
Abstract
We have previously developed a cell delivery and transfer technology for delivering autologous keratinocytes and melanocytes to patients with vitiligo. However, for this technology to benefit many patients geographically distant from the cell culture facility transportation issues need to be overcome. In this study we begin to investigate this by looking at what role surface chemistry and medium supplements, including fetal calf serum, CO2 gassing, and temperature, play in influencing cell viability. Cells were maintained on carriers for up to 48 h outside of a CO2 incubator at 37°C and their subsequent ability to adhere and become organized into a new epithelium with appropriately located melanocytes was assessed. Consistently good viability and performance on an in vitro wound bed model was achieved by maintaining cells for 48 h adherent to a 20% acrylic acid coated carrier at lower (around 23°C rather than 37°C) temperatures in the medium preperfused with CO2 before transport. Under these circumstances fetal calf serum was not required. In summary, the surface chemistry of the transport substrate and an appropriately CO2 buffered medium at near room temperature can extend the effective performance life of these cultured cells to at least 48 h from when they leave standard incubator conditions.
Introduction
The aim of this study was to examine the next key issue of how to transport viable cells to patients across the whole of the United Kingdom and possibly to Europe. Currently, a prominent barrier to the widespread distribution of tissue-engineered medical products is the lack of published transport technologies capable of maintaining high cell viability and integrity during several days of transport until the point of use. Due to the difficulty in successfully transporting cultured human cells, patients with vitiligo have historically traveled to specialized clinics that culture autologous skin cells to receive effective treatment. This can be inconvenient, time consuming, and expensive for the patient. Essentially for lack of good transport protocols the technology has not advanced to become something that could benefit many patients routinely. This problem is not unique to culturing cells for vitiligo patients. There is an unmet need in the tissue engineering market to establish and publish conditions for transporting cells for significant distances and over several days. This will include developing packaging that enables cells to withstand mechanical trauma and often extreme and varying temperature conditions. As yet there is little published on the basic influence of temperature and media on cell survival and subsequent performance on arrival.
Transportation of cells requires them to leave the enclosed, highly regulated, and optimized conditions of the culture laboratory. Cells are normally cultured in a constantly regulated environment (37°C and with 5% CO2 and 95% O2) with bicarbonate buffering to maintain pH at around 7.4. These are the gold-standard conditions used by tissue engineers worldwide. Once removed from this environment, cultured cell products can very quickly lose viability. Therefore, optimizing the transport conditions for cells can dramatically increase the time they remain viable during transportation.
This laboratory in conjunction with CellTran Ltd. has several years' experience of developing a tissue-engineered product consisting of autologous keratinocytes on a carrier surface, MySkin™, which has been used clinically for treating severely burned patients and also patients with nonhealing chronic foot ulcers.5–7 The technology involves using a plasma-polymerized silicone surface on which autologous human keratinocytes can be cultured and then transferred to a wound bed. Although this technology has been used clinically with considerable success, cells have until recently been delivered to patients mainly within the same city and occasionally to cities within a 50-mile radius. Under these circumstances, Myskin™ was either transported within conventional tissue culture Petri dishes containing liquid Green's medium (minus cholera toxin and serum) or placed cell side down on the solidified agar transport medium (based on the above Green's medium). Transport of Myskin™ under these conditions has proved adequate for a 24-h transport period (unpublished data).
To date, there is no one definitive published method for worldwide transport of cell and tissue therapies while maintaining viability for long periods. This is surprising taking into account the potential revenues of a growing tissue engineering market. For a tissue-engineered product to be able to be distributed locally, the product should be able to survive a minimum of 24 h in transport conditions. If that time is increased to 48 h, it could achieve intercontinental delivery and if increased further to 72 h, it could potentially have worldwide delivery.
In this study we investigated what role cell substrate, medium supplements (fetal calf serum [FCS]), CO2 gassing of the medium, and temperature play on cell viability during and post-transport (in this study cells were not transported outside of the laboratory but simply maintained in the laboratory outside of the incubator) and also cell function post-transport. The latter is of practical importance as delivery of not only viable but functionally active cells from a specialist cell culture center would enable cell-based treatments to benefit many patients.
Materials and Methods
Ethical approval
Informed written consent was obtained from all patients enabling the use of tissue that was surplus to requirement to be used for research. The study was approved by the Regional Committee for Research Ethics and by the University of Sheffield Research Office.
