
Abstract
In a search for the optimal nonviral gene transfer technique in epidermal and dermal supportive extracellular matrix studies, we investigated the efficiency of late generation liposomal transfection reagents and nucleofection of fibroblasts (FBs), endothelial progenitor cells (EPCs), and keratinocytes (KCs) as essential indicators of healing skin wounds. FBs, KCs, and EPCs were grown under serum-reduced conditions and manipulated according to optimized in vitro manufacturer protocols. Fugene HD, Effectene, PEI, and Lipofectin were compared to Amaxa Nucleofection. A green fluorescent protein (GFP)-encoded reporter gene plasmid was transfected, and transfection efficiencies were determined by green-fluorescence-activated cell sorting. Normal cell morphologies were observed after either transfection or nucleofection. For KC cell cultures, Fugene HD resulted in the highest transfection efficiency in human (41%) and porcine (42%) KCs. For EPCs, Effectene was optimal for human-derived cells (42%), whereas nucleofection was optimal (32%) for porcine cells. For FBs, however, nucleofection resulted in the highest transfection rates in human (46%) and porcine (60%) FBs. For specific epidermal cell studies, Fugene HD was the preferred gene transfer method, whereas Effectene appeared to be the optimal reagent for pro-angiogenic studies. Nucleofection in combination with FBs is the best combination to achieve the highest overall transfection rate and is thus the optimal combination for use in ex vivo gene transfer strategies of wound healing or skin tissue engineering.
Introduction
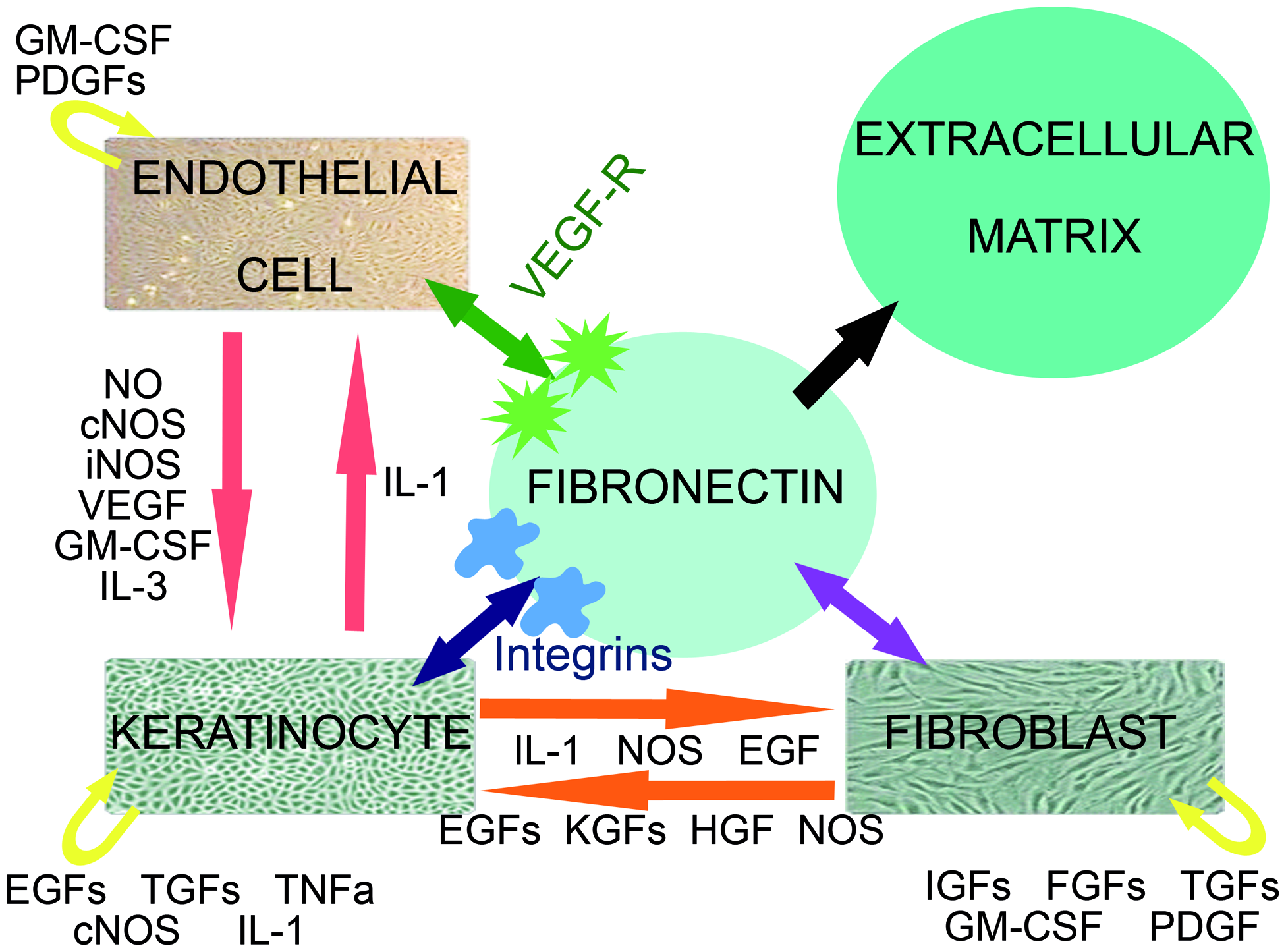
Interactive cross-talk between KCs, FBs, and endothelial cells during wound healing. The endothelial cell population represents both EPCs as differentiated ECs present in the wound. Schematic representation of the close relationship between KCs, FBs, and endothelial cells and the interacting growth factors and cytokines. cNOS, constitutive nitric oxide synthase; EGF, epidermal growth factor; ECs, endothelial cells; FBs, fibroblasts; FGF, fibroblast growth factor; GM-CSF, granulocyte macrophage colony stimulating factor; HGF, hepatocyte growth factor; IGF, insulin-like growth factor; IL, interleukin; iNOS, inducible nitric oxide synthase; KCs, keratinocytes; KGF, keratinocyte growth factor; NO, nitric oxide; PDGF, platelet-derived growth factor; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF-R, vascular endothelial growth factor receptor. Color images available online at
