
Abstract
Genetic modification of stem cells could be applied to initiate/enhance their secretion of therapeutic molecules, alter their biological properties, or label them for in vivo tracking. We recently developed a negatively charged gene carrier (“anioplex”) based on pullulan-spermine, a conjugate prepared from a natural polysaccharide and polyamine. In rat mesenchymal stem cells (MSCs), anioplex-derived reporter gene activity was comparable to or exceeded that obtained using a commercial cationic lipid reagent. Transfection in the growth medium with 15% serum and antibiotics was approximately sevenfold more effective than in serum-free conditions. Cytotoxicity was essentially indiscernible after 24 h of anioplex transfection with 20 μg/mL DNA, in contrast to cationic lipid transfection that resulted in 40%–60% death of target MSCs. Anioplex-derived reporter gene activity persisted throughout the entire 3-week study, with post-transfection MSCs appearing to maintain osteogenic, adipogenic, and chondrogenic multipotency. In particular, chondrogenic pellet formation of differentiating human MSCs was significantly inhibited after lipofection but not after aniofection, which further indicates the biological inertness of pullulan-spermine/DNA anioplexes. Collectively, these data introduce a straightforward technology for genetic engineering of adult stem/progenitor cells under physiological niche-like conditions. Moreover, reporter gene activity was observed in rat spinal cords after minimally invasive intrathecal implantation, suggesting effective engraftment of donor MSCs. It is therefore plausible that anioplex-transfected MSCs or other stem/progenitor cells with autologous potential could be applied to disorders such as neurotrauma or neuropathic pain that involve the spinal cord and brain.
Introduction
Genetic modification of MSCs has conventionally been performed using viral constructs in experimental settings. 6 However, the clinical applicability of virally transduced stem cells is limited by safety concerns of immune reactivity and oncogenesis,6,7 as well as by challenges in large-scale manufacturing. 7 Nonviral approaches based on cationic polymers and lipids tend to avoid these drawbacks but are also restricted by low efficacy and relatively high in vitro toxicity to target cells.
We recently reported the development of a unique type of negatively charged (ζ-potential −42.23 mV) gene delivery particle, termed “anioplex,” that was prepared by mixing plasmid DNA with pullulan-spermine, a conjugate of pullulan (a naturally occurring polysaccharide) and spermine (a polyamine), at a pullulan-spermine nitrogen: DNA phosphate (N:P) ratio of 1:1. 8 These materials innately offer a favorable safety profile; pullulan is already commercially used in oral healthcare and pharmaceutical coating applications and spermine is ubiquitously found at high concentrations in human cells, where it is thought to naturally interact with nucleic acids. 8 Incorporation of such materials may also allow the “anioplexes” we assemble from pullulan-spermine and plasmid DNA to have properties that are more physiological. We subsequently reported that application of pullulan-spermine anioplexes to primary sensory neuronal cultures resulted in efficient, serum-compatible transfection without detectable cytotoxicity, even at high DNA concentrations and long incubation times. 8
Given the safety requirements for clinical application as well as the notable sensitivity of MSCs to specific biological demands (e.g., growth factors and qualified sera) of in vitro culture conditions, 9 we hypothesized that the inertness of the anioplex system could provide particularly favorable ex vivo transfection of mesenchymal progenitors, allowing efficient gene delivery in the niche-like (i.e., serum- and growth factor-containing) growth medium without discernible toxicity or loss of stem cell properties.
Materials and Methods
Preparation of pullulan-spermine
Pullulan-spermine was prepared as previously described. 8 Briefly, 47.3 kDa pullulan (Hayashibara) was reacted for 5 min with N,N′-carbonyldiimidazole (Sigma) in dimethyl sulfoxide (DMSO) at a ratio of 3 mol N,N′-carbonyldiimidazole per mole of available pullulan OH groups. This mixture was then added dropwise to excess spermine (Sigma) in DMSO and incubated at 35°C for 20 h. Conjugates were then dialyzed against ultra-pure water, lyophilized, and resuspended in ultra-pure water.
Preparation of plasmid DNA
Beta-actin promoter-driven plasmid DNA expressing luciferase (pCAGGS-luciferase, a generous gift from Dr. Jyunichi Miyazaki, the Graduate School of Medicine at Osaka University) was amplified in DH5α-competent cells (Invitrogen) and then purified using an endotoxin-free Megaprep kit (Qiagen) according to the manufacturers' protocols. Plasmid DNA was dissolved in ultra-pure water and only used when A260/A280 ratios were over 1.9.
Preparation of pullulan-spermine/DNA complexes
The appropriate amount of DNA was diluted to half the desired transfectate volume in ultra-pure water and 10% glucose/40 mM HEPES such that the final concentration was 5% glucose/20 mM HEPES. The appropriate amount of pullulan-spermine required to generate the desired N:P ratio was similarly diluted. To generate the transfectate, the pullulan-spermine solution was added to the DNA solution and carefully triturated 10 times via pipetting. Complexes were then incubated at room temperature for 30 min.
Isolation and culture of rat bone marrow stromal cells (BMSCs)
BMSCs were collected as previously described by Takamoto et al. 10 Briefly, femurs and tibias were removed from 3-week-old Wistar rats (Shimizu Laboratory Supplies). The epiphysis of each bone was removed, followed by collection of the bone marrow using 1 mL of 9.6 mM phosphate-buffered saline (PBS; pH 7.4). The bone marrow was then syringe-minced with a 21 gauge needle. The cells were dispersed in α-minimal essential medium (MEM) medium supplemented with 15% fetal bovine serum (FBS) (vol/vol) and 1% penicillin–streptomycin solution, followed by seeding into two 25 cm2 culture flasks (Corning). After incubation at 37°C under a 5% CO2 atmosphere for 3 days, nonadherent cells were removed. Adherent cells were further cultured by exchanging the medium every 3 days for 7–10 days. The cells were detached with 0.25% trypsin (wt/vol), containing 0.8 mM ethylenediaminetetraacetic acid in PBS and subcultured in 225 cm2 cell-culture flasks (Iwaki) at a density of 2 × 104 cells/cm2. Cells were passed 1:2 at 80%–90% confluence, and used at passage 3.
Isolation and culture of rat adipose-derived stem cells
Subcutaneous fat pads were extracted from the backs of 3-week-old Wistar rats (Shimizu Laboratory Supplies), and then subjected to the isolation procedure described for adipose-derived stem cells (ASCs) by Kimura et al. 11 Briefly, adipose tissue was carefully washed with PBS solution (pH 7.4) to remove all blood cells, then minced, and digested with 520 U/mL collagenase (Nitta Gelatin) in a water bath at 37°C for 60 min with shaking. The digested tissue was suspended in the growth medium (Medium 199 containing 10% fetal calf serum), followed by centrifugation (200 g, 5 min at 4°C) and removal of the supernatant. After two washes in the growth medium, cells were cultured in a 75 cm2 cell culture flask (Corning 430720; 1 × 103 cells/cm2) in the growth medium containing 0.1 μg/mL basic fibroblast growth factor at 37°C under a 5% CO2 atmosphere. The cells were passed 1:2 at 80%–90% confluence and used at passage 3.
In vitro transfection
Cells were first seeded in 24-well plates at 1 × 104 cells/cm2. On the following day, the culture medium was replaced with 500 μL OPTI-MEM or growth medium. Pullulan-spermine/DNA complexes containing 1 μg DNA were added to each well in a 50 μL volume. After 6 h incubation at 37°C under a 5% CO2 atmosphere, the medium and transfectate were replaced by 1 mL fresh medium. Cells were harvested for analysis at 1 day after seeding. These parameters were used for all experiments except where otherwise specified. Transfection with Lipofectamine 2000 (Invitrogen) was performed as described above for pullulan-spermine/DNA, with the exception that complexes were formed in a total of 100 μL of OPTI-MEM medium (Gibco) according to the manufacturer's protocol at a Lipofectamine:DNA ratio of 2.5:1. For asialofetuin studies, cells were preincubated with asialofetuin (Sigma) for 1 h before transfection; the asialofetuin-containing medium was then replaced by the transfection medium without washing. For other inhibitor studies, cells were pretreated with chlorpromazine, methyl-beta cyclodextrin, or colchicine (all from Sigma) at the given concentration for 1 h before transfection and also during transfection. For colchicine studies, cells were additionally incubated at the given concentration from transfection until harvesting for analysis. At 5 mM methyl-beta-cyclodextrin, 28 mM chlorpromazine, 1 mg/mL asialofetuin, and 65 μM, we did not observe cell detachment or altered morphology suggestive of lethal cytotoxicity (please see WST-8 assay section below for systematic evaluation of cell viability).