Cell culture
Human keratinocytes
Human skin keratinocytes were isolated as detailed previously. 4 Briefly, skin samples obtained during routine plastic surgery breast reductions and abdominoplasties were cut into 0.5 × 2 cm pieces using a scalpel blade and incubated overnight (18 h) at 4°C in 10 mL 0.1% w/v trypsin (BD Biosciences). FCS (GlobePharm Ltd.) was added to neutralize the trypsin, the epidermal and dermal layers were carefully separated, and a scalpel blade was used to gently scrape keratinocytes from the epidermis and the papillary surface of the dermis. The cells were collected and centrifuged at 200 g for 5 min. Cells were resuspended and seeded in either Greens medium 4 at a cell density of 1–2 × 106 cells (along with 1 × 106 irradiated mouse 3T3s) or at 2–4 × 106 cells in the melanocyte culture medium (M2- Promocell GmbH).
Human melanocytes
Melanocytes were isolated in exactly the same manner as for human keratinocytes (described previously). Cell suspensions for melanocyte isolation were collected, centrifuged, and seeded at 1 × 105 cells per flask in either MCDB-153 or M2 medium. After several passages in both media, pure cultures of melanocytes were achieved. 4 Melanocyte cultures were fed twice weekly and were used between passages 2 and 6.
Preparation of DED
Split-thickness skin grafts suitable for use as dermal substrates were stored at 4°C in PBS containing 100 IU/mL penicillin, 100 μg/mL streptomycin, and 0.625 μg/mL amphotericin B for a minimum of 7 days. The split-thickness skin grafts were incubated in sterile 1 M sodium chloride for 18 h at 37°C. After 18 h, the epidermis was peeled away from the dermis and the dermis was washed repeatedly with PBS. The dermal substrates (DED) were then stored at 4°C in Greens medium.
Production of plasma-polymerized surfaces
A plasma-polymerized surface was prepared from gaseous acrylic acid (Sigma-Aldrich), which was deposited onto the silicone carrier. The apparatus for plasma polymerization comprised a 50-cm-long glass reactor with stainless steel flanges and an external copper coil electrode. 8 The reactor was evacuated with a rotary pump with liquid nitrogen cold trap and the base pressure was ∼10−3 mbar. A flow rate of 10 cm3STP min−1 and 1 W of plasma power was used for the 20% carboxylic acid surfaces. Depositions were carried out over 10 min.
Production of agar medium gels
Agar medium gels were made using a sterilized 10% agar (Sigma-Aldrich) solution (5g agar in 50 mL PBS), which was liquefied and quickly added (1 part to 9 parts) to heated Dulbecco's minimal essential medium (DMEM)/Hams F12 (1:1) medium containing 0.625 μg/mL amphotericin B, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 30 mM sodium hydrogen carbonate (NaHCO3). This resulted in a final 1% agar medium solution. About 13.5–15 mL of this heated agar medium was then quickly added to Petri dishes containing 0%, 1% (0.15 mL), 5% (0.75 mL), or 10% (1.5 mL) FCS. The FCS and 1% agar medium solution was quickly mixed, lids were replaced on the Petri dishes, and they were cooled for 10–20 min on a level surface to allow solidification. Excess Petri dishes not required for immediate use were stored at 4°C until needed.
Seeding cells onto carrier dressings
For experimental studies, individual cell cultures were combined to achieve a 2:1 coculture of keratinocytes (6 × 104 cells)–melanocytes (3 × 104 cells). Cells were seeded onto the plasma-polymerized carriers inside 0.8-cm-diameter stainless steel rings and incubated for 24 h at 37°C.
Transport of cell carriers on agar medium gels
After 24 h incubation, carriers were placed cell side down on transport medium agar plates (warmed to 37°C) ± FCS and placed under the after maintenance conditions for 24 or 48 h. For maintenance at 37°C, samples were placed back in the incubator at 37°C containing a constant 5% CO2 gassed environment. For transport at 23°C, samples were placed in a sealed polystyrene box out of direct sunlight on a laboratory work surface for 24 or 48 h. For transport at 23°C+CO2, samples were placed in a sealed plastic container with tubes for CO2 perfusion on either side. After gassing of the box for 30 s with 5% CO2, the tubes were closed off, thus retaining a gassed environment within the sealed box. This box was then placed in the larger polystyrene box alongside the other 23°C samples.
After maintenance conditions for 24 or 48 h, a third of the samples were removed from the agar plates and were subjected to 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)–eluted stain assay (MTT-ESTA; Sigma Aldrich) to assess how viable the cocultures were post-transport. Briefly, each carrier was removed from the agar gel, placed in a 12-well plate and 1 mL/carrier of MTT (0.5 mg/mL in PBS) was added. To assess if any cells had transferred to the agar transport gel, 6 mL MTT was also added to the agar plates. All samples were incubated for 40 min at 37°C. During this incubation time, dehydrogenase activity in the cells converts MTT to a colored formazan product. After 40 min, it was evident that no cells had transferred from the carriers onto the agar transport gel. All cells remained on the carrier dressings in the 12-well plates. The MTT from these samples was removed and 500 μL ethylene glycol monoethyl ether (Sigma Aldrich) was added to elute any colored formazan product formed within the cells. Once eluted 100 μL of the formazan product (triplicate samples) from each well was added to a 96-well plate and the plate was read at 540 nm with a reference filter of 630 nm. Control values for ethylene glycol monoethyl ether alone were subtracted from all readings and mean optical densities were calculated.