Most gene therapy approaches with KCs, FBs, or EPCs have been performed successfully with viral vectors. However, nonviral gene transfer may be preferable because wound healing is a time-limited process, and temporary expression is not disadvantageous in wound repair. 8 Ex vivo gene therapy using Lipofectin and KCs as gene carriers for wound repair leads to low transfection efficiencies5,9 that require optimization. Improved reagents and techniques, however, are now available. Promising strategies should achieve high transfection efficiencies without deteriorating the migration and proliferation capacities of KCs, FBs, and EPCs. We selected four ex vivo gene transfer methods to compare to a standard reagent, Lipofectin. 9 Three promising reagents (Fugene HD, Effectene, and PEI) were chosen according to transfection efficiency assays that have been performed successfully on different cell lines.10–14 Several reports have indicated higher transfection rates than conventional agents for nonviral ex vivo gene transfer by nucleofection.15,16 Therefore, we added the Nucleofection technology for ex vivo gene transfer of our three cell lines compared to the before mentioned three promising agents and Lipofectin as a control. The present study was designed to evaluate the transfection efficiency of nonviral ex vivo gene transfer using KCs, FBs, and EPCs as carriers. Optimization of these gene transfer strategies in vitro may facilitate the use of ex vivo gene transfer protocols for full-thickness cutaneous wound repair and skin tissue engineering.
Materials and Methods
Cell cultures
KCs were isolated and pooled from a 6 cm2 skin graft that was harvested from a 2-month-old Yorkshire pig under general anesthesia. Fresh skin grafts were washed successively with 70% ethanol (VWR International, Haasrode, Belgium) and phosphate-buffered saline (PBS; Invitrogen, Merelbeke, Belgium) and then incubated for 3 h with 2.4 U/mL Dispase II (Roche Applied Sciences, Vilvoorde, Belgium). The dermis and epidermis were separated, and basal KCs were harvested from the epidermal fraction with 0.05% trypsin-ethylenediaminetetraacetic acid solution (Invitrogen). KCs were seeded in the KC medium containing L-glutamine, 50 μg/mL bovine pituitary extract, 5 ng/mL human recombinant epidermal growth factor (Invitrogen), and 2.5% Chelex 100-treated, calcium-depleted (Bio-Rad Laboratories, Nazareth Eke, Belgium) fetal bovine serum (FBS; Lonza, Verviers, Belgium). Single cells were seeded at a density of 50,000 cells/cm2 on collagen type I-coated culture dishes and incubated at 37°C and 5% CO2. Second- and third-passage cells were used for experiments. The medium was changed every second or third day.
