
Abstract
Adult salivary gland stem cells are promising candidates for cell therapy and tissue regeneration in cases of irreversible damage to salivary glands in head and neck cancer patients undergoing irradiation therapy. At present, the major restriction in handling such cells is their relatively limited life span during in vitro cultivation, resulting in an inadequate experimental platform to explore the salivary gland-originated stem cells as candidates for future clinical application in therapy. We established a spontaneous immortal integrin α6β1-expressing cell line of adult salivary progenitor cells from rats (rat salivary clone [RSC]) and investigated their ability to sustain cellular properties. This line was able to propagate for more than 400 doublings without loss of differentiation potential. RSC could differentiate in vitro to both acinar- and ductal-like structures and could be further manipulated upon culturing on a 3D scaffolds with different media supplements. Moreover, RSC expressed salivary-specific mRNAs and proteins as well as epithelial stem cell markers, and upon differentiation process their expression was changed. These results suggest RSC as a good model for further studies exploring cellular senescence, differentiation, and in vitro tissue engineering features as a crucial step toward reengineering irradiation-impaired salivary glands.
Introduction
In previous studies, we have shown the ability to grow and manipulate a human submandibular gland (HSG) cell line on a variety of scaffolds, 7 grow primary cultures of salivary murine cells 8 and rat α6β1-expressing salivary epithelial stem cells, 9 and enhance protein secretion for use in artificial salivary glands. 10 We have also shown the ability to isolate adult stem cells with mesenchymal characteristics from human parotid gland tissue. 11
Nevertheless, a major drawback in these research platforms is the limited survival of salivary gland-originated cells in their undifferentiated stage, implying difficulty in obtaining enough cells for in vitro experimental modalities.12,13 In this study, we established a novel spontaneous immortal cell clone based on integrin α6β1-expressing cells and further characterized its cellular and differentiation potential.
Materials and Methods
Animals and heat-stress conditioning
Male Sprague-Dawley rats (200–300 g) were used as the animal model. All animals were treated according to procedures approved by the Animal Care and Use Committee at our institute and were monitored continuously for any signs of distress. Animals were kept at 22°C ± 2°C. For heat-stress (HS) conditioning, rats were placed in a heat acclimation room with an ambient temperature of 34.5°C for 48 h as previously described. 9 All other environmental conditions, including food, fluids, light, and space, were similar to those of the untreated control group. Immediately after HS conditioning, the animals were weighed and euthanized, and the submandibular salivary glands were excised under aseptic conditions.
Immunomagnetic isolation and cultivation of integrin α6β1-positive cells
The excised salivary glands were rinsed three times in 10% penicillin, streptomycin, and amphotericin (PSA; Biological Industries). Glands were then minced into small fragments, which were enzymatically dissociated with Roswell Park Memorial Institute (RPMI1640) digestion solution (Biological Industries) containing 0.5 mg/mL collagenase (Worthington) and 5 mM calcium chloride. After stirring for 30 min at 37°C, the supernatant was collected, and the sediment was rediluted with the same digestion solution and stirred for two more cycles.
The supernatant was then centrifuged (1300 g) for 15 min at room temperature. The cell-containing pellet was washed in RPMI 1640 supplemented with 1% PSA and 2% fetal calf serum (FCS; Biological Industries) and recentrifuged for 15 min at room temperature. To obtain purified parenchymal cells, the reverse Percoll (Amersham Biosciences) discontinuous gradient method was used as previously described, with minor modifications. 14 Briefly, cells were gently layered onto Percoll at three solution concentrations: 75%, 40% and 30%, creating a three-phase Percoll gradient. The Percoll gradient was then centrifuged (800 g) for 20 min at room temperature and the upper phase, containing the parenchymal cells, was collected and centrifuged (9000 g) in RPMI 1640 solution supplemented with 1% PSA and 2% FCS for an additional 5 min at room temperature.
The isolated parenchymal cells were subjected to the magnetic affinity cell sorting immunomagnetic separation system according to the manufacturer's specifications (Miltenyi Biotec). Cells were incubated with mouse anti-rat integrin α6β1 immunoglobulin (Ig) G1 monoclonal antibody (Chemicon) for 30 min at room temperature. Rat anti-mouse IgG1 microbeads were used as a secondary antibody (Miltenyi Biotec). 9 The α6β1 integrin-expressing cells isolated were termed integrin α6β1-expressing (SGIE) cells.
Cell culture
SGIE cells were inoculated into 25-cm2 Falcon flasks (BD Labware) precoated with 10 mg/cm2 of rat collagen type I (Sigma). The cells were maintained in a 3:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 supplemented with 10% FCS, 2 mM glutamine, penicillin (100 unit/mL), streptomycin (100 unit/mL), and amphotericin (0.25 mg/mL) (Biological Industries). The medium was supplemented with epidermal growth factor (10 ng/mL), insulin (5 mg/mL), transferrin (5 mg/mL), triiodothyronine (T3; 2 × 10−9 M), hydrocortisone (0.4 μg/mL), adenine (1.8 × 10−4 M), and cholera toxin (10 × 10−10 M) (Sigma). After passage 30 (P-30), rat salivary clone (RSC) cells were grown on DMEM supplemented with 10% FCS, 2 mM glutamine, penicillin (100 units/mL), streptomycin (100 units/mL), and amphotericin (0.25 mg/mL) (Biological Industries).
PC3, a murine prostate cancer cell line, was a generous gift from the Laboratory of Experimental Surgery, Hadassah University Hospital, Jerusalem. Cells were grown on DMEM supplemented with 10% FCS, 2 mM glutamine, penicillin (100 units/mL), streptomycin (100 units/mL), and amphotericin (0.25 mg/mL) (Biological Industries).
Passaging, limiting dilution, and clonogenic capability procedures
Cells were passaged once a week, when they reached 80% confluence. Cells were released with a solution of 0.25% trypsin and 0.05% ethylenediaminetetraacetic acid (EDTA) (v/v, 1:1; Biological Industries), and replated.