Transfer of cells from carrier dressings onto human DED and formation of skin composites
The rest of the samples (two-thirds), after the previously described maintenance conditions, were transferred cells downward onto human DED for a further 48 h. Small stainless steel grids were placed on top of the carriers to hold them in place. The DED plus carriers were then flooded with the medium. After 24 h at 37°C, the grids were removed and the samples were incubated for a further 24 h. At this point a third of the carriers and dermis were separated and the number of viable cells transferred from the carriers to the DED was assessed using MTT-ESTA analysis. The remaining third of carriers were also removed from the DED, but instead of being sacrificed at this stage, these DED samples plus transferred cells were placed on stainless steel grids in new six-well plates. The medium was added to each well so that it lapped over the edges of the grid and dermis but left the transferred cells exposed to air (air–liquid interface). These skin composites were then cultured for a further 10 days with medium changes every 3–4 days.
At the end of this culture period, cell proliferation was assessed using a nontoxic, nondestructive stain, AlamarBlue® (AbD Serotec). When added to cells, the oxidized form of AlamarBlue is converted to the reduced form by mitochondrial enzyme activity. This redox change is accompanied by a shift in color from indigo blue (no cell proliferation) to fluorescent pink (proliferative cultures). Briefly, 1.5 mL of AlamarBlue (diluted 1:10 in PBS) was added to each skin composite and incubated for 2 h at 37°C. After 2 h, the AlamarBlue was removed, composites were photographed, and then fixed in 10% buffered formalin (Genta Medical) for immunohistochemical analysis.
Immunohistochemical analysis
Paraffin wax sections (4 μm) were dewaxed, rehydrated, and washed and antigenic sites were unmasked using heat-induced epitope retrieval. In more detail, sections were microwaved on high power for 12 min in Tris/EDTA (pH 9) buffer (Sigma-Aldrich). Sections were blocked with 1:25 swine serum (Harlan Sera-Lab Ltd.) for 10 min, incubated with primary rabbit derived S100 antibody (1:2500; Dako) for 60 min, washed with Tris-buffered saline/Tween (BDH; Merk Ltd.), and incubated with secondary antibody (biotinylated anti-rabbit antibody; Vector Laboratories) for 30 min. Antibodies were observed with an alkaline phosphatase Avidin Biotin Complex kit and alkaline phosphatase substrate kit (Vector Laboratories). After S100 immunostaining, melanocyte numbers within each section were counted.
Statistical analysis
Statistical significance was determined using the two-tailed, unpaired Student's t-test and analysis of variance (multiple comparisons of the means).
Results
The functional biology of keratinocyte:melanocyte cocultures was investigated after maintenance of the cells on a surface engineered carrier for 24 and 48 h at room temperature. Three different media and the contribution of FCS were investigated.
The choice of medium and inclusion of FCS
Two clinically relevant media, Greens and M2,2,4 were chosen, as well as MCDB-153 medium. (Although MCDB-153 was included in this study as it represents the gold-standard medium for culturing melanocytes, it is not being considered for taking into the clinic because of the inclusion of bovine pituitary extract, tumor promoters, and cholera toxin.) Previously, autologous keratinocyte cultures used to treat burns and chronic ulcer patients6,7 were transported on an agar-based version of Greens medium (minus cholera toxin and serum). In this study we used a similar transport medium, but explored the contribution of FCS to the transport agar gels.
Effect of medium and FCS on viability of cells cultured on carrier for 24 h
Maintenance of cocultures of keratinocytes with melanocytes expanded in M2 and MCDB-153 medium were compared on both untreated silicone carriers and 20% acrylic acid-coated carriers. After 24 h culture (37°C) with cells in contact with the agar transport medium containing either 0%, 1%, 5%, or 10% FCS, the most striking result was that cell viability on the 20% acid coated carrier was significantly better under all transport conditions compared with the uncoated carrier (Fig. 1). This was apparent for both melanocytes expanded in M2 (Fig. 1a) and those expanded in MCDB-153 (Fig. 1b) based on nonpaired Students t-test.

Effect of 0%, 1%, 5%, and 10% serum (added to the agar medium gel) on the transport of keratinocyte:melanocyte (2:1 ratio) cocultures at 37°C+CO2 for 24 h. Cell viability was assessed using MTT staining.
