturated humidity). The hES cells were passaged as small clumps by enzymatic dissociation and subcultured on fresh HFF feeder layers every 6 days. In brief, the colonies were treated with 0.1 mg/mL collagenase IV (Invitrogen, Carlsbad, CA) in mTeSR at 37°C for 30 min, followed by TrypLE Select (Invitrogen) at room temperature for 2 min, gently rinsed with 5 mL Dulbecco's phosphate-buffered saline (DPBS; Invitrogen), followed by addition of 2 mL culture medium and gently pipetting them several times, and then broken into small clumps and passaged at a 1:3 ratio.
Bioreactor maintenance of hES cells
H9 cells passaged in static on Matrigel or using feeder layers were pretreated for 1 h with 10 μM Y-27632 (Sigma) and then dissociated as described earlier. The hES cells were then inoculated into 125-mL bioreactors (NDS Technologies; Fig. 1A) at 1.8 × 104 cells/mL. The bioreactors containing 100 mL of mTeSR medium supplemented with 10 μM Y-27632 and 0.1 nM rapamycin (Sigma) were agitated at 100 rpm and were incubated under standard conditions (37°C, 5% CO2, saturated humidity). After 24 h in suspension culture, the Y-27632 was removed and replaced with mTeSR medium containing 0.1 nM rapamycin. Every 6 days, the hES cell aggregates were passaged by removing the entire contents of the bioreactor and dissociating the cells using Accuatase™ (Millipore, Billerica, MA) in the presence of 10 μM Y-27632 until a single-cell suspension was achieved. The single-cell suspension of hES cells was split 1:5 and inoculated into a new bioreactor containing 100 mL of mTeSR medium supplemented with 10 μM Y-27632 and 0.1 nM rapamycin for 24 h. Media changes were performed as necessary; however, most commonly, media need not be changed until the point of passaging (every 5th day for cells treated with collagenase and every 6th day for cells treated with Accuatase). For sampling, one 2 mL sample was taken from the bioreactor immediately after it was removed from a stir plate (to maintain a homogeneous cell suspension in the culture). The sample was spun-down and dissociated using Accuatase, and two 50 μL samples were taken from this suspension and counted separately. The average of these counts was taken to determine the cell density in the culture.
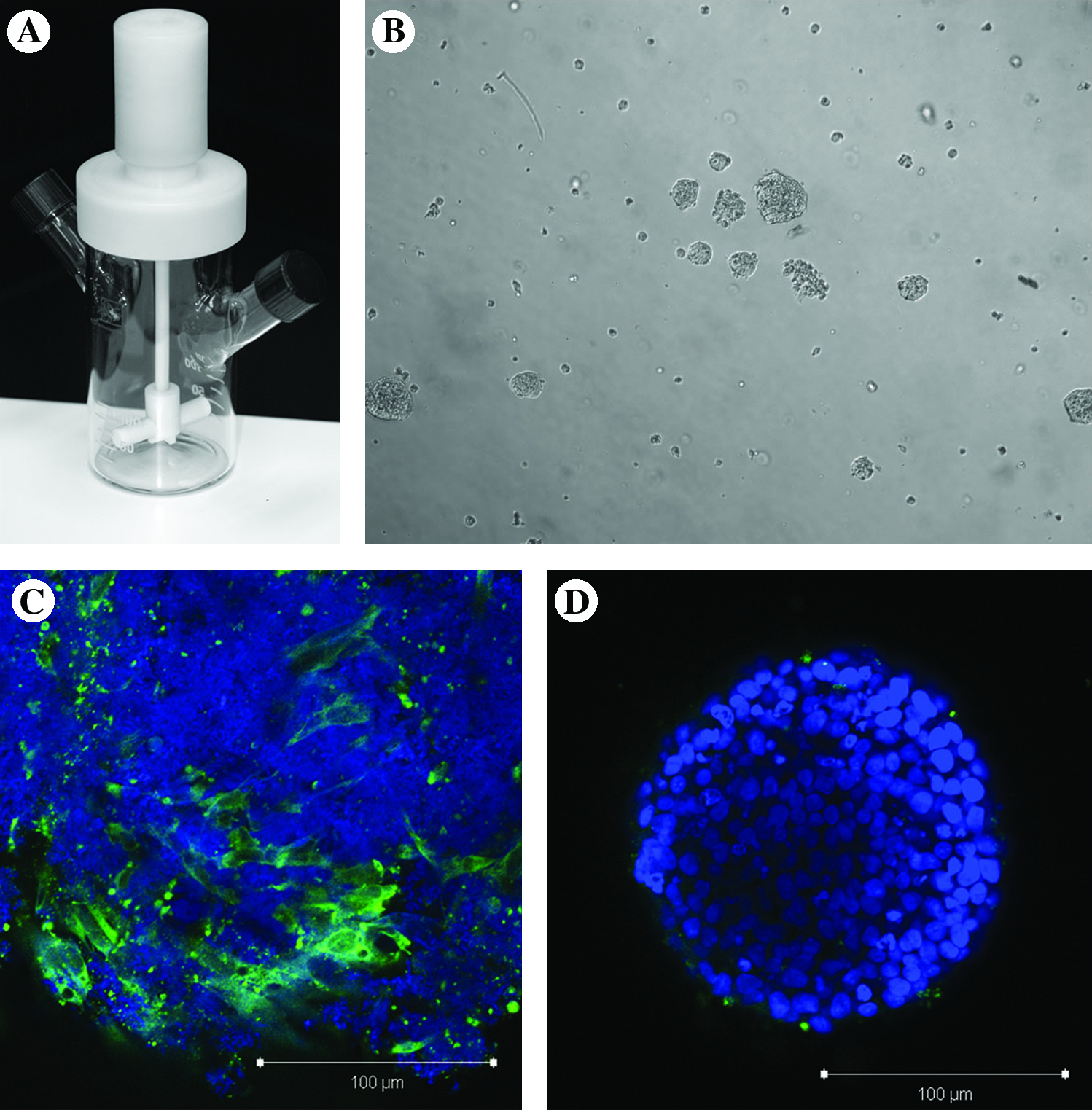
Effects of rapamycin on hES cells grown in suspension bioreactor. (
Karyotype analysis