FBs were pooled out of the dermal skin fraction following Dispase II treatment, and a collagenase type 1A (Sigma Aldrich, Bornem, Belgium) digestion was performed overnight. Single cells were washed, centrifuged, and filtered through a 70-μm cell strainer and plated on conventional culture plates at 50,000 cells/cm2. FBs were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS (Lonza) and 1% antibiotic/antimycotic solution (Sigma Aldrich) at 37°C and 5% CO2. Second- and third-passage cells were used for experiments. The medium was changed every second or third day.
For porcine EPCs, 50 mL of whole blood was harvested from a pig's internal jugular vein under general anesthesia. The blood was collected in a 60-mL syringe with 600 mg dalacin (Pharmacia, Puurs, Belgium) and 500 U heparin (Leo Pharma NV, Wilrijk, Belgium) and then diluted 1:4 with PBS. Diluted blood samples were centrifuged over a Histopaque 1083 layer (Sigma Aldrich) at 450 g for 20 min. The buffy coat was harvested, the cells were washed in PBS and centrifuged at 450 g for 5 min, and the washing step was repeated twice. Mononuclear cells were seeded at 200,000 cells/cm2 in endothelial basal medium 2 supplemented with an EGM-2 bullet kit (epidermal growth factor), GA-100 (gentamicin and amphotericin A), 2% FBS, vascular endothelial growth factor, basic fibroblast growth factor, insulin-like growth factor, ascorbic acid, and heparin without hydrocortisone (Lonza) and grown on collagen type I-coated culture dishes at 37°C and 5% CO2. The conditions for human EPC isolation and growth were slightly different than for porcine EPCs. Venous blood samples (40 mL) were obtained from healthy human volunteers, aged 28–35, and anticoagulated with 100 IU/mL heparin. The blood was diluted 1:1 in PBS containing 2% FBS and 1% penicillin/streptomycin/amphotericin. This mixture was layered onto 25-mL Ficoll-Paque (GE Healthcare, Diegem, Belgium) and centrifuged at 750 g for 30 min. The buffy coat was aspirated and washed three times in PBS, and cells were resuspended in endothelial basal medium 2 supplemented with the EGM-2 bullet kit (including hydrocortisone; Lonza) and 1% additional penicillin/streptomycin/amphotericin, plated on collagen type I-coated dishes (Greiner Bio One GmBH, Frickenhausen, Germany) at 200,000 cells/cm2 and incubated at 37°C and 5% CO2. Second- and third-passage EPCs were used for these experiments. The medium was changed daily until the second week and every second day afterward.
Plasmid preparation
The PureLink™ HiPure Plasmid DNA Purification Kit (Invitrogen) was used according to the manufacturer's instructions to purify porcine green fluorescent protein (pGFP) encoding for green fluorescent protein. DNA was measured photometrically at 260, 280, and 320 nm for concentration determination and purity control.
Transfection reagents
Five commercially available nonviral transfection reagents were used. The transfection reagents were chosen for their powerful transfection efficiencies and subtle but specific differences.
Lipofectin (Invitrogen) is a water-based 1:1 (w/w) liposome formulation of the cationic lipid N-[1-(2,3-dioleyloxy)propyl]-n,n,n-trimethylammonium chloride and dioleoyl phophotidylethanolamine in membrane-filtered water. Fugene HD (Roche Applied Sciences) is a proprietary blend of lipids and other components in 80% ethanol. PEI or polyethylenimine (Polysciences Inc., Eppelheim, Germany) is a highly branched polycationic polymer of the monomer ethylene-imine with a high charge density. Effectene (Qiagen, Venlo, The Netherlands) is a unique nonliposomal lipid formulation based on the conjunction of an enhancer and a DNA condensation buffer for high transfection efficiency. Nucleofection (Amaxa Inc., Cologne, Germany) is a DNA electroporation technique using cell-line-specific media and electric shock.