To isolate colonies derived from a single cell, a limiting dilution assay was utilized. Briefly, a limited number of cells in P-25 were plated in a 96-well plate. The cells were diluted in the medium (as described above) to 0.5 cells/well. The plates were monitored daily under an inverted microscope (model IX71, Olympus Corporation), and photos were taken with a CFW-1310C camera (Scion Corporation).
To test colony-forming efficiency, 1 × 103 cells were seeded on a 35-mm Petri dish for 10 days. The colonies were then fixed with 4% (v/v) formaldehyde (Sigma) for 10 min at room temperature. After counterstaining with 1% (w/v) rhodamine B solution (Sigma), colonies were manually counted under a light microscope.
Fluorescence-activated cell sorter (FACS) analysis
Flow cytometric analysis was performed according to the manufacturer's instructions (BD Biosciences) as previously described by us. 9 Briefly, cells were divided into two aliquots and washed with phosphate-buffered saline (PBS). One aliquot of cells was further incubated with rat integrin α6β1 IgG1 monoclonal antibody (Chemicon) for 30 min at room temperature. Then, the cells were incubated for 15 min with secondary antibody, Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). As a control, the second aliquot of cells was incubated with an isotype control antibody followed with the secondary antibody. After washing with PBS, the labeled cells were analyzed using a FACScan (BD Biosciences).
Soft agar test and karyotype analysis
A base layer was prepared with DMEM supplemented with 8% FCS and 0.6% (w/v) preheated nutrient agar. The mixture was heated to 45°C and then poured into sterile 60-mm dishes and left to cool down. Cells (1 × 105) were mixed in 1.5 mL of a 0.4% agar base layer and seeded on top of the gel base layer. Cultures were maintained in a humidified 37°C incubator for 6 days, and then photographed under an inverted microscope (model IX71) with a CFW-1310C camera. For karyotyping, 1.5 × 104 cells were seeded on a cover slide placed in a 35-mm dish with the growth medium. When cells reached 70% confluence, karyotype assay was performed as previously described. 15 Briefly, cells were exposed to colcemid (10 mg/mL; Gibco) for 24 h at 37°C, harvested using trypsin/EDTA (Biological Industries), and centrifuged (3 min, 1300 g). The pellet was treated with hypotonic trisodium citrate (0.8% w/v; Biological Industries) for 30 min at 37°C. Cells (P-3) were fixed in methanol:acetic acid solution (3:1, v/v) and spread on slides. The slides were incubated overnight at 60°C. The cells' chromosome complement was then stained according to the conventional Giemsa technique (0.4% v/w buffered methanol solution, pH 6.9; Sigma).
Matrigel, alginate, growth factor handling, and histochemical staining
Matrigel (200 μL; BD Biosciences) was added to 24-well tissue-culture dishes and incubated for at least 30 min at 37°C to form a gel. Cells were plated at a density of 1 × 105 cells/well and were cultured in a 37°C, 5% CO2 incubator. 16
To evaluate cell behavior factors, 10% FCS, 0.07% (w/v) bovine pituitary extract (BPE; Sigma), and 0.05% (w/v) hepatocyte growth factor (HGF; Sigma) were added to the culture medium. Cells were photographed under an Olympus inverted fluorescent microscope (model IX71) with a CFW-1310C camera.
Cells cultured in Matrigel were fixed in 4% formalin solution (Sigma). The samples were transferred to preheated liquid 1% agarose solution. Then, the samples were embedded in paraffin and 5-μm-thick sections were cut and mounted on slides. The sections were stained with hematoxylin–eosin. Periodic acid-Schiff (PAS) staining was used to identify acinar cells. 17
Macroporous alginate hydrogels were fabricated as we previously described 18 with slight modifications. Briefly, alginate hydrogels were prepared from 2% (w/v) sodium alginate (NovaMatrix; FMC Biopolymer) solution (55 mM EDTA and 10 mM HEPES, pH 7.4) and thereafter cross-linked with 0.2% (w/v) of CaCl2 solution (102 mM CaCl2 and 10 mM HEPES, pH 7.4). Alginate hydrogels were then cast on wells and gradually chilled (2h at 4°C and overnight at − 20°C). Before use, alginate hydrogels were thawed and washed twice with 0.5 mL CaCl2 solution and twice with culture media at room temperature. Cells were then plated upon the surface (5–50 × 104 cells/well) and incubated in a 37°C, 5% CO2 incubator. Alginate hydrogel RSC cultures were observed as described above for Matrigel cultures.
Immunofluorescence and confocal microscopy
About 25,000 RSC cells were plated on eight-compartment Lab-Tek chamber slides (Nelge Nunc International) and covered with 10 mg/cm2 collagen type I (Sigma) or with 30°μL Matrigel (Becton Dickenson, Franklin Lakes, NJ, BD). Cells were incubated with the growth medium for 3 days and then washed three times with PBS (Biological Industries). Cells were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature and then permeabilized for 5 min with 0.1% Triton X-100 (v/v in PBS; E. Merk). Nonspecific antibody binding was blocked by 60 min incubation with 5% FCS in PBS (v/v) at room temperature. The antibodies for amylase (200 μg/mL, mouse monoclonal IgG; Santa-Cruz Biotechnology), p63 (200 μg/mL, rabbit polyclonal IgG; Santa-Cruz Biotechnology), Cystatin C (1 mg/mL, rabbit polyclonal IgG; Chemicon), and α6β1 (mouse monoclonal, Chemicon) were diluted 1:100 with 5% FCS in PBS (v/v) solution and incubated for 60 min at room temperature. Alexa 488- and Alexa 647-conjugated secondary antibodies against rabbit and mouse IgG (Molecular Probes) were added to samples at a dilution of 1:500 and incubated for 60 min at room temperature. For nuclei stain, 4′,6-diamidino-2-phenylindole (DAPI; 100 μg/mL; Chemicon) was added to samples at a dilution of 1:1000 with PBS (v/v) and incubated for 10 min at room temperature. The stained preparations were evaluated using a confocal laser scanning microscope (Model 710; Zeiss).