Karyotype analyses of hES cells were carried out using G-banding method. In brief, cells were incubated with 0.2 μg/mL colcemid at 37°C for 30 min, trypsinized, resuspended, and incubated in 0.068 M KCl for 25 min at 37°C, then rinsed with 3:1 methanol:glacial acetic acid three times and dropped to make the spread of chromosomes on the slides. The dried slides were baked overnight at 55°C, treated with 0.05% trypsin for 30 s to 2 min, and stained with Giemsa and Leishman's solution.
Immunofluorescence
Aliquots of bioreactor-generated aggregates were washed in PBS and fixed in 4% paraformaldehyde (PFA) in PBS at 4°C overnight. The aggregates were then permeabilized in 0.5% saponin in PBS at 4°C overnight, rinsed once in PBS, and then blocked in 3% bovine serum albumin again at 4°C overnight. Primary antibodies (fibroblast growth factor [FGF]-R1, Oct4, Nanog, Tra-1-60, Tra-1-81, and SSEA-4; all from Santa Cruz Biotechnology, Santa Cruz, CA) were diluted 1:50 in 3% bovine serum albumin, added to the cell samples, and incubated overnight at 4°C. The aggregates were then washed three times with PBS and blocked again overnight at 4°C. Following the block, the aggregates were incubated with an appropriate alexa-fluor 488 secondary antibody (Molecular Probes, Carlsbad, CA) and Toto-3 (Molecular Probes) overnight at 4°C. After incubation, the aggregates were washed three times with PBS and mounted on slides with mountant (9:1 glycerol:PBS). Spacers (270 μm) were adhered to slides prior to mounting to avoid aggregate compression. The slides were analyzed using a Zeiss 510 confocal microscope with 488, 568, and 633 nm filters. Images were prepared using (Carl Zeiss AG, Oberkochen, Germany) LSM image browsing software.
Teratoma formation
Fox Chase CB-17 SCID mice were obtained from Charles River (Wilmington, MA) and housed in the single-barrier animal facility of the Faculty of Medicine, University of Calgary. Mice were fed ad libitum with a standard diet and water. One-million cells were injected into the skin fold of the inner thigh of six mice per group. The animals were killed and the emerging tissue material was dissected. Animal protocols were carried out as approved by the University of Calgary animal protocol no. M03038. Excised tissues were fixed overnight in 4% PFA at 4°C and then embedded in paraffin. The sections were stained in hematoxylin and eosin according to standard procedures.