Adipogenic and osteogenic differentiation assays
Cells were seeded and transfected as described above and then incubated in the adipogenic, osteogenic, or growth medium for 4 weeks with medium changes every 3 days. The adipogenic medium consisted of Medium 199 supplemented with 10% fetal calf serum and 0.5 mM isobutylmethylxanthine, 200 μM indomethacin, 1 μM dexamethasone, and 5 μM troglitazone. The osteogenic medium was as described by Takamoto et al., 10 and consisted of α-MEM supplemented with 10% FBS and 50 μg/mL ascorbic acid, 10 mM β-glycerophosphate, and 10 nM dexamethasone. After 4 weeks, cells were fixed in 4% paraformaldehyde for 30 min. For Von Kossa staining, fixed cells were washed in distilled water and then incubated in 5% silver nitrate under an ultraviolet light for 1 h. The silver nitrate solution was replaced by 5% sodium thiosulfate for 3 min, and cells were then washed with distilled water and photographed. For oil red O staining, fixed cells were washed in distilled water and then in 60% isopropanol. Fixed cells were then incubated in 0.21% oil red O in 60% ethanol for 15 min, washed with distilled water until excess dye was removed, and then photographed.
Chondrogenic differentiation assay
Human BMSCs were obtained from the BMSC distribution center at Texas A&M Health Science Center (
At passage 4, micromass cultures were generated by placing one 5-μL drop of 1.6 × 107 cell/mL BMSCs suspended in the growth medium in the center of each well of a 24-well culture plate. BMSCs were allowed to adhere to the plate for 2 h, after which 500 μL growth medium was added and transfection proceeded for 6 h as described above. Groups included nontransfected cells and cells transfected with pullulan-spermine/DNA anioplexes or Lipofectamine 2000/DNA as described above. After 6 h of transfection, the transfection medium was replaced with either fresh growth medium or chondrogenic differentiation medium (StemPro Chondrogenesis Differentiation Kit; Invitrogen). Cells were then cultured for 21 days with medium changes performed every 3 days. After 21 days, chondrogenic pellets and undifferentiated micromass cultures were fixed in 4% paraformaldehyde in PBS for 30 min and washed and stored in PBS.
Chondrogenic pellets and micromass cultures were stained in 1% Alcian Blue in 0.1 N HCl for 24 h, washed with 0.1 N HCl until all unbound dye was removed, and then washed and stored in PBS. The micromass culture, chondrogenic differentiation, and Alcian Blue protocols described above were slightly modified from the manufacturer's instructions for the chondrogenesis differentiation kit. Stained pellets were dark-field imaged using a Zeiss Axiovert 200 microscope and camera with Axiovision software (Carl Zeiss Microimaging). Pellet size was quantified by using the “draw line” function of ImageJ software (
The PBS was then removed, and the Alcian Blue dye was leached from chondrogenic pellets and micromass cultures by addition of 300 μL DMSO (Sigma). The absorbance of the leached Alcian Blue dye at 620 nm (Labsystems MultiSkan MCC 340) was recorded after subtraction of the absorbance of a DMSO-alone blank. Leaching and absorbance measurement of Alcian Blue in DMSO was previously described by Pfander et al. 12 The absorbance of the Alcian Blue from the chondrogenic pellets and the undifferentiated micromass cultures was normalized against that of the respective nontransfected controls.
Chondrogenic pellets were then subjected to a graded series of sucrose solutions in PBS (10%, 20%, and 30%; overnight for each) for cryoprotection and sectioned at 20 μm. Pellet sections were stained with Alcian Blue as described above but for 1 h, coverslipped, and imaged in phase contrast using the microscope system described above.
Luciferase assay
Cells were assayed for luciferase expression by addition of 40 μL of lysis buffer (Promega Corp.) per well. After scraping, lysis buffer was transferred from each well into microcentrifuge tubes. Tubes were centrifuged at 14,000 rpm for 20 min, after which 20 μL of the supernatant from each tube was applied to a 96-well plate. The relative luminescence of each well was measured by a luminometer (Berthold) after addition of 100 μL of luciferase assay reagent (Promega Corp.). An additional sample of supernatant (1 μL) was taken for determination of total protein concentration by bicinchonic acid assay (Pierce) according to the manufacturers' instructions.
WST-8 assay
WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium monosodium salt] measures mitochondrial activity that can then be taken as an indicator of cellular viability or proliferation. This is done via reduction of the tetrazolium salt by NADH produced in viable mitochondria. The WST-8 assay was performed using a commercially available kit (Nacalai Tesque). Briefly, 10% of the culture volume of WST-8 reagent was added to each well and incubated for 4 h, at which point 150 μL of the medium was withdrawn from each well and applied to a 96-well plate for analysis in a microplate reader (Molecular Devices). The absorbance at 650 nm was subtracted from the absorbance at 450 nm to give the final measurement.
Substrate-mediated transfection
Succinylated gelatin and E50 gelatin were prepared as previously described. 7 PI9 gelatin was obtained from Nitta Gelatin. For each well, substrates were prepared by mixing 30 μL of 1 mg/mL succinylated, E50, or PI9 gelatin with 60 μL of 1 mg/mL Pronectin F (Sanyo Chemical Industries) and 210 μL of PBS. Three hundred microliters of this mixture were then added to each well and incubated at 37°C for at least 1 h. Pullulan-spermine/DNA containing 1 μg DNA was prepared as described above in a final volume of 300 μL. The substrate solution was then aspirated from the plate and replaced with the pullulan-spermine/DNA solution. Plates were then incubated at room temperature for at least another 30 min, and the pullulan-spermine/DNA solution was aspirated immediately before cell seeding. ASCs were isolated as described above and then plated. Cells were assayed at 2 days after seeding.
The subfection and conventional transfection conditions used in this study both utilized thin coatings of soluble molecules for neuronal attachment, and both systems represented two-dimensional environments. Thus, we deemed that cell viability following subfection and conventional transfection could be analyzed and directly compared using the WST-8 assay. 8
In vivo lumbar puncture injection
Animal procedures were approved by the ethics committee at Kyoto University. For lumbar puncture injection, cells were cultured and transfected as described above, with ~40 μg anioplex-condensed DNA applied per 1 × 106 cells. Cells were then resuspended in the culture medium at 4 × 104 cells/μL and maintained on ice for up 2 h before implantation. For each injection, 30 μL of the cell suspension (1.2 × 106 cells) was loaded into the injection apparatus, which consisted of a 100 μL Hamilton syringe connected to a 25 gauge needle via a 12 inch catheter. Female Sprague–Dawley rats (300 g) were anesthetized with 3% isoflurane. A small incision was made over the lumbar spinal column, and muscle and connective tissue over the junction between the L5 and L6 vertebrae was cleared away to provide optimal visibility. The needle of the injection apparatus was then placed between L5 and L6, pointing rostrally with the beveled side facing dorsally. The needle was advanced rostrally such it punctured the ligament immediately caudal to L5, and its tip rested closely underneath L5. A mild tail-flick response was typically observed upon penetration of the intrathecal space. The 30 μL cell suspension was then slowly injected, the needle left in place for 2 min, and the skin incision sutured.
Tissue luciferase assay
Four days after intrathecal injection, rats were anesthetized with sodium pentobarbital (60 mg/kg intraperitoneal (i.p.)) and spinal cords were exposed by laminectomy. Spinal cords were harvested from L1 to L3 as well as from L3 to L6, and L4 and L5 dorsal root ganglia (DRG) were also harvested. All tissues were immediately snap-frozen in liquid nitrogen and then stored at −80°C. For each sample, 4 μL lysis buffer (Promega Corp.) was added per mg tissue. Samples were then disrupted using a Qiagen TissueLyser for 15 min at 25 oscillations per second. Lysed tissues were subsequently centrifuged and assayed for luciferase activity as described above for cell samples.
Statistical analyses
The normal distribution of all data sets was first assessed using the Shapiro–Wilkes test, with p < 0.05 accepted as a significant deviation from normal distribution. Student's t-test was then used for comparisons of two means. For comparisons of more than two means, one- or two-way analysis of variances (ANOVAs) were used with Neuman-Keuls, Tukey-Kramer, or Bonferroni post-hoc tests as appropriate.
Results
Transfection of rat bone marrow stromal cells by pullulan-spermine/DNA anioplexes
Initial experiments in the serum-free medium revealed a significant general increase in anioplex-derived luciferase reporter gene activity in rat BMSCs as compared to those transfected with Lipofectamine 2000 (Fig. 1A; p = 0.01, two-way ANOVA). When transfection conditions were broken down by DNA concentration, increases were observed at all concentrations tested (3-, 2.8-, and 2-fold for 2, 10, and 20 μg/mL DNA, respectively); however, when considered individually none of these increases reached statistical significance (Fig. 1A; p = 0.12, p = 0.15, and p = 0.060, respectively, Neuman-Keuls post-hoc). For both Lipofectamine 2000 and anioplexes, reporter gene activity tended to increase as the concentration of DNA increased; however, this effect did not reach statistical significance (Fig. 1A; p = 0.11, two-way ANOVA).