Videomicroscopy
About 1 × 105 RSC cells were mixed with 600 μL medium and were seeded in a 24-well tissue culture dish (Jet Biofil) precoated with 200 μL Matrigel. Cells were kept at 37°C in a humidified 5% CO2 atmosphere in a Chalmide microscope stage incubator (Live Cell Instrument). The incubator was attached to an Olympus inverted phase-contrast microscope (model IX71). Images were obtained at 10-min intervals for the first 6 h and then at 15-min intervals for an additional 18 h, using a CFW-1310C camera. The automated time-lapse imaging was controlled using NIH ImageJ software.
RNA isolation and real-time PCR analysis
RNA was extracted from 1 × 106 RSC in P-79 and SGIE cells in P-1. Extraction of total RNA and synthesis of complementary DNAs were performed using TaqMan Cells to CT kit (Applied Biosystems) according to the manufacturer's instructions. cDNA was amplified using the 7300 real-time PCR system (Applied Biosystems) with TaqMan gene expression assay for α-amylase, aquaporin 5, p63, cystatin C, cytokeratin 17, and glyceraldehyde 3-phophate dehydrogenase (Applied Biosystems).
Statistical analysis
The results were statistically analyzed by paired Student's t-test. p < 0.05 was considered statistically significant.
Results
Isolation of spontaneous immortal integrin α6β1-expressing cell clone
SGIE cells were immunomagnetically isolated from 21 HS rats. In each cell preparation, about 1 × 106 cells were typically isolated and grown in separate collagen-coated culture dishes until senescence. The cells survived an average of 19.72 ± 2.61 passages until senescence. Figure 1A shows the isolation of a clone originating from a single cell by the low-density culture system at P-25 from animal no. 4, where cells continued to divide. This clone was termed RSC. To examine the RSC's dividing potential, cells were seeded at three different densities (5 × 105, 5 × 104, and 1 × 104 cells/flask) for 50 weekly passages beginning at P-30 (Fig. 1B). The average number of divisions per week was 1.66 ± 0.84, 5.21 ± 0.78, and 7.43 ± 0.94, respectively, and the accumulated number of divisions (from P-1) was 117, 353, and 407, respectively. Figure 1C shows the clonogenic capacity of 1 × 103 RSC cells from each seeding level in P-66 cultured for 10 days and counterstained with rhodamine B. Cells derived from a seeding level of 1 × 104 cells produced fourfold the number of colonies produced by SGIE cells (333 ± 16.6 and 87 ± 7 colonies, respectively, p = 0.00008). Moreover, a seeding level of 1 × 104 RSC cells produced ∼1.5-fold more colonies than RSC cells originating from seeding levels of 5 × 104 and 5 × 105 (333 ± 16.6, 232 ± 8, and 218 ± 11.45, respectively, p = 0.0007). To examine whether the RSC cells maintained integrin α6β1 expression, cells in two passages, P-56 and P-84, were examined by FACS and immunofluorescence, respectively; 96% of the RSC cells were positive for integrin α6β1 (Fig. 2) that was detected in the outer membrane area of RSC (Fig. 7A-α6β1).
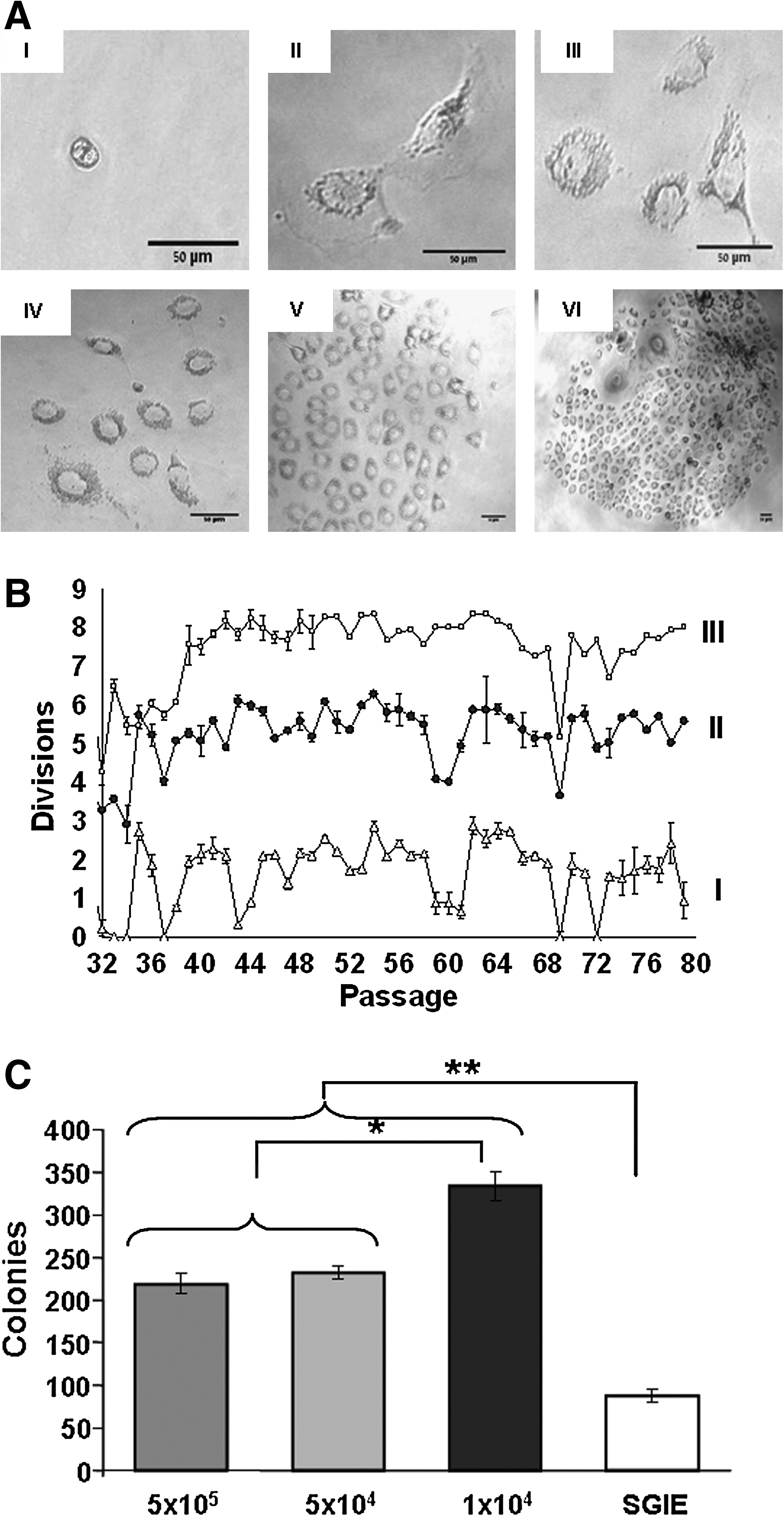
Colony formation, number of divisions, and clonogenic capability.