Flow cytometry
hES cells were dissociated into a single-cell suspension and subjected to fluorescence-activated cell sorting (FACS) using a FACS Calibur instrument and the CellQuest software from Becton Dickinson (Franklin Lakes, NJ). Ten-thousand events were registered per sample, and analysis of whole cells was performed using appropriate scatter gates to avoid cellular debris and aggregates. The cells were stained using the following antibodies: Oct4, Nanog, Tra-1-60, Tra-1-81, and SSEA-4 (all from Santa Cruz). All primary antibodies were directly conjugated to R-phycoerythrin or Alexa Fluor 488 using Zenon conjugation kits (Invitrogen) following the manufacturer's instructions. Anti-mouse or anti-goat IgG R-phycoerythrin or fluorescein isothiocyanate was used as isotope control depending on the primary conjugation molecule used.
In vitro differentiation
Cells were replated on HFF feeder layers. The resulting multilayer colonies were cut into small clumps using a stem cell passaging tool (Invitrogen) and cultured in 35-mm agar-coated dishes (Nunc). The differentiation medium consisted of 80% Dulbecco's modified Eagle's medium (Invitrogen), 20% fetal bovine serum (Invitrogen), 1 mM
Results
Effect of rapamycin on hES cell expansion in suspension bioreactors
H9 hES cells do not survive when transitioned directly from static tissue culture flasks into stirred-suspension culture (data not shown). Aggregates were not formed, and no viable cells remained after 2–3 days in suspension. To increase cell survival in the bioreactors, we added the ROCK inhibitor Y-27632 to the culture medium at 10 μM. The cells were exposed to the inhibitor for the first 4 days, at which point the medium was replaced with fresh mTeSR. Under this treatment, small aggregates were present in the culture after the first 24 h; however, after ROCKi was removed the aggregates spontaneously dissociated and few viable cells were present by day 6 (Fig. 1B). We examined the aggregates using confocal microscopy to determine why viability was lost after day 4 of suspension culture. We stained the aggregates with FGF-R1, which is expressed in hES cell-derived fibroblast cells (Fig. 1C), and found that fibroblasts were indeed present in the aggregates. As fibroblasts are substrate-dependent cells that do not aggregate in suspension, we suspected that these hES cell-derived fibroblasts destabilized the hES cell aggregates, leading to loss of aggregation and therefore a decrease in cell viability.
Through previous work with rapamycin in static hES cell cultures, we have determined that this inhibitor blocks the spontaneous differentiation of the hES cell-derived fibroblasts (data not shown); however, concentrations approaching 100 nM completely arrest the growth of hES cells. Therefore, we applied 0.1 nM rapamycin to the bioreactor to inhibit the appearance of these fibroblasts. When we stained aggregates grown in the presence of 0.1 nM rapamycin, we no longer observed FGF-R1–positive cells (Fig. 1D) and were able to maintain high cell viability beyond day 6.
Expansion of hES cells in suspension bioreactors without passaging
In the bioreactor systems, hES cells (collected from static cultures containing either Matrigel or HFF feeder cells) were inoculated at 1.8 × 104 cells/mL into 100 mL mTeSR medium (StemCell Technologies) and agitated at 100 rpm. Here, cells were cultured in the presence of 10 μM ROCKi for the first 4 days and had constant exposure to 0.1 nM rapamycin. hES cell aggregates were dissociated into smaller clumps every 5 days using collagenase (Fig. 2A, vertical dotted lines). Under these conditions, the hES cells formed tight aggregates (Fig. 2A), similar to that observed with the mouse ES cells, 6 and an overall expansion of 67-fold was achieved over 20 days (Fig. 2B). When ROCKi was excluded from the bioreactor cultures, noticeably fewer aggregates were present; mainly single cells were observed by day 5 and little expansion was achieved (data not shown). Moreover, when the concentration of rapamycin was reduced to 0.05 nM, hES cell aggregates became irregular and no viable cells were present by day 6 (data not shown), even in the presence of ROCKi.