More extensive between groups analysis was undertaken by analysis of variance, which showed considerable variance between groups. Subsequent comparisons between groups (t-test) are summarized in Table 1. Essentially for cells grown on the uncoated carrier, the addition of FCS had no or negligible effect on improving cell viability. For cells grown on 20% acrylic acid in M2 medium the addition of 1.5% and 10% FCS improved cell viability. For cells in MCDB, 1% and 5% FCS improved cell viability, but 10% FCS did not. For this reason all future experiments were carried out with cells on coated carriers comparing results obtained with 0% and 5% FCS.
Multiple groups were compared by ANOVA. F-values indicated significant variance between groups, and individual differences were then analyzed by T-tests.
p < 0.05.
p < 0.001.
p < 0.01.
AC, acrylic acid; ANOVA, analysis of variance; FCS, fetal calf serum; M2, melanocyte medium; NS, not significant.
Effect of serum, temperature, and CO2 on cell viability post-transport
Keratinocyte/melanocyte cocultures were placed on 0% and 5% FCS agar medium and left for 24 or 48 h under different transport conditions. These consisted of keeping cells in a CO2 incubator at 37°C (used as our reference throughout) or maintaining cells on carriers at 23°C (room temperature in a polystyrene box) or at 23°C+5% CO2 (room temperature in a sealed gassed box within a polystyrene box). MTT-ESTA was then used to assess how metabolically active the cocultures were after the above transport conditions (Table 2). These results are summarized in Tables 2 and 3 for both 24 and 48 h transport conditions, but only the 48 h data are illustrated in Figure 2.
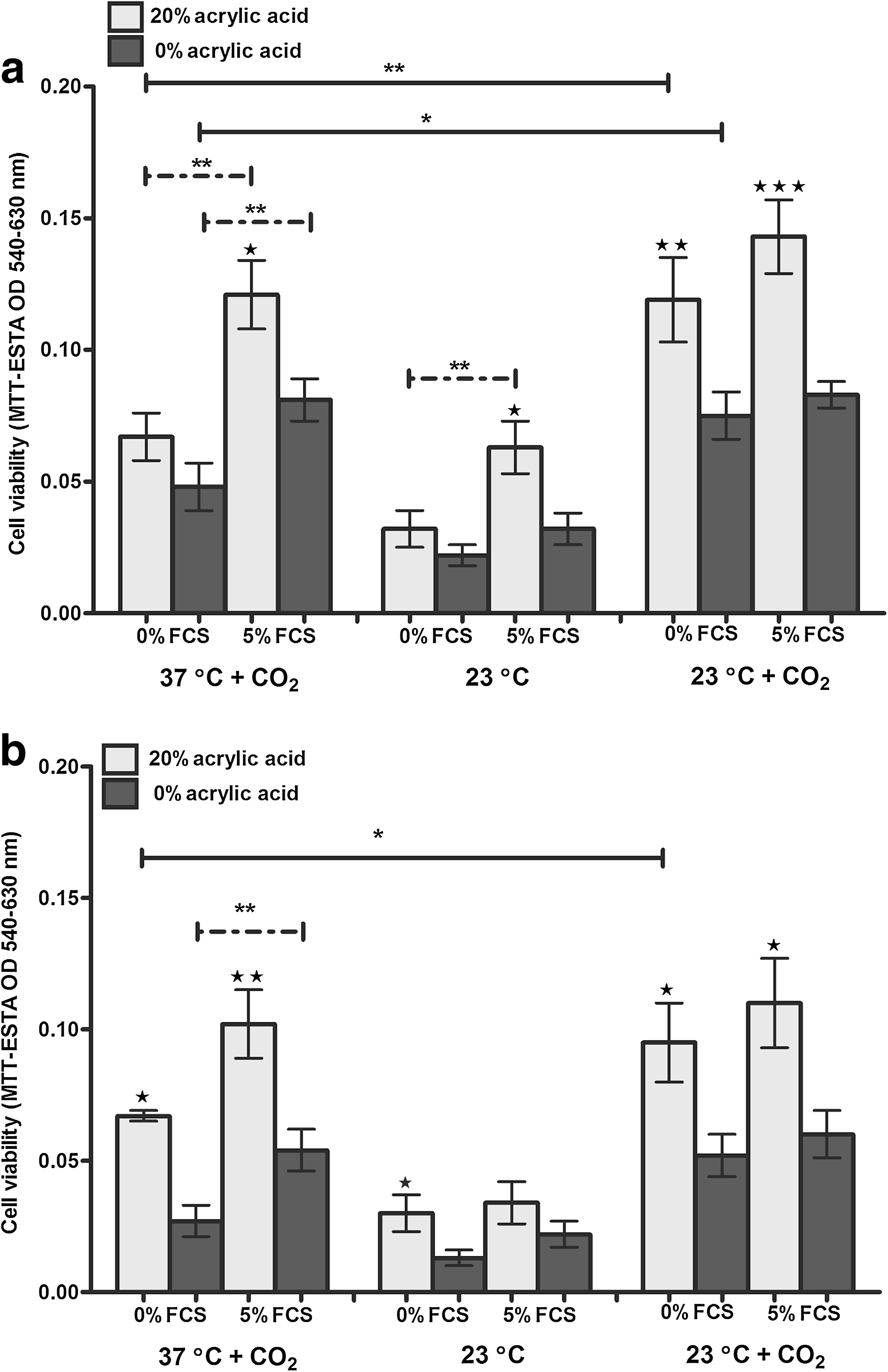
Effect of 0% and 5% serum on the transport of keratinocyte:melanocyte cocultures at 37°C+CO2, 23°C, and 23°C+CO2 for 48 h. Cell viability was assessed using MTT staining.
MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide.
MCDB, melanocyte medium.
In most circumstances cells on the acid carrier significantly outperformed the uncoated carriers for both melanocytes established in M2 medium (Fig. 2a and Table 2) and those established in MCDB medium (Fig. 2b and Table 3), after transport conditions for 24 and 48 h. Inclusion of 5% FCS also significantly improved cell viability in most circumstances (coated and uncoated carriers) after maintenance at 37°C+CO2 and 23°C. Although FCS appeared to have no beneficial effect on cell viability after maintenance at 23°C+CO2 (Fig. 2), the inclusion of 5% CO2 did prove beneficial. Metabolic activity of the cocultures (after 48 h) was also significantly higher after maintenance at 23°C in a sealed box with 5% CO2 compared with culture at 37°C in a constant 5% CO2 environment (Fig. 2).
Effect of temperature and CO2 on cell viability after maintenance and transfer onto human DED
The next experiments assessed the biological function of the keratinocytes and melanocytes postmaintenance under the different conditions. For these studies cocultures of keratinocytes and melanocytes established in M2 medium were maintained on 20% acid carriers and on 0% FCS agar medium for 48 h at 37°C+CO2, 23°C, and 23°C+CO2. Carriers plus cells were then transferred onto DED and left for a further 48 h. After transfer, while some of the samples were sacrificed for MTT-ESTA analysis, the remaining samples were raised to an air–liquid interface for 10 days to enable the transferred cells to form a fully stratified epithelium. At this point cell proliferation was assessed using AlamarBlue staining followed by fixation in formalin for S100 immunohistochemistry.