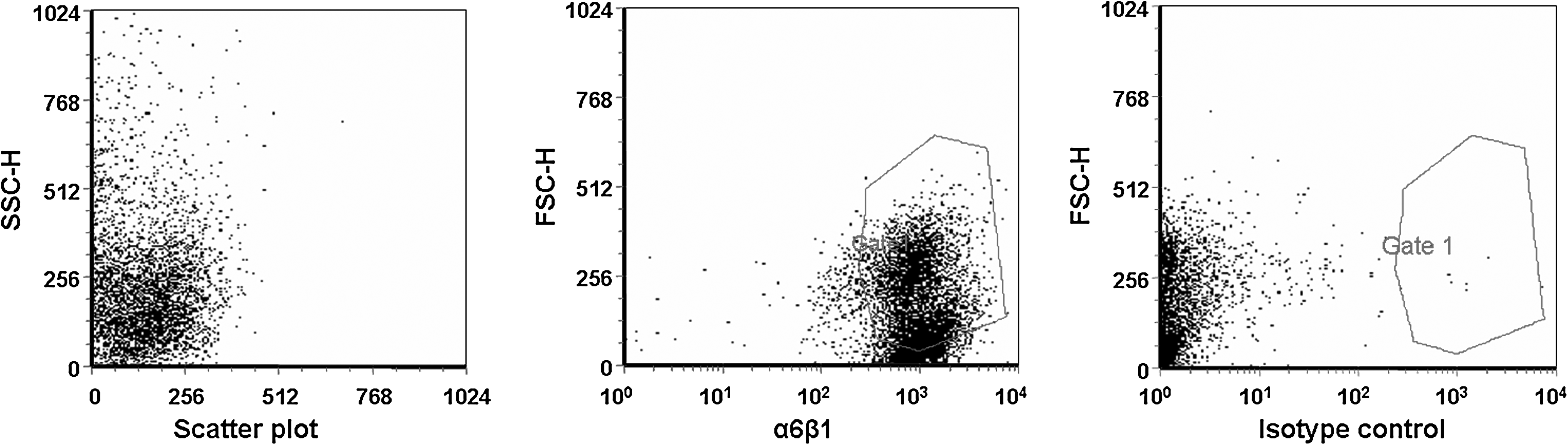
Expression of integrin α6β1 stem cell marker. FACS analysis for RSC cells (P-56) on plastic.
Tumorigenic potential of RSC
To test the cells' ability to form colonies under anchorage-independent conditions in a semisolid medium, a soft agar assay was carried out. RSC (1 × 105) and PC3 (1 × 105) murine prostate cancer cells (serving as a positive control) were mixed into liquid agar medium mixture and seeded on top of a base layer for 6 days. RSC cells from P-70 succeeded to grow (Fig. 3AII), but formed far less colonies than PC3 (Fig. 3AI).
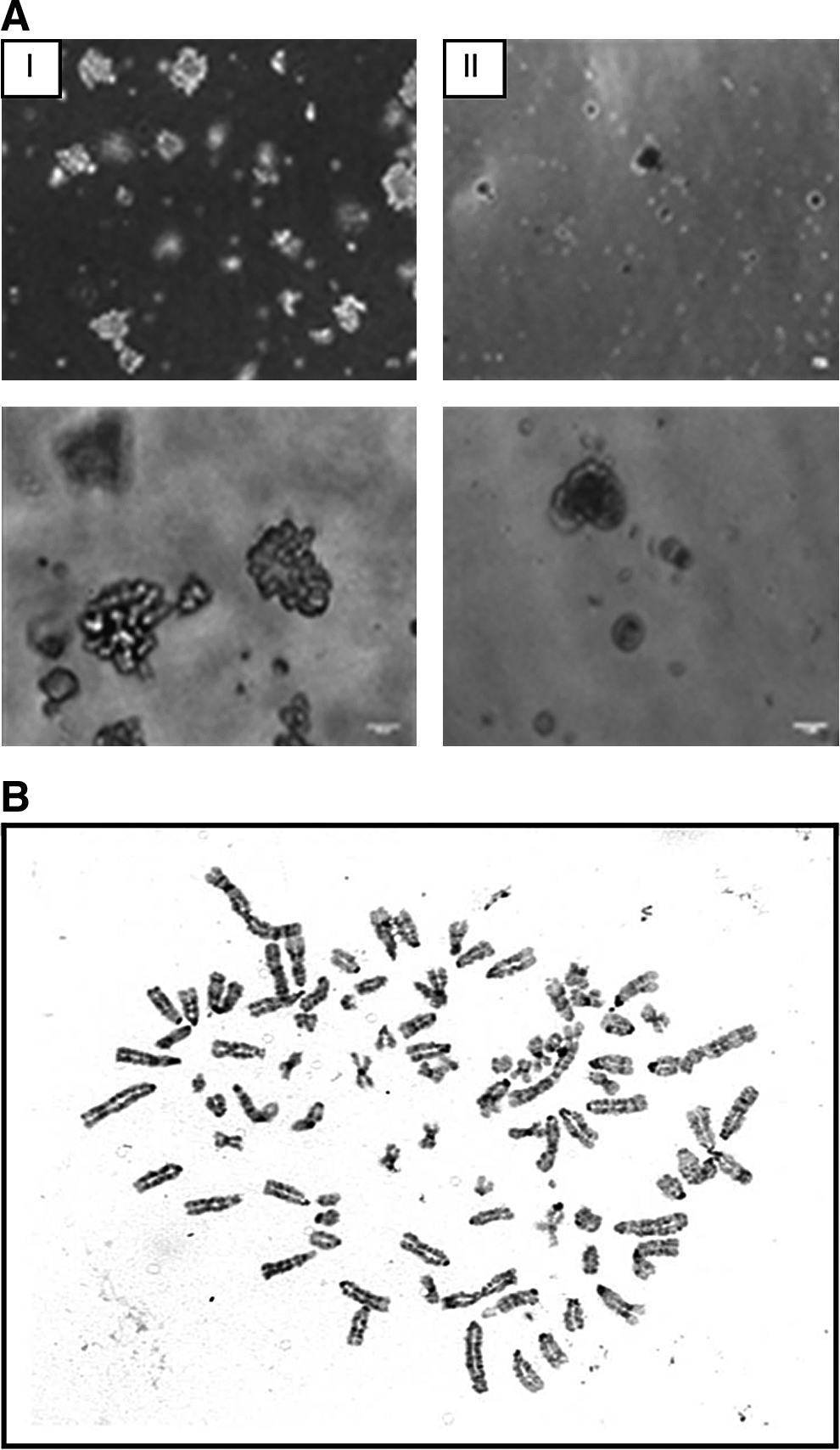
Cell growth and karyotype.
Karyotype
A representative karyotype of a metaphase cell (P-77) is shown in Figure 3B. RSC displayed an increase in overall chromosome number, resulting in a hypertetraploid karyotype.
Sustaining ductal and acinar differentiation multipotential of RSC in 2D and 3D scaffolds
RSC cells were cultured and differentiated in a 3D Matrigel scaffold for 3 days with the serum-free medium. Cells in 100% Matrigel formed net-like structures by day 2, including branches terminating with spheres (Fig. 4AII). PAS staining showed complex acinar and ductal-like structures (Fig. 4AIII). Cells in 25% Matrigel formed spherical structures by day 2 (Fig. 4BII); PAS staining showed acinar-like structures (Fig. 4BIII). The addition of 10% FCS to the medium induced the formation of strictly acinar-like structures (Fig. 4CII, III). The same pattern was observed upon addition of 0.07% BPE (Fig. 4DII, III). In contrast, the addition of 0.05% HGF resulted in a less prominent change, that is, the basic structure remained but with elongated ductal components (Fig. 4EII, III).