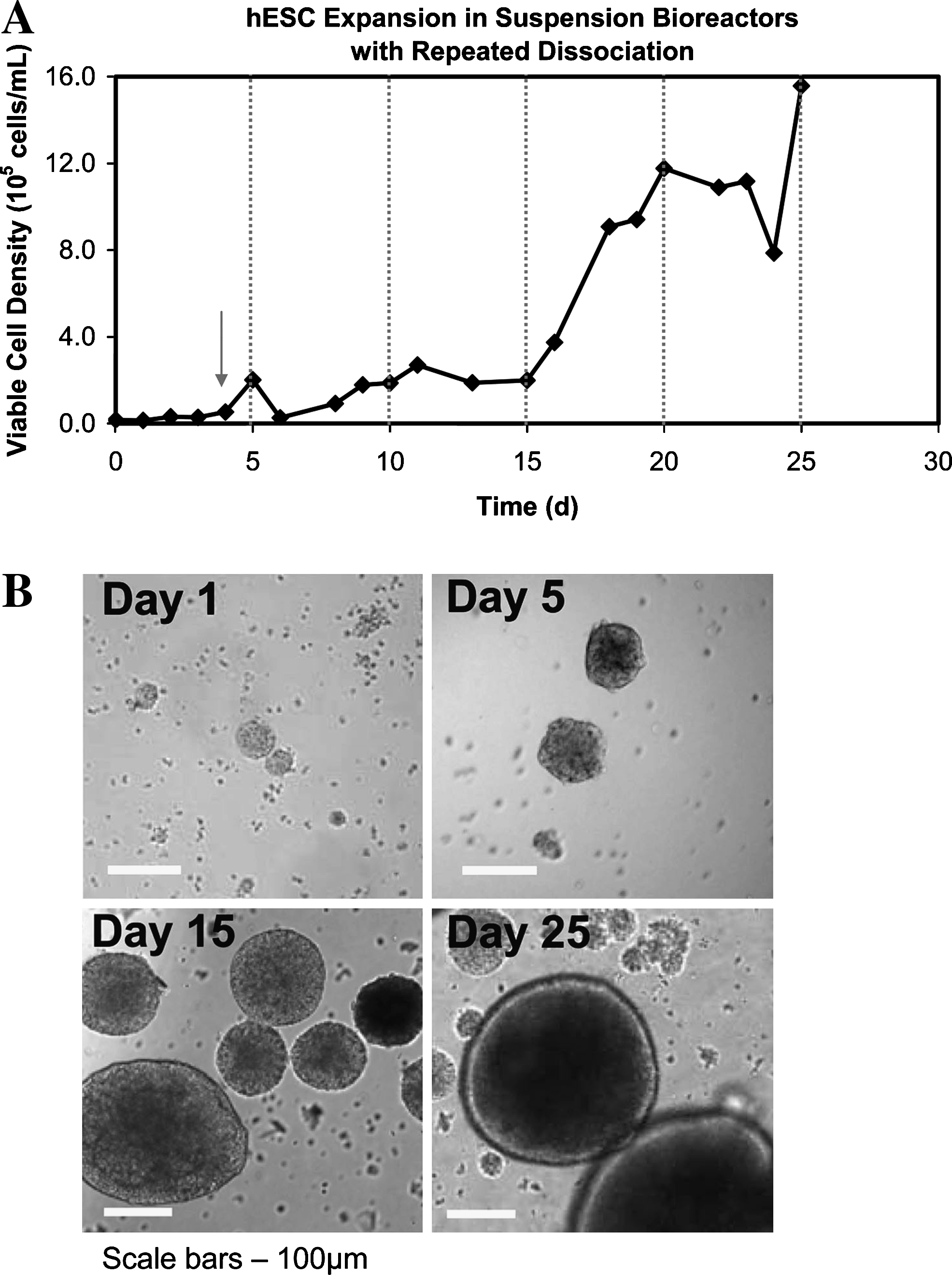
Growth kinetics and morphology of hES cells in suspension bioreactors without passaging. (
Although this method led to hES cell expansion, collagenase treatment was not effective for the dissociation of the hES cell aggregates and this resulted in aggregates that were overgrown. As a result, the cells possessed decreased expression of pluripotency markers (Oct-4, SSEA-4, TRA-1-60, and TRA-1-81) by day 20 (Fig. 3A). FACS analysis demonstrated that only 50% of the cells expressed SSEA-4, TRA-1-60, and TRA-1-81 by day 20, and 79% of the cells were positive for Oct-4 (Fig. 3A). To visualize the localization and expression pattern of the pluripotency markers within aggregates by day 20 of expansion in the bioreactors, we performed whole-mount immunofluorescence using confocal microscopy. Although there was a substantial drop in the number of pluripotent cells in the bioreactors by day 20, these cultures retained a normal karyotype (Fig. 3B). hES cells that were expanded in the bioreactor also retained the ability to form teratomas in vivo (Fig. 4A–C). Additionally, aggregates were taken from day 20 bioreactor cultures and transferred into static nonadherent dishes containing differentiation medium for an EB formation assay. After 16 days, an upregulation in the expression of several early differentiation markers was detected in the EBs using RT-PCR (Fig. 4D).