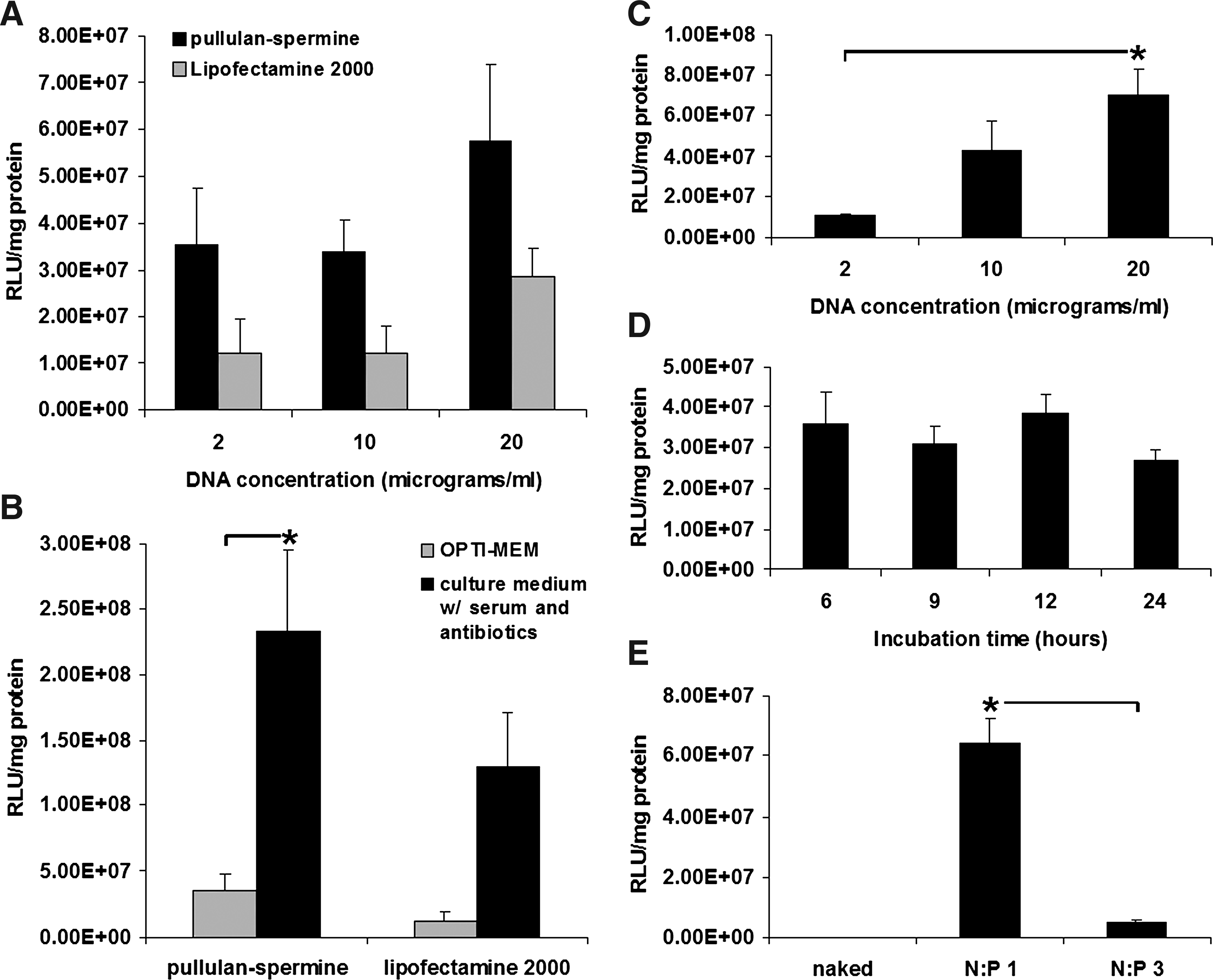
Transfection of rat bone marrow stromal cells (BMSCs) by pullulan-spermine/DNA anioplexes.
We previously observed that the negative charge of pullulan-spermine anioplexes allowed effective transfection in the presence of serum, which is normally detrimental to nonviral gene delivery due to sequestering interactions between positively charged vector complexes and anionic serum proteins. 8 In the present study, anioplex-derived transfection of BMSCs in the presence of serum resulted in significantly increased reporter gene activity (6.6-fold) as compared to serum-free transfection (Fig. 1B; p = 0.0064, Neuman-Keuls post-hoc after two-way ANOVA); a similar increase (11-fold) observed for Lipofectamine 2000 did not reach statistical significance (Fig. 1B; p = 0.058, Neuman-keuls post-hoc after two-way ANOVA). Lipofectamine 2000-derived luciferase activity was also 11-fold higher after transfection in 15% serum and antibiotics, although the difference did not reach statistical significance (p = 0.058). In the presence of serum, anioplexes also tended to show increased reporter gene activity in comparison to Lipofectamine 2000 (1.8-fold), although the difference was not statistically significant (Fig. 1B; p = 0.093, Neuman-Keuls post-hoc). Notably, the manufacturer's protocol for Lipofectamine indicates that transfection can be performed in the presence or absence of serum, but many groups use serum-free conditions13–16 ; our findings suggest that serum should be included for optimal Lipofectamine 2000 performance.
Our further analysis revealed that anioplex-derived transfection in the presence of serum was concentration-dependent (Fig. 1C; p = 0.030, one-way ANOVA), with significantly increased luciferase activity (6.3-fold) occurring at 20 μg/mL DNA as compared to 2 μg/mL DNA (Fig. 1C; p = 0.022, Neuman-Keuls post-hoc). However, reporter gene activity did not depend on anioplex incubation time (Fig. 1D; p = 0.49, one-way ANOVA); similar levels were observed for 6 and 24 h of incubation. In future studies, it may be interesting to test the effect of incubation time at higher anioplex concentrations.
To confirm the importance of the negative charge of the anioplexes for transfection of BMSCs in the presence of serum, we compared pullulan-spermine/DNA anioplexes (prepared in salt-free 20 mM HEPES at a pullulan-spermine N:P ratio of 1:1; these particles have a surface charge of −42 mV8) to positively charged pullulan-spermine/DNA polyplexes (prepared in 0.5 × PBS at an N:P ratio of 3:1; these particles have a surface charge of +16 mV 17 ). The negative charge of the anioplexes was most likely the critical factor for their effective transfection in serum, as positively charged particles formed from the same starting materials showed a significant reduction (92% decrease) of reporter gene activity (Fig. 1E; p = 0.0013, t-test). However, it remains possible that there could be some unknown effect of the larger size of the anioplexes (349.2 vs. 210 nm for N:P 3:1 polyplexes) or a conformational effect of the different N:P ratios or the use of salt-free 20 mM HEPES for anioplex condensation instead of the 0.5 × PBS used for N:P 3:1 polyplexes.
Uptake and intracellular trafficking of pullulan-spermine/DNA anioplexes in MSCs
Anioplex transfection was significantly inhibited (29% decrease) by pretreatment of MSCs with 1 mg/mL asialofetuin (p = 0.036, t-test), suggesting the utilization of glycoprotein interactions for endocytosis (Fig. 2A). Specifically, asialoglycoprotein receptor (ASGPR) is a likely target for receptor-mediated endocytosis, as the rat MSCs used by our group show strong ASGPR expression 18 and pullulan itself utilizes the ASGPR for internalization into liver parenchymal cells. 19 Transfection was not affected by 5 mM methyl-beta-cyclodextrin (p = 0.42, t-test), indicating that uptake did not utilize a cholesterol-dependent mechanism (i.e., lipid rafts or caveolae; Fig. 2B). However, anioplex transfection was significantly inhibited by 28 mM chlorpromazine (28% decrease; p = 0.030, t-test). This suggests a role played by clathrin-dependent endocytosis (Fig. 2C). This is logical if the ASGPR contributes to anioplex transfection, as ASGPR endocytosis occurs along two distinct clathrin-dependent pathways. 18 Transfection by gene carriers such as cationic lipids that primarily use diffusion for intracellular trafficking is typically enhanced by colchicine treatment because of the resulting cytoskeletal disruption and decreased cytoplasmic viscosity.20,21 However, anioplex transfection tended to be inhibited (37% decrease) by 65 mM colchicine, which destabilizes microtubules in the cytoskeleton, although this difference did not reach statistical significance (p = 0.069, t-test). This finding suggests that pullulan-spermine/DNA anioplexes at least partially utilize active transport along microtubules for gene delivery.