MTT-ESTA staining (Fig. 3a–c) and analysis (Fig. 3g) demonstrated that maintenance/transport conditions dramatically influenced cell transfer to human DED. Cells kept at 37°C+CO2 transferred well (Fig. 3a), those kept at 23°C showed poor transfer (Fig. 3b), but those kept at 23°C with CO2 gassing before maintenance did well (Fig. 3c). Further, cell proliferation analysis (Fig. 3d–f) after 10 days culture after transfer correlated well with metabolic ability post-transfer (Fig. 3a–c). Although there was little cell proliferation after maintenance of cells at 37°C+CO2 (Fig. 3d) or 23°C (Fig. 3e), cells maintained at 23°C+CO2 were able to proliferate and migrate over the 10 day culture period on DED (Fig. 3f). This is shown quantitatively in Figure 3g, where it can be seen that there was an approximately threefold increase in cell number post-transport in the presence of CO2.
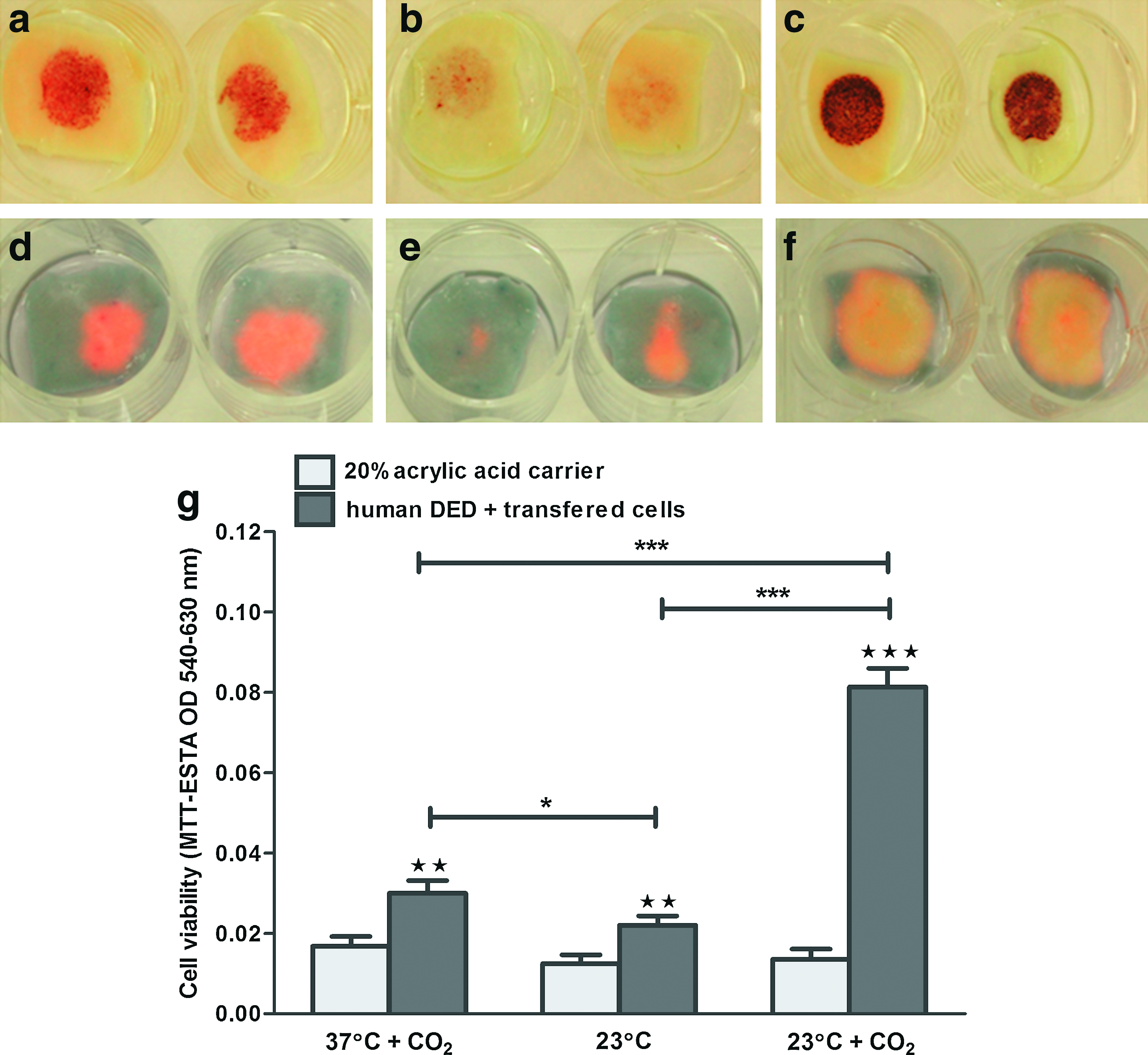
Effect of temperature on the transport and transfer of cocultures and resultant skin composites. Cocultures on 20% acid carriers were transported for 48 h on 0% FCS agar medium at 37°C+CO2
The final part of this study examined whether or not a fully stratified epithelium with correctly located melanocytes could be achieved after transport of cells. Immunohistochemical staining for S100 melanocytes demonstrated that under all maintenance/transport conditions an epithelium with melanocytes could be achieved (Fig. 4a–c). However, a consistently good stratified epithelia with a physiologically relevant number of melanocytes located along the basement membrane was observed most reliably after maintenance at 23°C+CO2 (Fig. 4c). A semiquantitative count of melanocytes present in these sections demonstrated that there were significantly more melanocytes present after maintenance at 23°C+CO2 compared with either maintenance at 37°C+CO2 or 23°C (Fig. 4d). Very few melanocytes survived maintenance at 37°C+CO2 or 23°C and those that did were often located suprabasally (Fig. 4a, b) in the epithelium and not, as they should be located, along the basal membrane (as illustrated in Fig. 4c).