Characterization of suspension bioreactor expanded hES cells. (
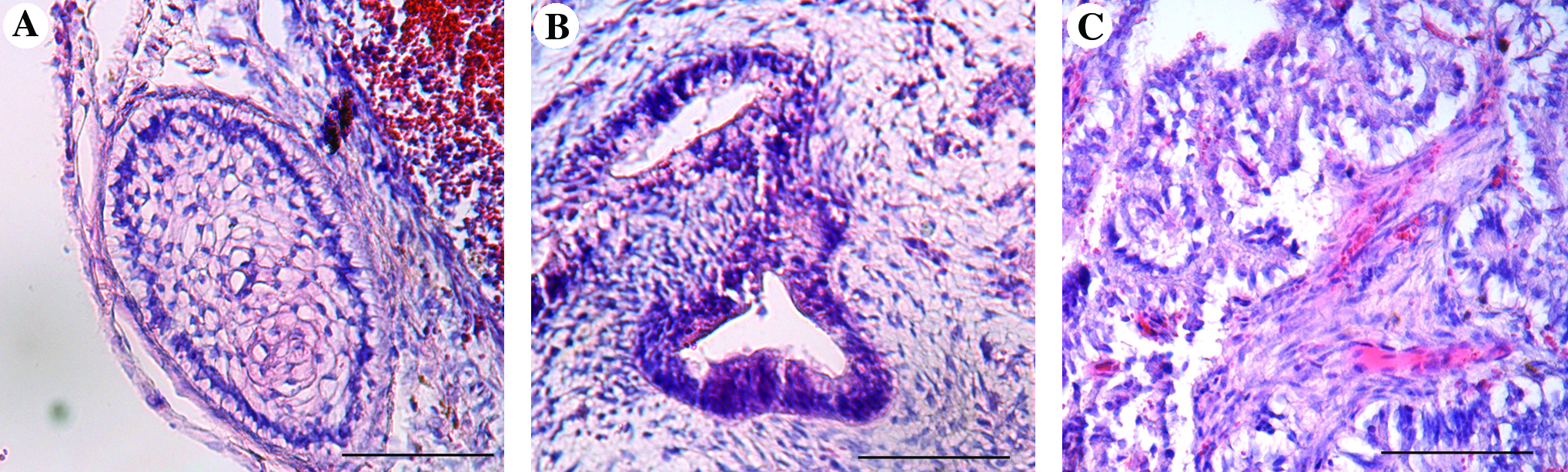
In vivo and in vitro differentiation of bioreactor expanded hES cells. (
Passaging of hES cells in suspension bioreactors
To achieve higher levels of pluripotency in the hES cell bioreactor cultures, smaller aggregate diameters were maintained by routine passaging of the cultures. Here, the cultures were dissociated into single cells every 6 days, using Accuatase (Millipore), and were split 1:5. During each passage, the cells were treated with ROCKi for the first 24 h only, following their dissociation into single cells (Fig. 5A). This method was used to expand hES cells that had been collected from static culture systems containing either Matrigel or HFF feeder cells. 16 The hES cells collected from static HFF cultures possessed reproducible growth characteristics over each passage in the bioreactors. Interestingly, the hES cells collected from static Matrigel cultures did not expand well over the first passage but over subsequent passages acquired a growth curve similar to that of the hES cells from the HFF cultures (Fig. 5A).
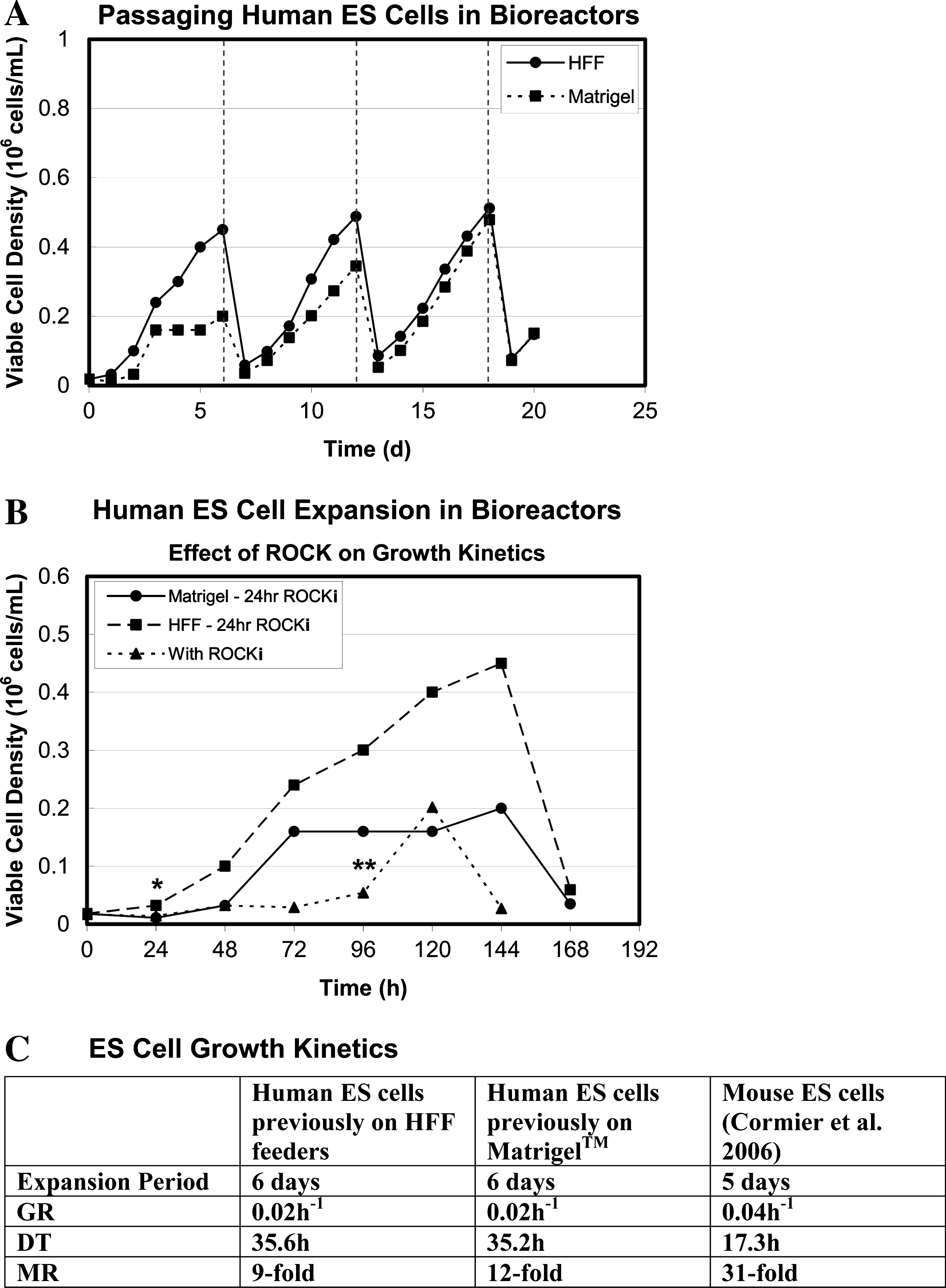
Changes in growth kinetics with ROCKi treatment duration. (
Although we found that ROCKi was a critical component for the transition from static to suspension culture, we also observed adverse affects of ROCKi treatment within the suspension bioreactors (Fig. 5B). Specifically, treatment with ROCKi delayed hES cell expansion, resulting in an extended lag phase in the growth curve. When ROCKi treatment was reduced from 4 days to 24 h, a more immediate expansion of the cells was observed.