Effect of Matrigel concentration and growth factors on RSC cell organization. RSC cells (1 × 105) from P-56 were cultivated for 3 days in
Kinetic analysis of RSC's structural appearance showed that the cell organization process begins as early as 3 h after seeding; by 24 h, cell organization is well established (Fig. 5B). In the Matrigel without FCS (Fig. 5A), 3 h after seeding, small ducts ending in spheres began to appear; however, by 18 h, the ducts had disappeared but the spherical structures were still observed. In contrast, in the Matrigel cultures with FCS, 3 h after seeding, ducts ending in spheres appeared and further developed into a complex of elongated ducts ending in spheres at 6–9 h. By 24 h after seeding, spheres were further condensed into major structures of the complex or acinar form, while maintaining the ductal component (Fig. 5B). In another set of experiments, we also found that structural organization is dependent on the type of Matrigel (regular or reduced, data not shown) and on initial seeding density (data not shown).
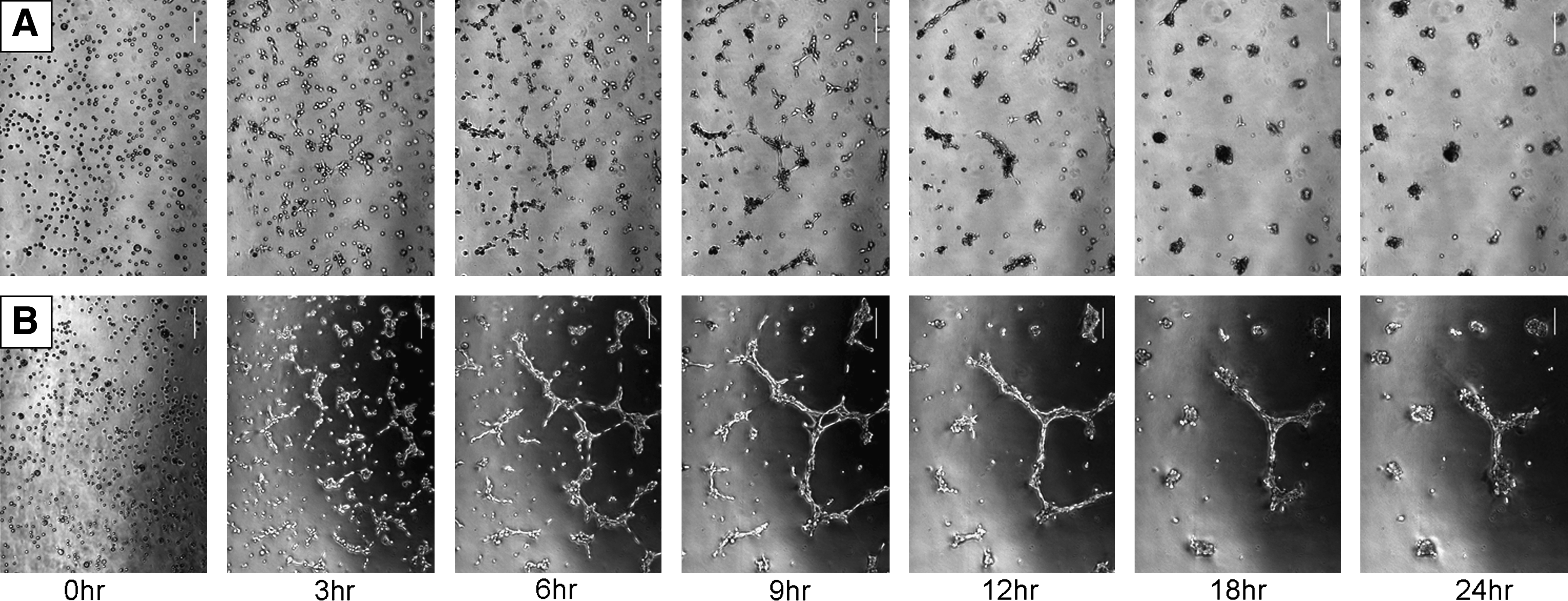
Kinetic development of RSC structures in Matrigel. RSC cells (1 × 105) from P-80 were cultivated for 3 days in
We next examined whether RSC cells would organize in 3D cultures without the use of the biological factor-enriched Matrigel scaffold utilizing 3D alginate hydrogel scaffold. We found that RSC could produce well-established spherical structures by day 3 (Fig. 6A) and their appearance was dependent on the initial cell density.