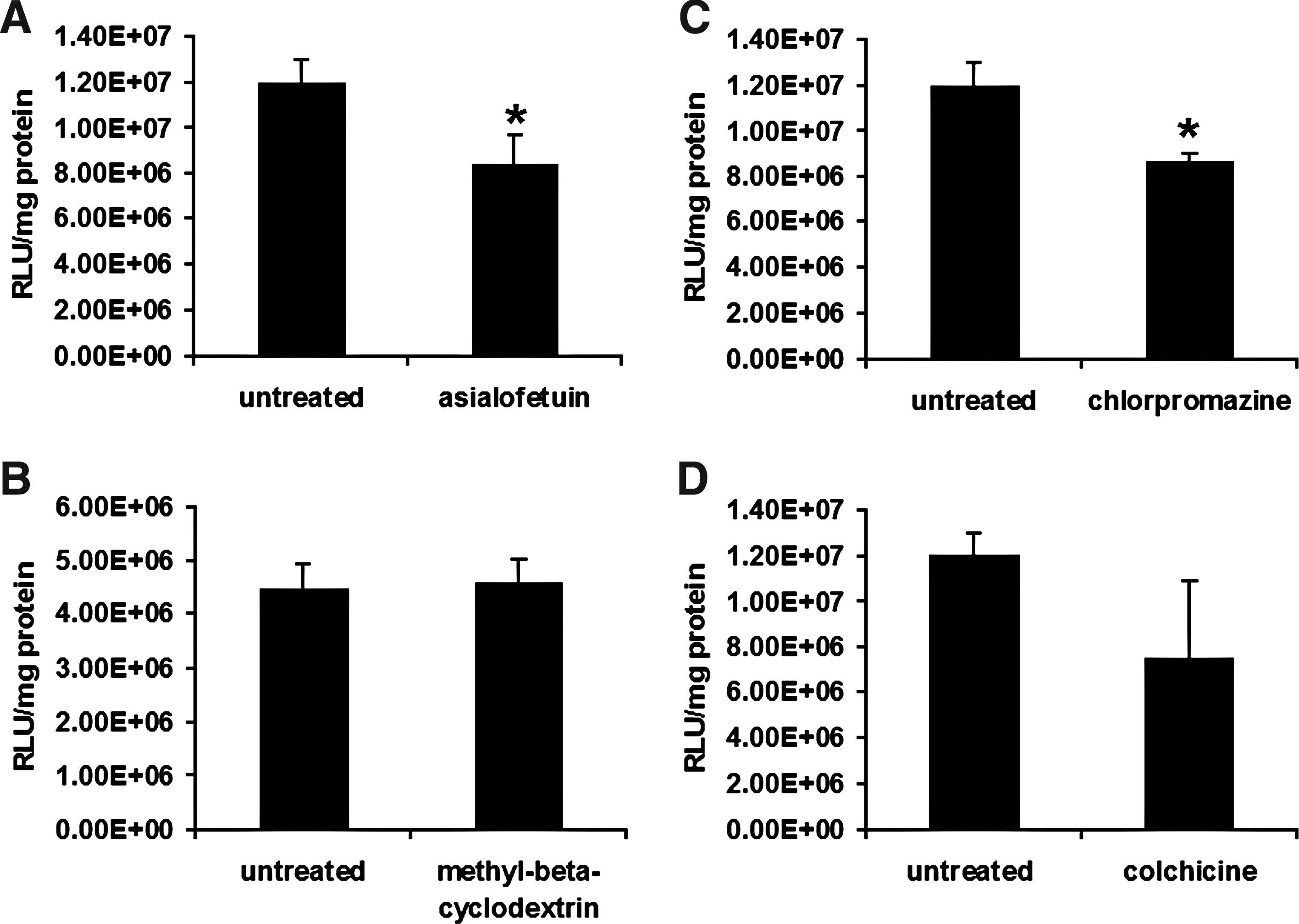
Uptake and intracellular trafficking of pullulan-spermine/DNA anioplexes in BMSCs.
Differentiation of rat and human MSCs after transfection by pullulan-spermine/DNA anioplexes
Rat BMSCs transfected with pullulan-spermine/DNA anioplexes and Lipofectamine 2000 still adhered to tissue culture plates. Upon exposure to osteogenic and adipogenic media for 4 weeks, they showed calcium deposition and formation of lipid vacuoles, respectively, which confirms their capacity for osteogenesis and adipogenesis (Fig. 3). Conversely, culture in the growth medium for 4 weeks after transfection did not generate such effects (Fig. 3), implying that the observed differentiations were indeed derived from the specific induction protocols and not from biological activity of the transfection agents.
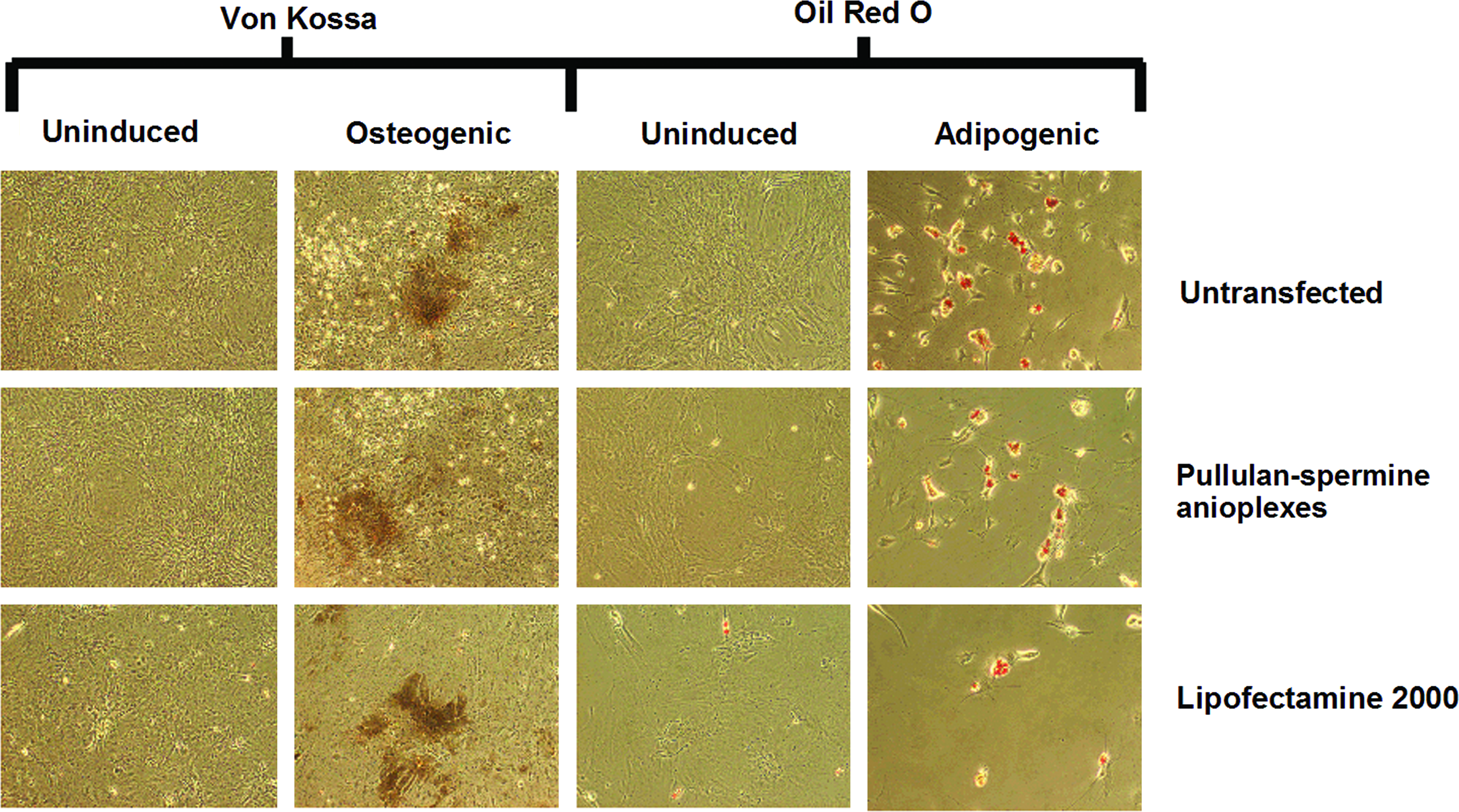
Osteogenic and adipogenic differentiation of BMSCs after transfection. Upon exposure to osteogenic and adipogenic media for 4 weeks, BMSCs transfected with anioplexes and Lipofectamine 2000 showed calcium deposition (Von Kossa staining) and formation of lipid vacuoles (oil red o staining), respectively, suggesting osteogenesis and adipogenesis. Culture in the growth medium for 4 weeks after transfection did not generate such effects, suggesting a lack of unintended differentiation. Color images available online at
We further subjected human BMSCs to chondrogenic differentiation. Within 24 h after exposure to the chondrogenic differentiation medium, nontransfected and anioplex-transfected human MSC micromass cultures showed rapid formation of chondrogenic pellets; chondrogenesis was confirmed after 3 weeks by Alcian Blue staining (Fig. 4A). However, for the first week after transfection, the morphologic appearance of Lipofectamine 2000-transfected cultures did not change. During the second week after transfection, Lipofectamine 2000-transfected cultures grew opaque, and they finally showed pellet formation after 2 weeks. This impaired pellet formation is reflected in the significantly larger size (i.e., reduced contraction) of the Lipofectamine 2000-transfected pellets after 3 weeks compared to nontransfected and anioplex-transfected pellets (Fig. 4B; p < 0.0001, one-way ANOVA; *p = 0.003 vs. nontransfected, *p = 0.0005 vs. pullulan-spermine 2 μg/mL, *p = 0.0013 vs. pullulan-spermine 10 μg/mL, Tukey-Kramer post-hoc). Notably, the size of anioplex-transfected pellets was not significantly different from that of nontransfected pellets, even when the high plasmid DNA concentration of 10 μg/mL was used (p > 0.05, Tukey-Kramer post-hoc).