Transfection techniques
Cell cultures (passage 3–4) were seeded at 75,000 cell/cm2 in six-well plates the day before transfection. The next day, the medium was replaced, and the transfections were performed according to the manufacturers' protocols. The transfection reagents Fugene HD (Roche Applied Sciences) and Effectene (Qiagen) were used at room temperature. Lipofectin (Invitrogen) and PEI (Polysciences Inc.) were preserved on ice. Plasmid DNA (pGFP for quantitative analysis using fluorescence-activated cell sorting [FACS]; Invitrogen) was stored on ice until further use. A small amount of DNA was used (1 μg for Fugene HD, Lipofectin, and PEI and 0.1 μg for Effectene) with no transfection reagent as a negative control. Analyses were performed in triplicate 48 h after transfection to be certain that DNA was incorporated and transcribed properly within the cells.
Fugene HD transfection
EPC, KC, and FB cell cultures were transfected following the recommended protocol, using ratios of 5:2 and 8:2 (μL Fugene HD transfection reagent per μg plasmid DNA) and varying concentrations of DNA (1 and 2 μg). Briefly, plasmid DNA was dissolved in 200 μL serum-free Waymouth medium. Fugene HD was added, mixed, and incubated for 15 min at room temperature. The transfection complexes were subsequently added in a drop-wise manner to the cultures. According to the manufacturer's recommendations, a medium change to serum-containing Waymouth media was not necessary. Analyses were performed 48 h after transfection.
Effectene transfection
EPC, KC, and FB cell cultures were transfected following the recommended protocol, using ratios of 10:1 and 25:1 (μL Effectene transfection reagent per μg plasmid DNA) and varying concentrations of DNA (0.1 and 0.2 μg). It should be noted that 0.1 and 0.2 μg DNA was used for Effectene, in contrast to the standard 1 μg DNA used for all other tested reagents. In previous experiments, we found that the use of 1 and 2 μg DNA resulted in low transfection efficiencies in our cell lines (Table 1). Therefore, in subsequent experiments, we followed the manufacturer's recommended concentrations. The kit provided the Effectene transfection reagent, an optimized DNA condensation buffer and an enhancer. Briefly, plasmid DNA was dissolved in 60 or 120 μL buffer EC and an eightfold higher concentration of enhancer. The complex was vortexed for 1 s and incubated at room temperature for 4 min. Next, Effectene was added to the diluent, mixed for 10 s, and incubated for 7 min at room temperature. A total of 500 μL of the growth medium was added to the transfection complexes, mixed, and immediately added in a drop-wise manner to the cell cultures. After an overnight incubation, the cell cultures were provided with a fresh growth medium and analyzed 48 h after transfection.
Data sheet representing different cell-lineage-specific transfection conditions with varying DNA concentrations and DNA-to-reagent ratios for Effectene in human cell lines under standard transfection conditions.
Lipofectin transfection
EPC, KC, and FB cell cultures were transfected following the recommended protocol, using ratios of 6:2 and 9:2 (μL Lipofectin transfection reagent per μg plasmid DNA) and varying concentrations of DNA (1 and 2 μg). Briefly, plasmid DNA was dissolved in 200 μL serum-free Waymouth Medium (Invitrogen) and simultaneously Lipofectin was dissolved in 200 μL Waymouth Medium. The two mixtures were combined and incubated at room temperature for 45 min. The DNA solutions were carefully combined and incubated for another 15 min. The complexes were added in a drop-wise manner to the cultures and incubated overnight. The growth medium was replaced, and the transfection results were analyzed 48 h after transfection.
PEI transfection
EPC, KC, and FB cell cultures were transfected following the recommended protocol, using ratios of 4:1 and 10:1 (μL PEI transfection reagent per μg plasmid DNA) and varying concentrations of DNA (1 and 2 μg). Briefly, plasmid DNA was dissolved in 200 μL serum-free Waymouth Medium, and PEI was added, mixed, and incubated for 10 min at room temperature. The transfection complexes were subsequently added in a drop-wise manner to the cell cultures. The cultures were incubated overnight and provided with a fresh growth medium. Analyses were performed 48 h after transfection.