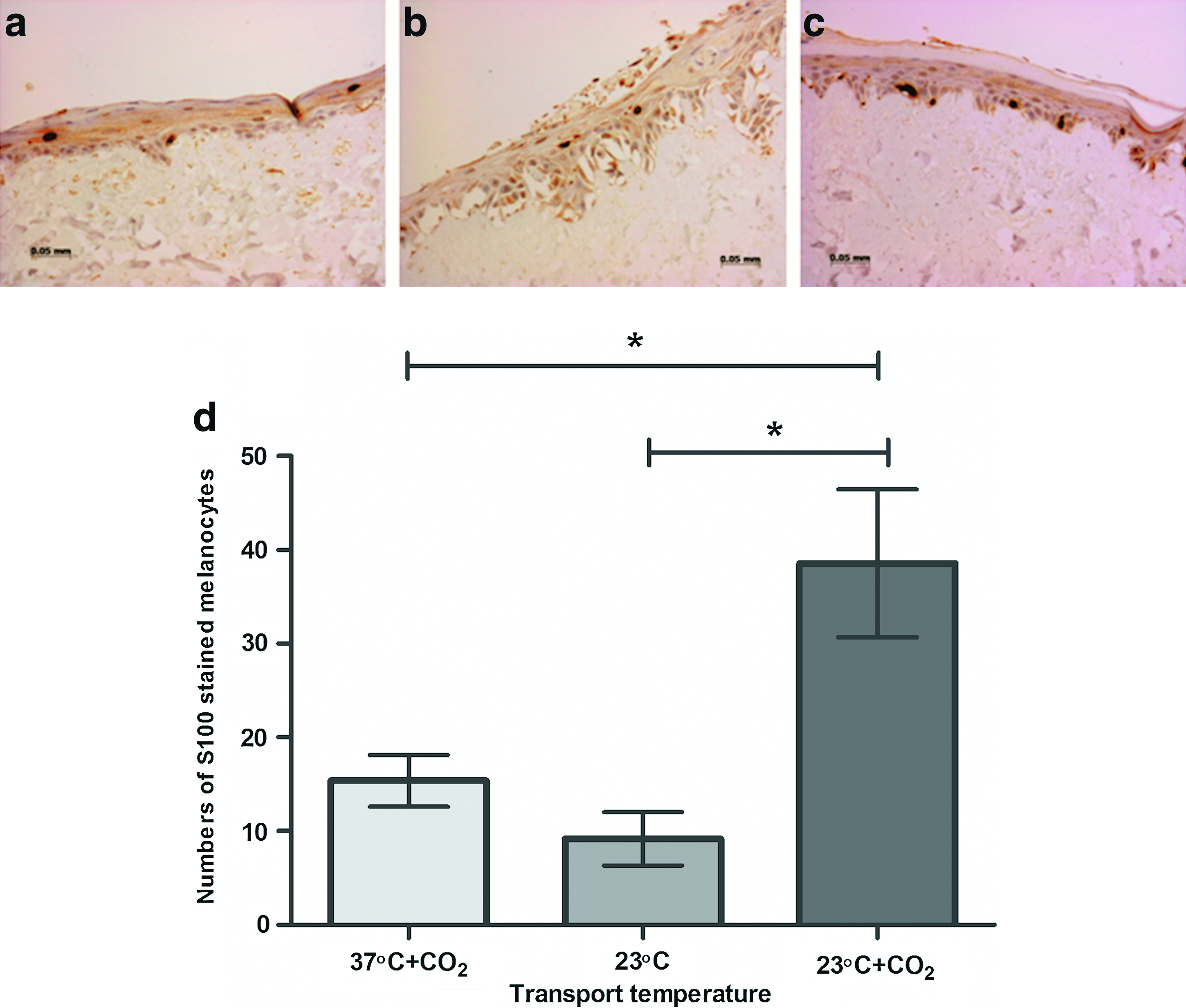
Effect of temperature on the location and numbers of melanocytes within resultant skin composites. Cells were mock-transported for 48 h on 20% acid carriers and 0% FCS medium gels, transferred onto human DED for 48 h, and cultured at an air–liquid interface for 10 days.
Discussion
Successful translation and eventual commercialization of tissue-engineered products requires live cells to be transported for up to several days while maintaining viability and the ability of the cells to deliver their desired biology post-transportation. In the case of epithelial cells most products are based on the ability of these cells to attach and survive (long term in the case of autologous products) on the patients wound bed. Accordingly, the aim of this study was to investigate what role the surface chemistry of the transport membrane, medium supplements, temperature, and CO2 gassing of the medium pretransport play in maintaining cell viability during and post-transport. In this study the cells did not leave the laboratory but were simply maintained on the laboratory bench under the conditions described. Issues of transport containers and mechanical trauma and the ability of containers to withstand variations in temperature were not tackled in this study—these are discussed briefly later.
In brief, the study demonstrated that a chemically defined coating (20% acrylic acid) significantly improved cell viability after different transport conditions and that while serum improves cell viability after maintenance/transport at 37°C+CO2 and 23°C without CO2, when cells were maintained at 23°C with CO2 they did well and there was no extra benefit seen from adding 5% FCS. The ability of cells to form a fully stratified epithelium with correctly located melanocytes was also affected by the temperature and pH (CO2 gassing of media) the cells experienced during transport. The best conditions for ensuring good cell performance post-transport in this study were with cells attached to a 20% acrylic acid-coated surface at room temperature (no serum required) in a CO2 bicarbonate buffered environment.
Patients with vitiligo currently have to travel to a specialized clinic that cultures autologous skin cells to receive effective treatment. This can be inconvenient, time-consuming, and expensive for the patient. There is a clear need to find a method for transport of cultured cell therapies significant distances, often under extreme and varying conditions. For our specific application, the challenge is to maintain cell viability and attachment to a carrier dressing during transportation, so that the autologous skin cells can be efficiently transferred onto the patient's dermis to facilitate the treatment of patients who live considerable distances from the laboratory. Also to repigment skin for the treatment of vitiligo, it is essential that functionality remains both in the keratinocytes to promote wound healing and minimize scar formation and in the melanocytes to enable melanin production and hence repigmentation. To the best of our knowledge, this is the first such study to demonstrate the extent to which basic cell transport conditions can affect cultured cell performance post-transport. (As cells did not leave the laboratory for this study, it is likely that cells that also have to withstand physical trauma and temperature excursions on transportation may be even more challenged.) We found many years ago that while keratinocytes poststorage might retain reasonable metabolic activity, they lost their ability to attach to a human wound bed model. 9 This shows that it is important to assess the desired functional biology of the cells post any manipulation such as storage or transport. For the current study it is necessary for these cells to be capable of attaching to the patients wound bed post-transport.