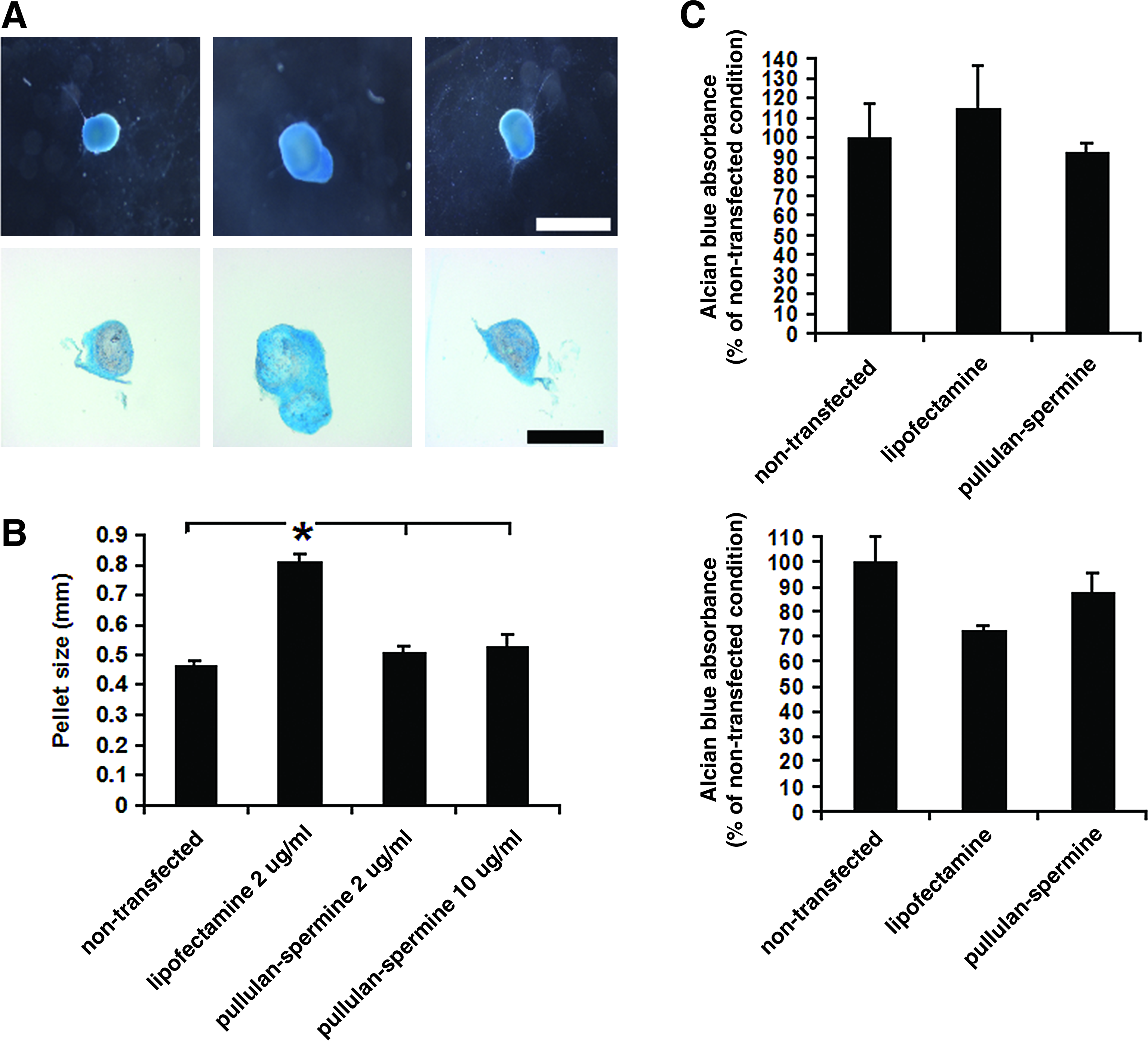
Chondrogenic differentiation of human BMSCs after transfection.
The secretion of chondral proteoglycans by chondrogenic pellets as determined by the absorbance of leached Alcian Blue dye was not significantly different among nontransfected pellets and those transfected with pullulan-spermine/DNA anioplexes or Lipofectamine 2000/DNA (Fig. 4C; p = 0.10, one-way ANOVA). Micromass cultures incubated in the growth medium did not form pellets, but after 3 weeks they still showed a limited extent of Alcian Blue staining; the absorbance of the leached Alcian Blue dye was not significantly different across transfection conditions (Fig. 4C; p = 0.94, one-way ANOVA).
Transfection of rat ASCs by pullulan-spermine/DNA anioplexes
Anioplex transfection of ASCs in the culture medium containing serum and antibiotics was both successful and donor gene concentration-dependent (Fig. 5A; p < 0.0001, one-way ANOVA). Pairwise comparisons revealed significantly increased luciferase expression at 20 and 10 μg/mL DNA compared to 2 μg/mL DNA (9.1- and 6.1-fold increases with p = 0.0008 and p = 0.0084, respectively, Neuman-Keuls post-hoc), increased expression at 20 and 10 μg/mL DNA as compared to 2.5 μg/well (5.7- and 3.8-fold increases with p = 0.0009 and p = 0.0067, respectively, Neuman-Keuls post-hoc), and increased expression at 20 μg/mL DNA as compared to 10 μg/mL DNA (1.5-fold increase with p = 0.044, Neuman-Keuls post-hoc). Similarly to findings obtained with MSCs, reporter gene expression did not depend on anioplex incubation time (Fig. 5B; p = 0.54, one-way ANOVA).