Nucleofection technique
The Amaxa nucleofection technology (Lonza) was performed following manufacturer protocol 11, which is specific for KCs (Human Keratinocyte Nucleofector Kit; VPD-1002; Protocol T-07, T-24), FBs (Human Dermal Fibroblast Nucleofector Kit; VPD-1001; Protocol A-24, Y-16, U-12, U-23, V-13), and EPCs (Basic Nucleofector Kit for Primary Mammalian Endothelial Cells; VPI-1001; Protocol M-03, T-05, T-23, T-27, U-11). The protocol includes the Nucleofector device to deliver a manufacture-known specific electrical shock and incubation time. In brief, cells were harvested, counted, and resuspended at 500,000 cells/100 μL Nucleofector solution supplemented with 1 or 2 μg DNA. Nucleofection was performed following the recommended Nucleofector program, and cells were plated on six-well plates with the appropriate medium. Analyses were performed 48 h after transfection.
FACS assay
Before performing the FACS analysis, a morphology assessment was conducted to verify cell survival and proliferation levels (at 70%–90% confluence) using a microscope under low power field magnification. FACS provides fast, objective, and quantitative recording of fluorescent signals from individual cells and was conducted with the following protocol. The growth medium or transfection medium was removed, and the culture plates were rinsed with PBS. Cells were trypsinized for 5–7 min at 37°C. The suspension was centrifuged for 5 min at 400 g at 4°C. Cells were rinsed with PBS once and centrifuged again. Next, the pellet was dissolved in 500 μL PBS and 0.5% bovine serum albumin solution (Sigma Aldrich) and transferred to FACS tubes with a cell strainer filter mesh (VWR International). The cell pellet solution was filtered through a mesh membrane and analyzed by FACS analysis. The transfection conditions were measured in triplicate.
Statistical analysis
The results for each transfection reagent were compared to the optimal conditions of the other reagents using an analysis of variance. A p-value of <0.05 was considered statistically significant. The data are depicted as averages and standard deviations.
Results
For human KCs, Fugene HD in a 2:8 DNA-to-reagent ratio and 1 μg of total DNA performed better than all other treatments (41.4%; p < 0.001). Similarly, for porcine KCs, Fugene HD in a 2:8 DNA-to-reagent ratio and 1 μg of total DNA (42.4%; p < 0.001) performed better than all other treatments except for Effectene at a 1:10 ratio and 0.2 μg DNA (p > 0.05) and Fugene HD at a 2:5 DNA-to-reagent ratio with 1 μg DNA (p > 0.05). The second best reagent for human KCs (but with a significantly lower transfection efficiency) was PEI (16.5%; p < 0.001) (Table 2 and Fig. 2). Both human and porcine FB cell populations had the highest transient GFP expression with Amaxa Nucleofector technology. The Nucleofector program V-13 and 2 μg DNA showed transfection rates of 46.2% for human FBs (p < 0.001 compared to all other treatments) and 60.1% for porcine FBs (p < 0.05 compared to Amaxa protocol U-23 and p < 0.001 compared to all other treatments).

Transfection efficacy of KCs, FBs, and EPCs. Representation of transfection of human- and porcine-derived KCs, FBs, and EPCs. Only the optimal condition for each reagent is shown and plotted against each other. EPCs, endothelial progenitor cells.
Data sheet representing different cell-lineage-specific transfection conditions with varying DNA concentrations and DNA-to-reagent ratios for five different nonviral gene transfer techniques in human- and porcine-derived keratinocytes. The overall optimal condition is indicated in bold; the optimal condition for each transfection reagent separately is underlined.
Transfection with all other reagents led to inconsistent outcomes, with peak expression levels of 12.7% (PEI) and 10.4% (Fugene HD) for human FBs. The peak expression levels for porcine FBs were 17.9% with Fugene HD and 9.5% with PEI (both 1 μg DNA) (Table 3 and Fig. 2). A difference in the EPC lineages was observed between human and porcine endothelial cells. Transfection with Effectene resulted in a maximal transfection rate of 41.9% for human EPCs with 0.1 μg DNA and a 1:25 ratio. There was no significant difference within Effectene conditions; however, there was a significant difference (p < 0.001) compared to all other transfection reagents. Porcine EPCs showed high transfection levels using Amaxa technology program T-05 with 1 μg DNA (31.8%). There was no significant difference within Amaxa protocols M-03 and T-05 treatments, but there was a significant difference (p < 0.001) compared to the other transfection reagents and ratios. Transfection of human ECs with 2 μg Fugene HD resulted in the second best condition, with a transfection efficiency of 28% (p < 0.001). For porcine ECs, Effectene was the second best reagent, but it resulted in a significantly lower transfection efficiency (14.5%) under conditions of 0.1 μg DNA and a 1:10 DNA-to-reagent ratio (p < 0.001) (Table 4, Fig. 2 and Fig. 3).