RSC cells (from P-74 or higher) cultured on alginate hydrogel
We further examined whether RSC would display organization behavior in 2D cultures. Indeed, RSC formed net-like structures from day 5, followed by a well-established net with spherical forms on day 9 (Fig. 6B).
Immunofluorescence and confocal microscopy of RSC grown on collagen type I for 3 days (Fig. 7), before structure formation, revealed the presence of the membrane integrins α6β1 and the nuclear transcription factor p63. It also demonstrated the cytoplasmic salivary-specific protein markers α-amylase and cystatin C. Confocal microscopy of RSC grown on Matrigel for 3 days, when acinar-like structures were already established (observed if Matrigel is cut to 2D preparations), revealed that both α6β1 and mainly p63 proteins expression were largely reduced, coupled with decreased expression of α-amylase except of structure margins where cells expressed α-amylase. Cystatin C expression remained high.
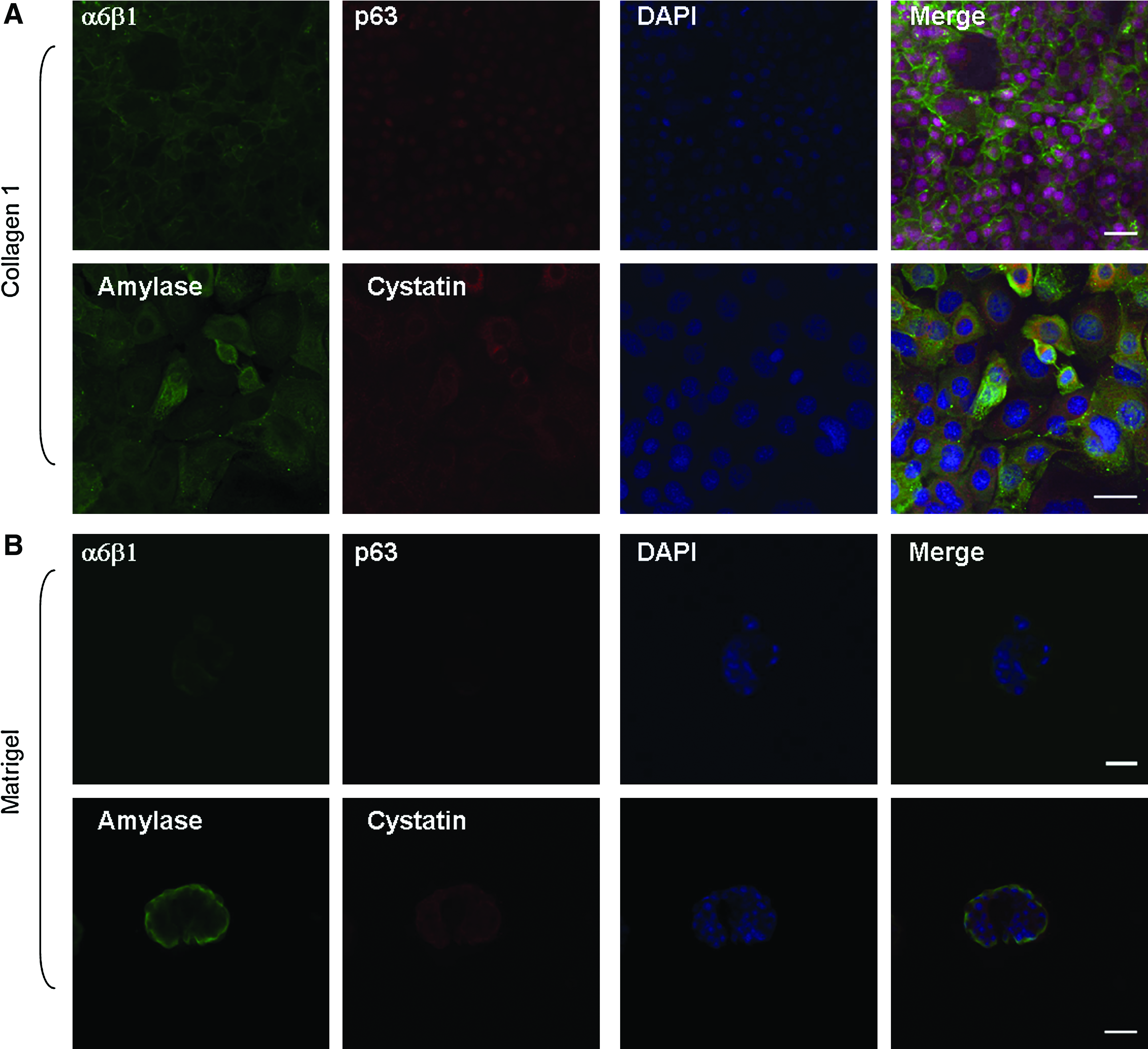
Confocal microscopy of organizing RSC cells. RSC cells from P-84 grown for 3 days on collagen type I
Gene expression of RSC
A comparison of mRNA expression between primary SGIE (P-0) and RSC (P-79) cells cultured on plastic revealed dramatically higher levels of the ductal marker CK17 and the progenitor cell marker p63 in the latter(14-fold, p = 3 × 10−5 and 15.2-fold, p = 6 × 10−4, respectively). The levels of the acinar and functional markers cystatin C and α-amylase were 90% lower (p = 6 × 10−6 and p = 8 × 10−6, respectively). Moreover, no aquaporin 5 expression could be detected in RSC cells grown on plastic (Fig. 8).