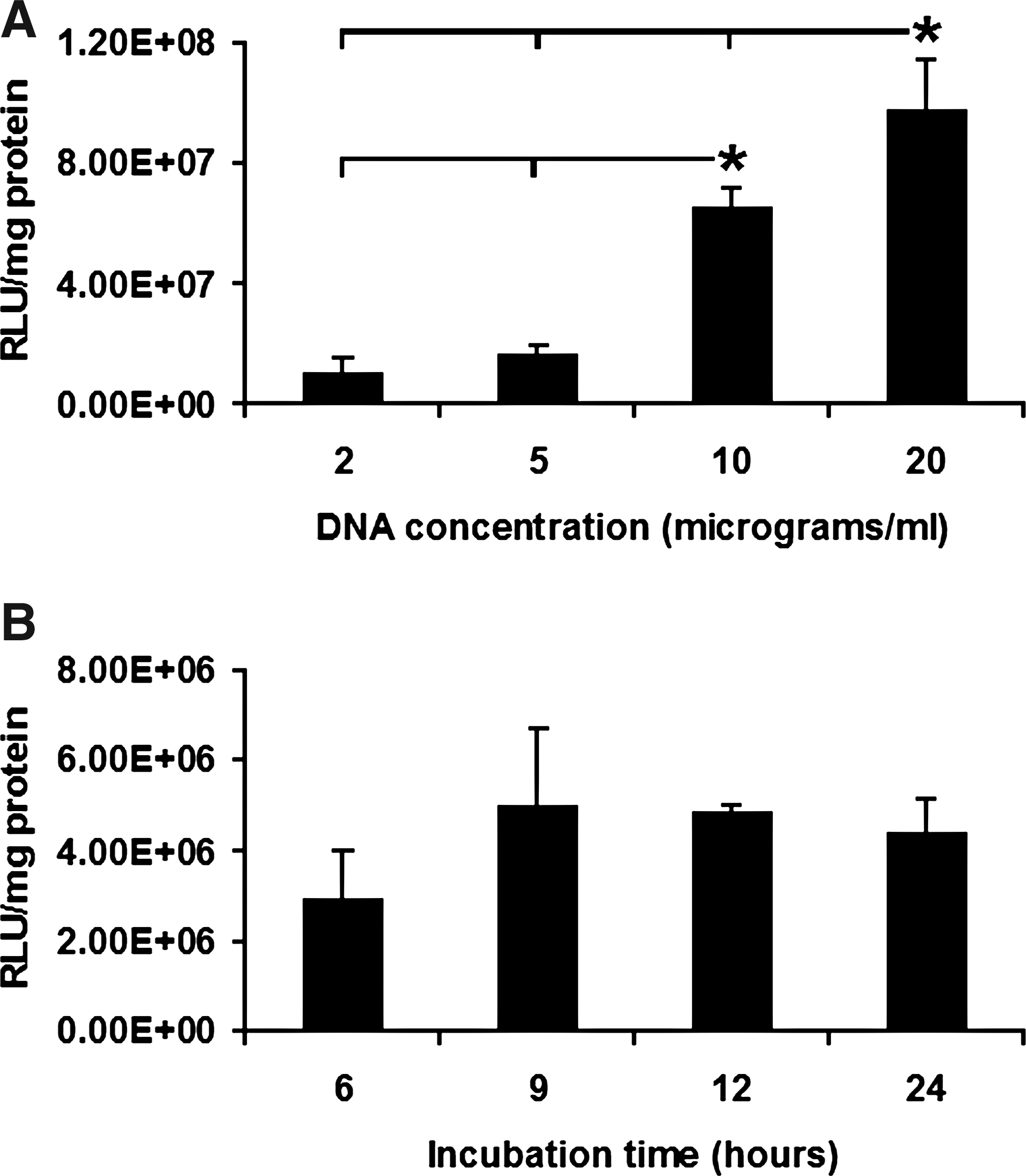
Transfection of rat adipose-derived stromal cells (ASCs) by pullulan-spermine/DNA anioplexes.
Minimal cytotoxicity in BMSCs and ASCs after transfection with pullulan-spermine/DNA anioplexes
Over the full range of concentrations (up to 20 μg/mL DNA) and incubation times (up to 24 h) tested, both BMSC and ASC viabilities were dramatically enhanced with anioplex transfection as compared to transfection with Lipofectamine 2000 (Tables 1 and 2). With anioplex transfection, no significant decreases in BMSC viability as compared to untransfected cells were observed at any concentration or incubation time. ASC viability was also not decreased at any incubation time, although a significant 20.2% decrease in viability was observed at 20 μg/mL DNA, the maximum concentration tested and 5–10-fold higher than concentrations conventionally used for transfection. In marked contrast, BMSC and ASC viabilities were significantly decreased by about 60% and 60%–90%, respectively, at all concentrations and incubation times tested for Lipofectamine 2000.
Percentage viability is presented with respect to untransfected cells. Displayed p-values are outcomes from two-way ANOVA analysis followed by Newman-Keuls post-hoc.
ANOVA, analysis of variance.
Percentage viability is presented with respect to untransfected cells. Displayed p-values are outcomes from two-way ANOVA analysis followed by Newman-Keuls post-hoc.
Substrate-mediated transfection
We recently developed a substrate-mediated transfection technique where pullulan-spermine/DNA could be electrostatically preadsorbed onto cell culture plates, allowing transfection to proceed directly from the solid culture support into subsequently seeded cells rather than using particles dispersed in the culture medium (Fig. 6A).8,22 In the present study, we applied this technique to anioplex transfection of ASCs.
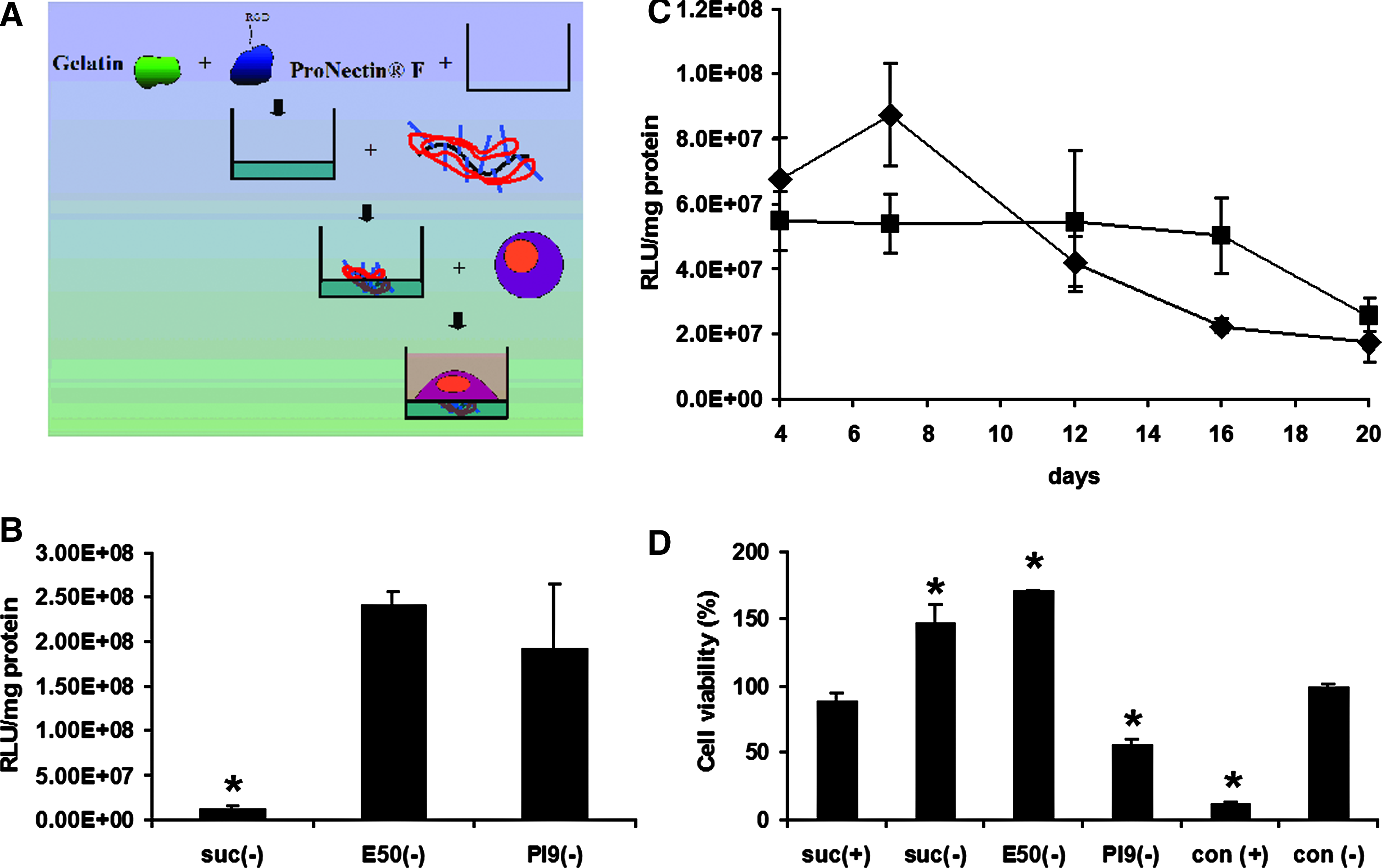
Substrate-mediated transfection.
Pronectin, a commercially available cell adhesion molecule, was mixed with gelatin that had been conjugated with ethylene diamine (E50 gelatin), processed to have an innate isoelectric point of pH 9 (PI9 gelatin), or succinylated to obtain surface coatings with a strong or weak positive charge, or a negative charge, respectively. Pullulan-spermine/DNA anioplexes were then plated onto the surface coatings before seeding of ASCs. Substrate-mediated transfection (subfection) gave rise to significantly more luciferase reporter gene activity when anioplexes were paired with E50 or PI9 gelatin than with succinylated gelatin (Fig. 5B, 18.5- and 14.8-fold increases, respectively; p = 0.020 and p = 0.022, Newman-Keuls post-hoc), presumably due to a lack of electrostatic binding of anioplexes to the negatively charged substrate. However, there was no significant difference between E50 and PI9 groups (Fig. 6B; p = 0.45, Newman-Keuls post-hoc).