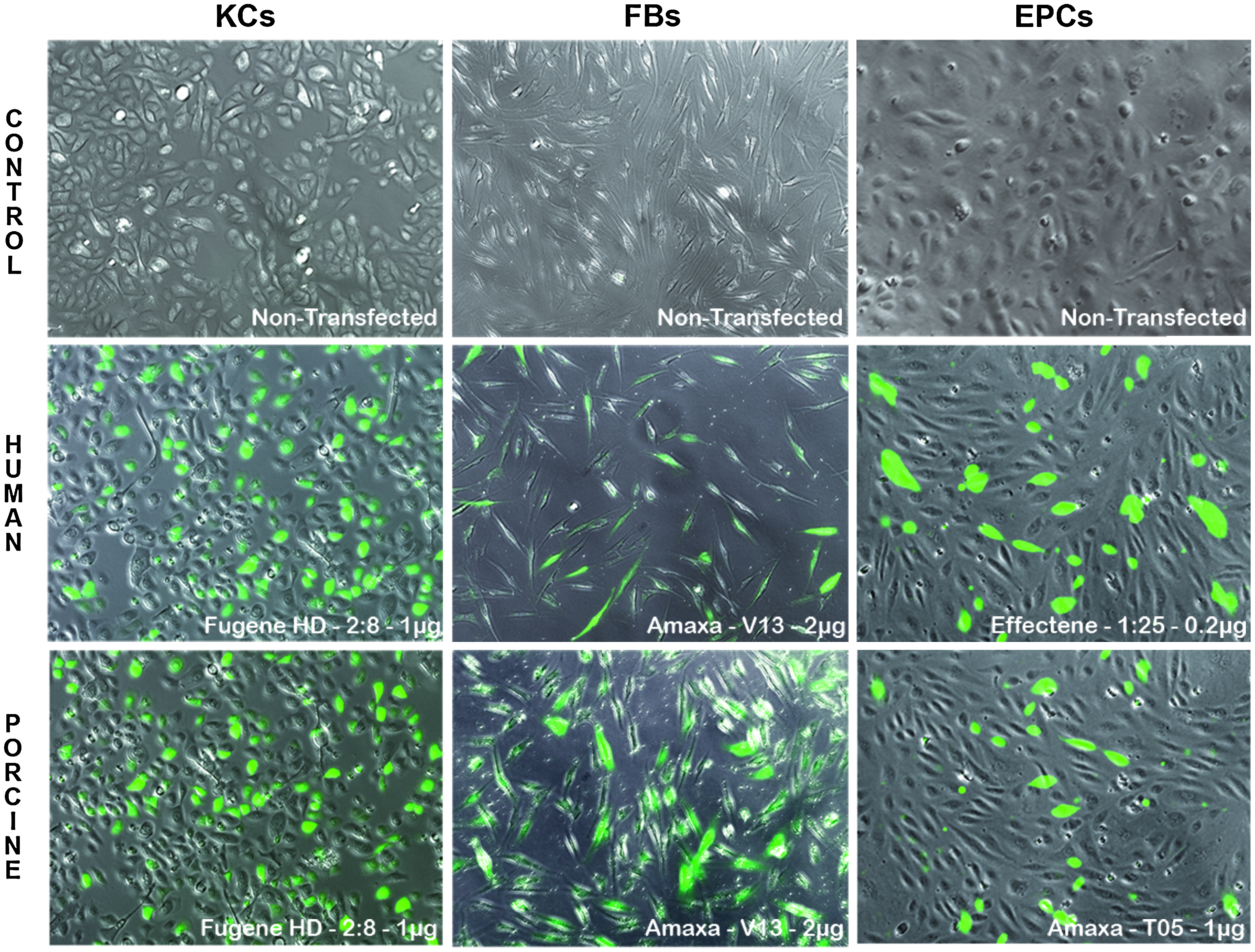
Transfection efficacy of KCs, FBs, and EPCs. Representation of the optimal transfection condition of human- and porcine-derived KCs, FBs, and EPCs using the optimal GFP transfection protocol compared to controls. Color images available online at
Data sheet representing different cell-lineage-specific transfection conditions with varying DNA concentrations and DNA-to-reagent ratios for five different nonviral gene transfer techniques in human- and porcine-derived fibroblasts. The overall optimal condition is indicated in bold; the optimal condition for each transfection reagent separately is underlined.