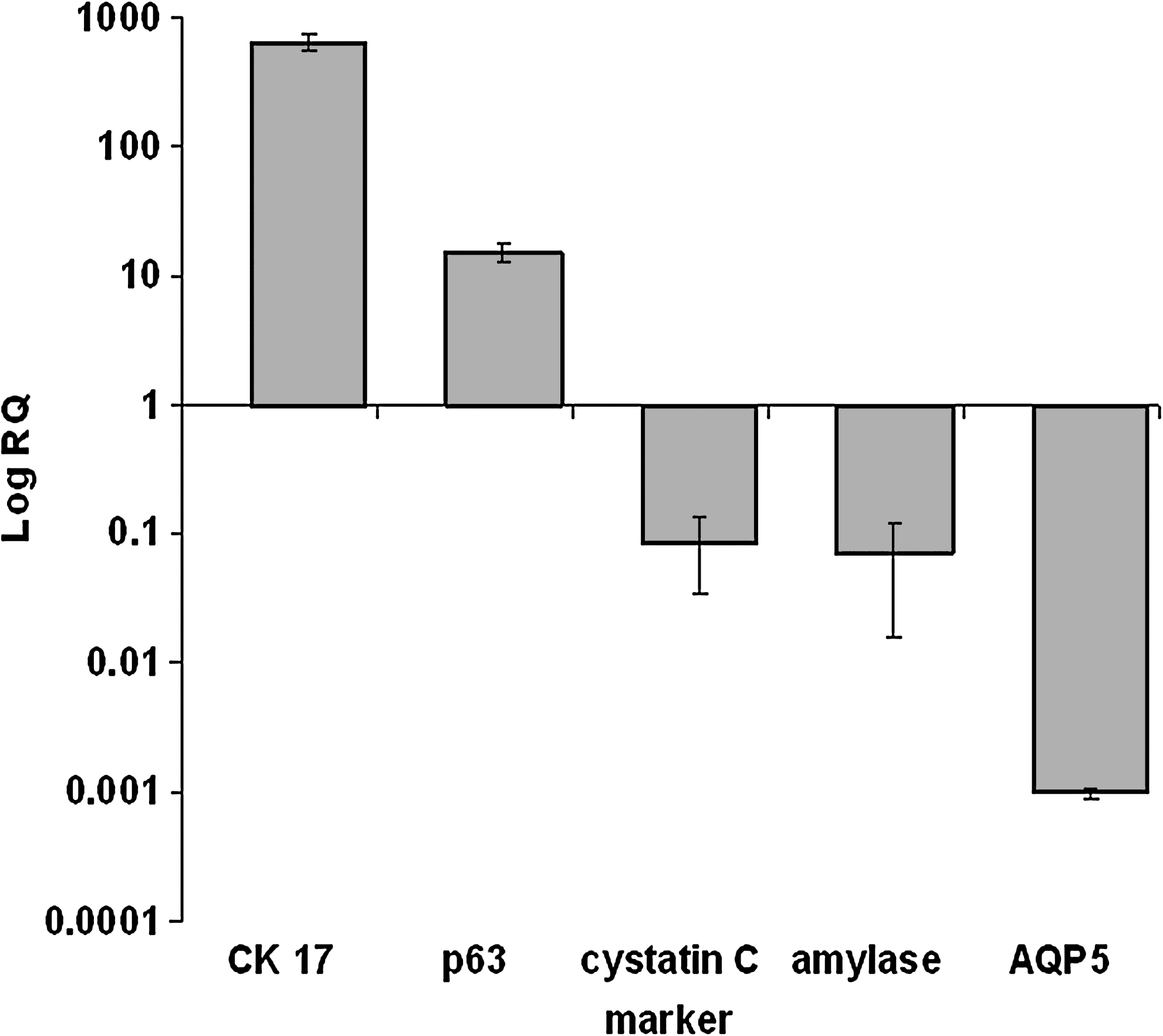
Relative mRNA expression of α-amylase, CK17, cystatin C, p63, and aquaporin 5 (AQP5) in RSC (P-77) versus SGIE (P-0) cells. mRNAs were analyzed by real-time RT-PCR. p < 0.0006 for all genes tested, n = 4.
Discussion
Regeneration of salivary glands' normal function in patients with head and neck cancer who have been treated with irradiation would make a major contribution to their quality of life. Such regeneration could be accomplished directly by reimplantation of autologous salivary gland cells 14 into the residual salivary tissue or by implantation of an artificial salivary gland device.6,7 Both strategies depend on the isolation of cells that can propagate and differentiate into functional salivary cells. Two cell types have been reported as potential salivary gland stem cells: SGIE cells9,16 and c-kit-expressing cells that lose their surface marker by 10 days in vitro. 12 Both types exhibit limited life span in vitro, a phenomenon which has also been demonstrated recently for primary cultures of human salivary cells. 13 The aim of this study was to establish a line of salivary stem cells with a prolonged life span that maintain parental cell differentiation properties. We succeeded in creating a clone based on the previously studied SGIE cells with the ability to propagate for over 80 passages in culture and termed it RSC.