With continuous passaging, the hES cells (from HFF cultures) possessed an average growth rate of 0.02 h−1 and expanded 25-fold over a 6-day passage period in the bioreactors (Fig. 5C). In contrast, Cormier et al. showed that mouse ES cells possessed a growth rate of 0.04 h−1 and expanded 31-fold over 5 days. 6 The slower growth rate of the hES cells compared with the mouse ES cells in the bioreactor cultures mimics the difference in growth kinetics observed in static culture systems.
Although there were differences between the growth kinetics of the Matrigel versus the HFF-cultured hES cells over the first passage in the bioreactors, we found no appreciable difference in the expression of all pluripotency markers on day 6. We found that under regular passaging conditions (every 6 days), uniform expression of all the tested pluripotency markers were maintained within the aggregates by day 21 of bioreactor culture (Fig. 6A). FACS analysis of the days 6 and 21 bioreactor cultures showed higher positive expression levels of the pluripotency markers in the passaged versus the nonpassaged bioreactor cultures, with many of the markers being expressed at levels greater than 90%. Interestingly, however, hES cell aggregates generated from Matrigel static cultures contained greater numbers of pluripotent cells on days 6 and 21 (Fig. 6B) compared with hES cells that were first expanded on HFFs. There was no detectable difference in the localization of the pluripotency markers within the aggregates when the cells were first cultured on either Matrigel or HFFs in the static systems when assayed at the protein or mRNA levels using RT-PCR (Fig. 6D); moreover, no abnormalities when detected when the karyotypes were analyzed (Fig. 6C). Finally, both groups produced teratomas in vivo (Fig. 7A–C) and spontaneously differentiated in tissues from all three germ layers in vitro (Fig. 7D).