Data sheet representing different cell-lineage-specific transfection conditions with varying DNA concentrations and DNA-to-reagent ratios for five different nonviral gene transfer techniques in human- and porcine-derived endothelial progenitor cells. The overall optimal condition is indicated in bold; the optimal condition for each transfection reagent separately is underlined.
Discussion
The U.S. Food and Drug Administration has previously approved FB and KC cell cultures for clinical use. Both FBs and KCs have been integrated into commercially available skin replacement products, including Epicel, Dermagraft, Apligraf, Orcel, and PermaDerm. Using an ex vivo gene transfer strategy, cell cultures can serve as gene carriers and function as a temporal local production unit of de novo synthesized growth factors within the wound or skin replacement.5,8,9 Growth factors that are secreted by these cells will stimulate matrix formation, angiogenesis, and remodeling by intensive crosstalk and interactions.5,17 In this study, we selected KCs, FBs, and EPCs as gene transfer carriers because they represent three major cell populations in skin engineering protocols3,18–21 (Fig. 1 and Fig. 3).
In addition to the Nucleofector technology, four reagents (Lipofectin, Fugene HD, Effectene, and PEI) were selected because they have been used in previously reported transfection efficiency assays.10–14 These reagents can be divided into three subgroups: cationic lipids (Lipofectin, Fugene HD), cationic polymers (PEI), and nonliposomal lipids (Effectene). Amaxa nucleofection is a nonviral transfection technology that is based on a unique combination of electrical parameters and cell-type-specific solutions. Cationic liposomes were the first nonviral transfection reagents described 21 and have since become the most frequently used nonviral transfection reagents in vitro. 23 They lack the biohazard of viral transduction, are safe and reliable drug carriers in vivo, and are approved for clinical use as a drug delivery system.24–27 The mechanism of liposomal transfection is based on the condensation of DNA by positively charged compounds.23–28 Positively charged liposomes and negatively charged DNA spontaneously form complexes due to the electrostatic and hydrophobic interactions between DNA phosphate groups and the reagents,22,23 resulting in condensed complexes of DNA, liposomes (Lipofectin and Fugene HD), or micelles (Effectene). The major difference between Lipofectin, Fugene HD, and Effectene is the manner of condensation.
Lipofectin and Fugene HD act through cationic polar head groups, whereas Effectene acts through a specially formulated, highly condensed enhancer system. PEI represents a third mechanism; complexes are condensed using synthetic polymers (PEI) through electrostatic interactions by means of amines and ammonium ions. Nucleofection 13 is a novel nonviral transfection technology that is designed for primary and difficult-to-transfect cell lines. It allows transfected DNA to enter the nucleus directly by an electric shock and does not rely on cell division for transfer.13,29 Therefore, nucleofection provides the ability to transfect nondividing cells such as neurons and quiescent blood cells. Nucleofection can achieve viral-mediated transfer efficiencies of up to almost 100% for each of our three cell lines.
In research protocols for clinical-based wound healing, a 100% transfection efficiency is not mandatory because wound healing is a time-limited process, and temporary expression is not disadvantageous in wound repair. 8 In addition, transient gene therapy is of particular interest for wound repair and regeneration because a temporary increase in growth factors, such as vascular endothelial growth factor or basic fibroblast growth factor, are required at a particular point in the wound regeneration process; when the next phase is reached, other growth factors become more important for matrix and scar remodeling.6,7 Moreover, viral approaches may lead to significant inflammatory responses in clinical settings. Because wound repair is intrinsically linked to a significant inflammatory response, a viral approach that elicits this response is undesirable. 9
In most of our transfection protocols, we noted that KCs and FBs do not undergo great morphology changes when cationic liposomes are used, but do slightly change in the first 3–24 h following a transfection protocol using Lipofectin and Lipofectamine 2000.9,17 After an initial distress phase, cells begin to proliferate and migrate again. This cell revival, at ∼48 h, marks the ideal time point to harvest and transfer transfected cells to full-thickness skin wounds in our clinical porcine wound model5,8,9,17 to elicit a maximal effect on the wound repair and regeneration process. Therefore, we analyzed the transfection rates and cell morphologies at 48 h, a clinically relevant time point in the wound-healing environment.