We found that RSC cells could proliferate for more than 400 doublings and that the doubling rate was inversely dependent on seeding cell density; that is, lower density resulted in higher doubling rate, reaching up to 7.4 doublings per week. This finding was supported by tests of their clonogenic capability. Consequently, we found that more colonies were established at the lower cell density seeding level. Indeed, comparison of SGIE and RSC clonogenic potential showed the latter's much higher ability to form clones, demonstrating its proliferation stability. Nevertheless, immunohistochemical and FACS analyses showed that RSC maintains integrin α6β1 expression similar to the SGIE parental cell.
Soft agar test and karyotype analysis showed that RSC immortalization is a result of spontaneous transformation. In the second part of this study, we therefore examined the effect of cell transformation on the cells' differentiation abilities.
Tissue engineering combines cells, growth factors, and scaffolds for repair and regeneration of biological tissues. We therefore examined RSC cell organization on two 3D (Matrigel and alginate hydrogel) and one 2D (collagen type I) scaffolds with two supplemented growth factors (BPE and HGF). Matrigel, a mouse sarcoma extract, is a well-established scaffold for salivary cells in vitro differentiation assays and contains high amounts of growth factors. Alginate hydrogel and collagen type I do not contain growth factors, are inert, and are well-accepted scaffolds for clinical use. In previous studies, an HSG cell line was shown to be able to differentiate into acinar structures when grown on a 3D Matrigel scaffold in the medium containing serum. 16 Culturing HSG on a collagen I-coated plastic dish (2D) or in Matrigel without serum did not result in good propagation and, further, resulted in poor differentiation (unpublished data and Hoffman et al. 16 ). We adopted the same model to examine whether RSC could be induced to differentiate in vitro and found that it could differentiate in both 2D and 3D scaffolds, but that the time course in Matrigel and alginate hydrogel was faster than and structural morphology superior to 2D culturing.
Interestingly, RSCs cultured on inert nonadhesive alginate hydrogels were coaxed to reorganize and form compact spherical structures consistent with the results previously reported for hepatocytes, which are also epithelial-originated cells18,19 (and were shown previously being able to differentiate from α6β1-expressing salivary progenitor cells. 20 The microporous structure and nonadhesive nature of alginate hydrogels induces cell–cell interactions, thus providing a constructive milieu for salivary glad cell organization much like Matrigel. However, opposed to Matrigel, alginate does not contain growth factors and soluble inducers, indicating that RSC possess the independent capacity to differentiate in vitro and create acinar-like structures even without scaffold-based biochemical stimulation and suggests the possible use of alginate hydrogel as scaffold for artificial salivary glands or cell transplantation.
Another intriguing result was the ability of RSC cells to grow and differentiate in the serum-free medium, thus enabling the performance of future in vitro differentiation analyses without the masking effect of serum. Interestingly, kinetic analysis of RSC organization in both serum-free and serum-containing media followed the same time course but resulted in a different morphological pattern: only acinar structures versus complex acinar-ductal components, respectively. The time course of cell organization was rapid, indicating that cell proliferation is not mandatory for this process. Cell organization also included migration over long distances. These two parameters resembled other tissues' in vivo regeneration processes. Acinar and ductal RSC structural formation was also shown to be controlled by media additives, overcoming the enriched factors contained in the Matrigel 3D scaffold as previously shown for untransformed cells (BPE additive 21 and HGF additive 20 ). RSC differentiated into both ductal- and acinar-like structures when grown on 2D collagen scaffolds. This unique phenomenon took more than 5 days, indicating that cell proliferation is needed for this process. The results of the 2D and 3D scaffold experiments indicated that RSC organization is dependent on cell density, matrix properties, and biological additives.
To have an insight if differentiation processes occurred when cells were organizing, we examined by immunofluorescence and confocal microscopy both stem cells and specific salivary proteins markers before and after cell organization. RSC expression changed along structure formation, indicating that a differentiation process occurred. The expression levels of both stem cells markers as well as α-amylase were reduced, whereas expression of cystatin C remained high. This pattern of expression is typical to the submandibular glands where amylase protein activity is not found 22 and from which RSC was derived.
Importantly, RSC grown on plastic continued to express major salivary gland genes in P-77. These included increased levels of CK17—a ductal differentiation-oriented gene, and decreased levels of acinar-related genes (cystatin C, α-amylase, and aquaporin 5). Recently, in a mouse model, a population of multipotent progenitor cells, marked by expression of Ascl3, which is capable of generating both gland cell types, was found to originate from the ductal compartment. 23 These results support this notion and are in agreement with the increases found in mRNA of the keratinocyte stem cell marker p63. 24 Further studies are needed to explore gene expression patterns under a variety of conditions, including different scaffolds and different growth additives in the culture media. Such studies would help clarify whether the change in gene expression is a result of the immortalization process or of the natural dedifferentiation process occurring in other cultivated tissues such as liver, 25 cartilage, 26 and primary cultures of human salivary gland cells. 9
In summary, RSC cells were capable of differentiating into both acinar- and ductal-like structures and responded to their culturing conditions by changing their phenotype: this suggests their potential use to investigate differentiation mechanisms and morphogenesis of salivary structure development, and the possibility of directing regeneration of salivary gland tissue.
In conclusion, we established an immortal multipotent rat salivary progenitor cell line termed RSC. These cells were able to maintain their proliferation and differentiation potential, similar to SGIE cells, following numerous passages. Thus, RSC could provide us with a cellular model for experiments on salivary stem cells and tissue engineering and serve as a platform to establish a progenitor cell line that will assist in recovering saliva secretion of irradiated head and neck cancer patients.
Footnotes
Acknowledgment
This research was supported by the German Israeli Science Foundation (no. 1911-58.2/2006).
Disclosure Statement
No competing financial interests exist.