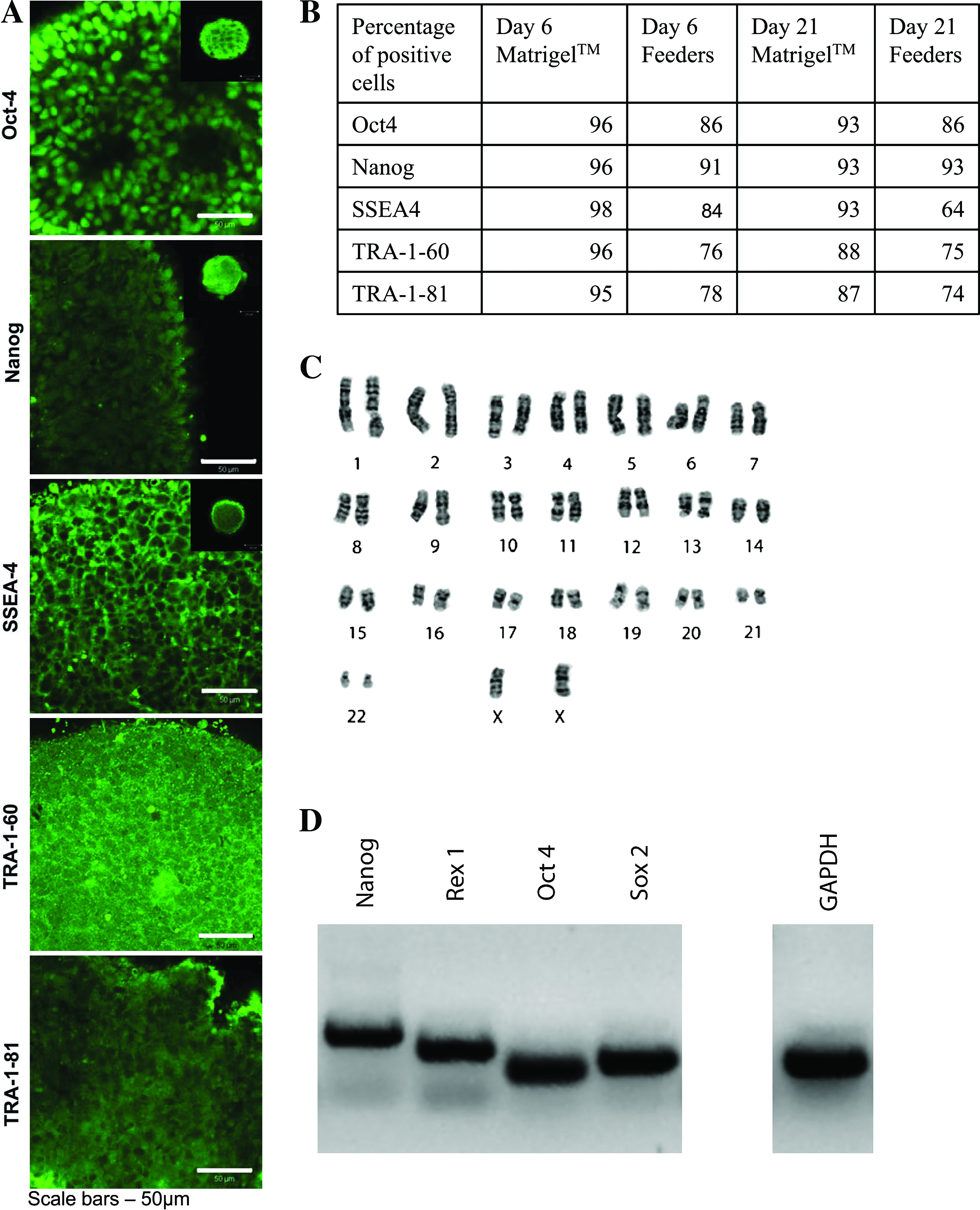
Characterization of bioreactor expanded and passaged hES cells. Characterization of hES aggregates from bioreactor cultures that were repeatedly passaged. (

In vivo and in vitro differentiation of bioreactor expanded and passaged hES cells. (
Discussion
In previous studies, we have demonstrated that mouse ES cells can be readily expanded as aggregates in suspension bioreactors.6,7 In the presence of leukemia inhibitory factor (LIF), cells maintained their pluripotency in long-term culture, as determined by marker expression, spontaneous and directed differentiation, as well as in vivo teratoma formation. However, our attempt to apply a similar approach to hES cells was initially fraught with significant difficulty. Like many other groups, we encountered significant difficulty with the viability of hES cells following enzymatic dissociation and were forced to expand cells using manual colony cutting. 20 This approach, however, made it impossible to generate sufficient numbers of cells to inoculate the smallest of suspension culture flasks.