There were no significant differences in reporter gene activity between PI9-based subfection and conventional transfection techniques, either overall (Fig. 6C; p = 0.37, two-way ANOVA) or at any individual time point out to 20 days (day 4, p = 0.788; day 7, p = 0.753; day 12, p = 0.877; day 16, p = 0.719; day 20, p = 0.840, Bonferroni post-hoc). There was an overall significant effect of post-transfection time point on reporter gene activity (Fig. 6C; p = 0.049, two-way ANOVA), with a significant decrease at day 20 as compared to day 7 (Fig. 6C; p = 0.005, Bonferroni post-hoc). However, when the subfection and conventional transfection techniques were considered individually, there were no significant differences between time points with the highest activity (Fig. 6C; 4 days for conventional transfection, 7 days for subfection) and those with the lowest activity (Fig. 6C; final 20 day time point for both transfection techniques; p = 0.522 for subfection, p = 0.672 for conventional transfection, Bonferroni post-hoc). Thus, although there was some attenuation over time, anioplex-derived reporter gene activity was still notable at least 20 days after transfection when using either the subfection or conventional technique.
On the basis of our previous report of reduced toxicity for subfection with positively charged pullulan-spermine, 8 we further evaluated cell viability at 24 h after subfection. Conventional anioplex transfection for 6 h in the presence of serum was used as the control, as this condition showed a lack of detectable toxicity when compared to untransfected cells (Table 2). Viability was not significantly changed (Fig. 6C; 10.6% reduction; p = 0.35, Newman-Keuls post-hoc) after subfection with succinylated gelatin and positively charged pullulan-spermine/DNA, but was greatly decreased upon conventional transfection with positively charged pullulan-spermine/DNA (Fig. 6D; 87.8% reduction; p < 0.0001, Newman-Keuls post-hoc); this is in line with our previous findings and further suggests that the negative charge of pullulan-spermine/DNA anioplexes plays an important role in their biocompatibility. 22 For anioplex subfection, substrates containing succinylated gelatin and E50 gelatin generated increased mitochondrial activity suggestive of proliferation (Fig. 6D; 1.5- and 1.7-fold increases, respectively, p = 0.0003 for both; Newman-Keuls post-hoc), whereas viability with the PI9-containing substrate was reduced by 43.9% (Fig. 6D; p = 0.0041; Newman-Keuls post-hoc). Given the apparent lack of difference between conventional and substrate-mediated transfection in terms of reporter gene activity and time course, we continued to use the conventional technique for subsequent ex vivo BMSC and ASC transfection.
Spinal cord implantation of anioplex-transfected BMSCs
We developed a protocol for minimally invasive spinal cord BMSC implantation via acute lumbar puncture-mediated intrathecal injection. At 4 days after injection with 1.2 × 106 BMSCs that had been transfected ex vivo with pullulan-spermine/DNA anioplexes carrying ~40 μg of DNA, luciferase reporter gene activity was detected in spinal cord tissue both in the vicinity of and rostral to the injection site (L3–L6 and L1–L3, respectively). Luciferase activity was 4.2-fold higher near the injection site, although this difference did not reach statistical significance (Fig. 7A; p = 0.14, t-test). Additionally, expression was not detected in L4–L5 bilateral DRGs.
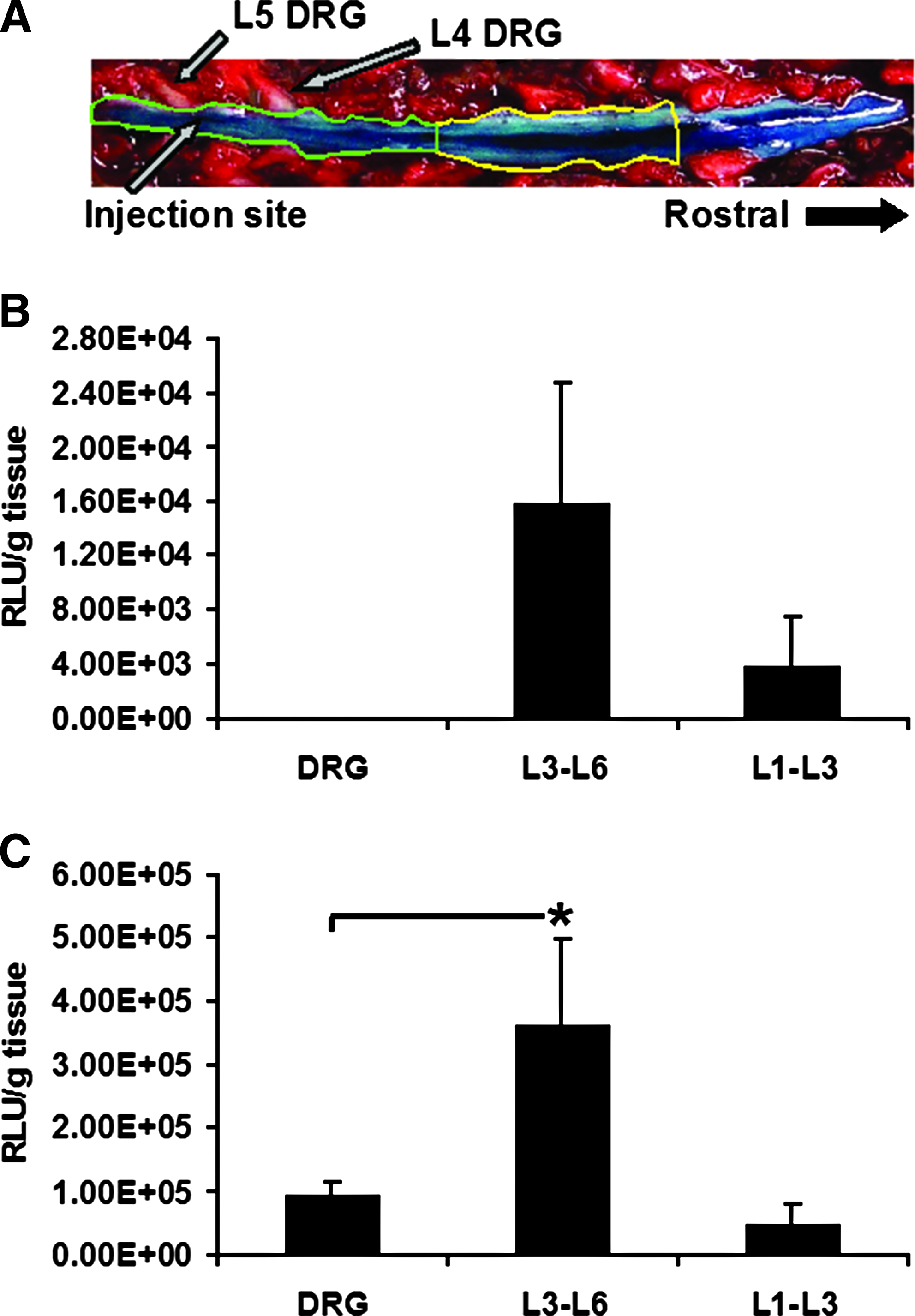
Spinal cord implantation of anioplex-transfected BMSCs.
For comparison, we also performed acute lumbar puncture injection of 100 μg naked plasmid DNA; this technique and dose have been previously applied for therapeutic direct overexpression of interleukin 10 in a neuropathic pain model. 23 In this case, luciferase activity at 4 days after injection was 7.2-fold greater near the injection site as compared to the more rostral region; however, the difference again did not reach statistical significance (Fig. 7B; p = 0.057, Newman-Keuls post-hoc). In contrast to results obtained with injected BMSCs, luciferase activity was observed in L4–L5 DRGs at a fairly similar level (1.83-fold increase) to that observed in the spinal cord rostral to the injection site (p = 0.72, Newman-Keuls post-hoc). Luciferase activity near the spinal cord injection site (L3–L6) was significantly greater than that observed in the DRGs (3.9-fold, p = 0.044, Newman-Keuls post-hoc). Thus, reporter gene activity derived from BMSCs appeared to be more localized than that from naked plasmid DNA. These findings suggest that transplantation of anioplex-transfected cells could provide a favorable alternative for gene therapy of the spinal cord.
Discussion
In the present study, we found that pullulan-spermine/DNA anioplex-derived reporter gene activity in MSCs was comparable to, if not greater than, that of Lipofectamine 2000, a gold standard previously found to have MSC transfection rates of ~5%–19.6%.13,14,24,25 Additionally, post-transfection reporter gene activity was observed throughout the entire 3-week study duration and anioplex-mediated transfection did not quantitatively affect MSC differentiation characteristics, in contrast to transfection by Lipofectamine 2000. We therefore conclude that the anioplex system may have substantial utility for nonviral gene delivery to stem and/or progenitor cell populations.