We have already demonstrated that viable and fully functional melanocytes and keratinocytes can be delivered from a flexible, easy-to-handle silicone carrier. 4 In this previous study, no significant differences were demonstrated between the 20% acid-coated and untreated silicone carriers. However, no transport issues were taken into consideration in this earlier study; cells were transferred directly from the carrier dressings after a culture period of 24 or 48 h at 37°C+CO2 in a constant 5% CO2/air atmosphere. In contrast, the current study demonstrates that the introduction of a further 24 or 48 h (after 24 h initial culture on the carriers) under different maintenance conditions results in significant differences in cell viability between 20% acid-treated and untreated and silicone carriers.
To help reduce mechanical trauma, cocultures of cells on the carriers were placed with cells downward in contact with solidified 1% agar medium. Once placed on these gels, the cell carriers did not move laterally on the gels. After subsequent transfer of cells from the carriers onto the human wound model, we confirmed that there were no viable cells remaining on the agar medium gel. This is an important point showing that the cell cultures remained viable on the agar medium gels for 24 and 48 h, but all remained attached to the transport carrier.
Another key aim for the development and commercialization of tissue-engineered products for treating patients has been to try and reduce the exposure of patients to animal serum proteins used for the in vitro culture of patient cells. Although we have been unable to fully eliminate the use of animal sera for the culture and expansion of autologous keratinocyte cultures (we source our serum from New Zealand to reduce the risk of exposure to bovine spongiform encephalitis), the data reported in this study suggest that the presence of serum during the transport process is not required providing cells are kept in a buffered CO2 environment.
The two remaining issues were temperature fluctuations and the relevance of a buffered CO2 atmosphere during transport. In specialized GMP facilities patient's cells are normally maintained in a constantly regulated environment of 37°C with 5% CO2. Once removed from this environment, our experience is that some cultured cells can very quickly lose viability—some cells withstand this more than others—for example, fibroblasts are relatively resistant, but melanocytes are not. Such observations are rarely published. The current study shows that maintenance of cocultures of keratinocytes and melanocytes for 48 h at 37°C+CO2 or 23°C without any CO2 gassing of the medium had adverse effects on the overall viability of the cocultures from which the cells did not recover very well even after 10 days culture on the human wound bed model. For the latter conditions, immunohistochemical analysis also showed the majority of melanocytes were located in the upper layers of the epithelium. In contrast, three times more viable cells were transferred after maintenance at 23°C+5% CO2, and these underwent rapid proliferation and migration after a 10 day culture period. These resultant skin composites consistently achieved a stratified epithelium with melanocytes correctly located amongst the basal keratinocytes. The latter result illustrates that optimizing conditions for transportation of particular cells is important to substantially increase the effective useful shelf-life of these products.
In summary, this study highlights the important role surface chemistry can play in helping maintain cell viability under transport conditions and demonstrates that transporting cells at lower (around 23°C) temperatures with a CO2 buffering system significantly helps protect the cells during transport and crucially extends their effective shelf-life to at least 48 h when they leave the laboratory. Under these circumstances FCS is not required.
Finally, there are many more questions to be answered that are beyond the scope of this study—how long can the cell viability and performance of these cells be maintained? In this study experiments did not extend beyond 48 h. Clearly, much more work is needed to explore how long the effective transport life may be. Transport for 72 h or more could enable cells to be sent between continents. There are other issues to consider what happens when cells are also subjected to mechanical trauma and to extreme variations in temperature. Here hopefully generic packaging solutions can be developed. Will these findings for adherent keratinocytes and melanocytes apply to other cells or will each cell type have different tolerances for transport? Clearly, many of these variables will impact on each other. We conclude that this study gives clear information on culture conditions for future transport of cocultures of adherent keratinocytes and melanocytes. Many more such studies on other cell types are needed to advance the tissue engineering communities' ability to benefit significant numbers of patients.
Footnotes
Acknowledgments
The authors would like to thank CellTran Ltd. and Yorkshire Forward for funding part of this work and Sheffield Burns Unit for funding Marta Baran. The authors also thank Mr. David Ralston Head of Sheffield Burns Unit for assisting with skin provision for this study.
Disclosure Statement
The authors state that no competing financial interests exist.