Following reports that the ROCKi, Y-27632, allowed for the enzymatic passaging of hES cells, we began to investigate its application toward the suspension culture of hES cells. We compiled evidences suggesting that ROCKi has no direct effect on apoptosis but instead increases the cell–cell and cell–substrate interactions of hES cells.21,22 In this study, the increase in cell–cell interactions may have facilitated the aggregation of the cells that was observed over the first 24 h following inoculation of the bioreactors. Importantly, by decreasing the exposure to ROCKi from 4 days to 24 h, a substantial decrease in the lag phase of the cultures was achieved (Fig. 5B). This suggests that ROCKi may impede the proliferation of the hES cells in suspension bioreactors and that exposure of the cells to this supplement should be minimized. Consistently, other research demonstrated that Y-27632 can inhibit the growth of certain cells types, while appearing to have no other negative consequences of cell viability. 23
Since rapamycin is crucial for hES cell expansion within the bioreactors, in part through regulating the spontaneous differentiation of fibroblasts, we are interested in studying the mechanism behind this observation; however, our ability to identify the signal transduction pathways is difficult because running parallel controls (i.e., running a culture that lacks rapamycin) is not possible. As an alternative, we have treated static hES cell cultures with various concentrations of rapamycin, ranging from 0.1 to 100 nM, to examine its effects. Overall, we observed that at high concentrations (100 nM), hES cell cultures no longer expand and at lower concentrations (0.1 nM) the spontaneous differentiation of hES cell-derived fibroblast-like cells no longer occurred (data not shown), as within the bioreactor. This observation was intriguing because previous groups have demonstrated the requirement of the insulin-like growth factor (IGF)/FGF signaling axis in hES cell maintenance. 18 Here, they showed that the hES cell-derived fibroblasts responded to the bFGF signal and in turn secreted IGFII, which is recognized by insulin growth factor receptor on the surface of hES cells. They also demonstrated that IGF signaling is capable of maintaining hES cells.
If rapamycin is regulating insulin signaling in the bioreactor, there must be another molecule maintaining hES cells in an undifferentiated state. Previous studies have shown that TGF-β signaling is necessary for the maintenance of hES cell pluripotency.18,24 Although hES cells may lack the supporting fibroblast cells when cultured in the bioreactors, we believe that TGF-β within the mTeSR™ medium is primarily responsible for the maintenance of pluripotency in the bioreactor cultures during long-term expansion. It has been demonstrated that rapamycin cooperates with TGF-β, potentiating the signaling cascade. 25 This might contribute to the observation that rapamycin increases the potential of stem cells under certain conditions. 26 Additionally, there is evidence that rapamycin inhibition of mTOR negatively affects ROCK expression in macrophages. 27 This observation requires further study to determine if there is indeed cross-talk between mTOR, ROCK, and TFG-β when hES cells are expanded within a stirred-suspension bioreactor culture system.
Utilizing a microcarrier-free approach in a stirred-suspension bioreactor culture system we have been able to produce a clinically relevant number of hES cells while expending a fraction of the hands on required to produce the same cell numbers using conventional static culture systems. Further, recent articles have demonstrated that hES cells can be expanded on microcarriers in stirred bioreactors with equal or reduced expansion rates that we demonstrate in this study;28,29 however, utilizing our system, hES cells do not have to be separated from microcarriers to be utilized in downstream applications.
In conclusion, we have developed a novel bioreactor expansion process for the large-scale production of highly pluripotent hES cell cultures. This process offers significant advantages over the use of static tissue culture flasks as it permits the generation of clinically relevant cell numbers with minimal labor in a controlled and reproducible culture system. Further, the protocols that we demonstrate here can facilitate the implementation of GMP level standards to hES cell culture and thus provide an important foundation for future studies that look to transition hES cells into clinical therapies utilizing next-generation technologies capable of producing cell-based products in a safe, robust, and cost-effective manner. 30
Footnotes
Acknowledgments
The authors thank the University of Calgary Flow Cytometry Facility, for their assistance and advice, and StemCell Technologies (Vancouver), for provision of media. J.T. was funded by NSERC and AHFMR scholarships. D.E.R. is an AHFMR Senior Scholar. This work was funded by an NSERC CHRP grant.
Disclosure Statement
No competing financial interests exist.