Previously, viral, especially retro/lentiviral, vectors were used to achieve high rates of sustained expression in MSCs, but safety and manufacturability concerns still exist. Cationic nonviral vectors were originally developed to allay such concerns6,7; however, they typically show reduced efficacy6,7 and increased cytotoxicity26,27 that may arise from cell membrane destabilization, formation of reactive oxygen species, and triggering of apoptosis via mitochondrial cell death pathways.26–28 Additionally, cationic polyplex transfection (i.e., that by polyethylenimine or polylysine) is hindered by serum incompatibility due to sequestering interactions with negatively charged proteins such as albumin.29,30 These factors could have important negative consequences for pretransplantation ex vivo transfection of donor stem/progenitor cells, which are known to be sensitive to culture conditions (i.e., serum/growth factor content) and whose low-passage yield after primary isolation might be too low to afford a high rate of cell death.
We therefore hypothesized that our negatively charged anioplex vector could be used to overcome drawbacks of polyplex-mediated transfection linked to positive surface charge. Indeed, anioplex-derived reporter gene activity was nearly sevenfold higher when transfection of MSCs proceeded in the presence of 15% serum and antibiotics. Additionally, anioplex transfection showed a lack of detectable cytotoxicity in both bone marrow- and adipose-derived MSC types, in marked contrast to Lipofectamine 2000. Although we previously showed ~30% viability for Lipofectamine 2000 in BMSCs (in line with the present study), 22 other groups reported 80%–90% BMSC and ASC viability after Lipofectamine 2000 transfection,14,16,24,31 and ~80% ASC viability after polyethylenimine transfection 16 ; these previous studies used lower lipoplex concentrations14,16 or incubation times,16,24 or a different viability measure (measurement of the proportion of cells with intact membranes). 14 Even taking methodological differences into account, the 101%–103% BMSC and 99%–107% ASC viability we observed is, to our knowledge, still higher than that reported for any previously described nonviral vector.
The biological inertness of anioplexes is further supported by phenotype analysis, which revealed that transfected cells adhered to tissue culture-treated plastic surfaces and differentiated along mesenchymal lineages (i.e., osteogenic, adipogenic, and chondrogenic) when exposed to appropriate differentiation media, thus meeting the functional definition of MSCs.32,33 Additionally, anioplex-transfected cells did not show adipogenic or osteogenic differentiation when maintained in the growth medium; this might be important, given the potential for phenotypic alteration as evidenced by spontaneous MSC adipogenesis over long culture periods. 34
Quantitative analysis revealed impaired contraction of chondrogenic pellets after transfection with Lipofectamine 2000. In contrast, no significant differences were observed between anioplex transfection and the nontransfected condition, even at a high DNA concentration. As such cellular aggregation or condensation is thought to be critical for chondrogenesis, 35 our findings highlight the particular importance of selecting nontoxic/biologically inert gene delivery systems for the engineering of physiological tissues from stem cells. Our findings also indicate the importance of quantitative analyses of stem properties in transfected/transduced cells; most studies to date only employed qualitative analyses of differentiation that could determine whether the transfected cells had differentiated but not whether the differentiation was actually different from that of naïve cells. Such a qualitative analysis would have missed the impaired contraction of chondrogenic pellets observed here for Lipofectamine 2000, which might have physiological relevance.
In addition to serum compatibility, lack of toxicity, and lack of effects on differentiation, we speculated that the negative charge of anioplexes might permit prolific transgene expression, as luciferase reporter activity was minimally attenuated even at 20 days. In contrast, luciferase activity decreased within 4 and 6–8 days, respectively, after conventional and substrate-mediated MSC transfection by positively charged pullulan-spermine/DNA. 22 Similarly, nearly complete attenuation (99.86%) of reporter enhanced green fluorescent protein (EGFP) mRNA expression was observed 4 days after transfection with Lipofectamine 2000. 14 Another study found that reporter EGFP-expressing cells decreased within 12 days after transfection with Lipofectamine 2000 or poly-L-lysine, 25 or 7 days after transfection with palmitic acid-modified poly-L-lysine. 13 Because the cytoplasmic half-life of luciferase is only ~3 h, 36 the longevity of luciferase activity in the present study likely reflects continued mRNA expression. Although we speculated that a relationship exists between negative charge and sustained transgene expression, differences in promoter37,38 or plasmid preparation protocol may also play a role. Thus, future studies should systematically investigate the permissive factor for long-term anioplex-derived transgene expression, as highly transient expression is a characteristic problem for nonviral gene delivery, and stable expression could be important for chronic applications such as nerve regeneration or long-term tracking of implanted cells.
The development of effective gene transfer strategies is particularly important for MSCs because of their potential clinical role as a substrate for tissue engineering and regenerative medicine. Compared to other types of stem/progenitor cells, MSCs can be isolated with low donor-site morbidity from multiple tissues and easily expanded in vitro, thus opening the door to autologous implantation. Indeed, human clinical trials have already commenced for a variety of diseases, including spinal cord injury.39–41 Gene transfer to MSCs could augment already-existing strategies via induction of differentiation for cell replacement,42,43 or labeling for noninvasive determination of in vivo localization and fate. However, the growing realization that MSCs and other types of stem/progenitor cells innately exert therapeutic effects via mechanisms other than cell replacement44,45 introduces a powerful new application: modulation of the secretome and interaction profiles of the MSCs for enhanced interaction with the host microenvironment.
In rat models of spinal cord injury and inflammatory pain, nerve growth factor (NGF)-, basic fibroblast growth factor-, brain-derived neurotrophic factor (BDNF)- and neurotrophin (NT)-3-overexpressing fibroblasts,46,47 and met-enkephalin-overexpressing adrenal chromaffin cells, 48 respectively, showed definitive therapeutic effects after intraspinal transplantation. Additionally, overexpression of the TrkC neurotrophin-3 receptor in neural stem cells resulted in enhanced survival and migration in the rat spinal cord. 49 As cellular substrates for secretome modification by gene delivery, MSCs offer the combined advantage of potentially autologous transplantation with intrinsic neurotrophic and neuroprotective effects 50 that may arise from synergistic coactivity with host regenerative cells (e.g., neural stem cells).50–54 We envision that anioplex-derived genetic modification might realistically be used to more subtly shape and augment these modulatory properties of MSCs, in addition to simply overexpressing therapeutic molecules.
In this study, we also established a prototype regimen for minimally invasive implantation of anioplex-transfected MSCs in the lumbar spinal cord and tracking of donor cell distribution using luciferase activity. Our preliminary findings suggest that future studies should use similar tracking techniques to systematically determine MSC graft stability and fate. Previous reports showed that retroviral overexpression of BDNF 55 and insulin-like growth factor 56 in MSCs before transplantation resulted in enhanced axonogenesis and neuroprotection, respectively, in rat models of spinal cord injury. Additionally, plasmid-derived overexpression of interleukin 10 after intrathecal injection led to reversal of neuropathic pain symptoms in rodents. 23 On the basis of these encouraging outcomes, our future studies will specifically focus on the minimally invasive injection of anioplex-transfected MSCs for ameliorating pathologies of spinal cord injury and neuropathic pain. We also envision the application of the substrate-mediated anioplex transfection technique described here for long-term gene delivery to MSCs incorporated into chemically engineered polymer scaffolds, which could then be implanted into the target region of the host. 45
Our results demonstrate that pullulan-spermine/DNA anioplexes allow efficient, biologically inert, and prolific gene delivery to either bone marrow- or adipose-derived MSCs, potentially under more physiological niche-like conditions optimal for their culture. Future application of this technology could likely enable direct gene transfer to MSCs from biomaterial scaffolds as well as ex vivo MSC labeling before minimally invasive implantation into the central nervous system. Together, these outcomes establish pullulan-spermine/DNA anioplexes as an effective platform for genetic engineering of stem/progenitor cells for both investigative and therapeutic applications.
Footnotes
Acknowledgments
D.K.T. has been supported by a foreign postdoctoral fellowship and grant-in-aid from the Japan Society for the Promotion of Science, as well as the Takanawakai, the Clinical Implant Society of Japan, and a VA Biomedical Merit Grant. Work at Y.D.T.'s labs are supported by VA, NIH, CIMIT-DOD, and the State of Massachusetts. We thank Dr. Jun-Ichiro Jo, Mr. Masataka Yoshida, and Mr. Norio Doi of Kyoto University for assistance in the preparation of pullulan-spermine. We thank Dr. Jyunichi Miyazaki of Osaka University for kindly providing luciferase. We thank Dr. Masaru Tobe of Gunma University for assistance with animal experiments. We thank Mr. Nirav Thakor for assistance with figure preparation.
Disclosure Statement
No competing financial interests exist.