During sequential wound healing stages, FBs are in close relation to epithelial cells. The interaction between these cell types results in closure by wound contraction or a combination of matrix formation, contraction, and reepithelialization. The latter process avoids a loss of function and is more desirable than wound contraction. Both cell types are easily obtained from thin split-thickness skin grafts and are easily cultured. Therefore, both cell types are readily available as gene carriers.
KCs multiply rapidly under culture conditions and represent a favorable population of cells for ex vivo gene transfer. However, traditional liposomal gene transfer of KCs has results in low transfection efficiencies.8,17 Zellmer and co-workers stated that Effectene had the highest transfection efficiency in KCs. 11 In our study, Effectene performed significantly better in the transfection of basal cell KCs compared to the Lipofectin control, but Fugene HD consistently resulted in the highest transfection efficiency (Fig. 3).
During regular skin wound healing, the proliferative phase is characterized by granulation tissue formation, angiogenesis, and epithelialization. FBs transform the post-trauma, pro-angiogenic temporary matrix into a structured extracellular matrix by producing connective tissue elements, such as fibronectin and collagens, which increase the strength of the wound. FBs play a role as coordinators of the constantly remodeling matrix. Therefore, FBs may also serve as ideal gene transfer carriers. However, gene therapy with FBs by viral transduction is very inconsistent. Chen et al. 14 observed long-lasting gene expression in rabbit-derived FBs by viral transduction but only in a very limited percentage of animals; traditional liposomal formulations also had low transfection efficiencies. Balestrin and colleagues 30 showed high transient β-galactosidase expression in FBs after nonviral transfection with Lipofectamine 2000 in vitro. We found that FBs are difficult to transfect using nonviral-based reagents, and we found no consistently superior transfection mechanism. We observed that Amaxa yielded consistently and significantly higher transfection efficiencies with FBs compared to other reagents.
Nucleofection of KCs and EPCs was significantly less efficient. These findings were in line with earlier work of Jacobsen et al. 13 In addition, Nakayama and co-workers 31 showed nucleofection rates of up to 60% using the Amaxa Nucleofection system in porcine embryonic FBs.
During the wound repair process, angiogenesis is critically important to provide oxygen to all of the cells involved and to fuel all cellular interactions. EPCs play a role in angiogenesis and vasculogenesis and are indispensable for wound regeneration. In addition, these cells may be used as efficient carriers for (proangiogenic) growth factors. Kiefer et al. 12 investigated the transfection of EPCs with three different transfection reagents, Lipofectin, Effectene, and Fugene 6 (the previous version of Fugene HD). Effectene was the most successful reagent, with a transfection rate of 10%. We found a transfection efficiency of 42% in human EPCs that were transfected with Effectene and 14.6% transfection efficiency in porcine EPCs. Effectene has an advantage compared to the other three reagents: 10 times less DNA is required for transfection (0.1 vs. 1 μg DNA) because of the intrinsic designed enhancer molecule for DNA condensation. The use of less DNA and less transfection medium decreases the cytotoxic side effects.
Nucleofection was the best transfer method for porcine EPCs and led to transfection efficiencies of up to 32%, whereas Effectene seems to be the most appropriate for human-derived EPCs (42%). This difference in outcome may be explained by the distinct isolation and proliferation protocols, different growth media, or differences related to intrinsic cell morphologies.
In conclusion, the selection of the most efficient transfection reagent is dependent on the carriers and the ultimate aim of gene transfer. Fugene HD is superior for topical, epidermal, nonviral gene therapy, whereas Effectene appears to be more efficient for transfection of EPCs. Nucleofection is favorable for all tested cell types, most notably for FBs as gene carriers. In the context of cutaneous ex vivo gene transfer therapy to full-thickness wounds, nucleofection, using FBs as the gene carrier, seems to be the best strategy.
Footnotes
Disclosure Statement
The authors report no conflicts of interest relevant to this article.