
Abstract
Stem cells are of widespread interest in regenerative medicine due to their capability of self-renewal and differentiation, which is regulated by their three-dimensional microenvironment. In this study, a computer-aided biofabrication technique based on laser-induced forward transfer (LIFT) is used to generate grafts consisting of mesenchymal stem cells (MSCs). We demonstrate that (i) laser printing does not cause any cell damage; (ii) laser-printed MSC grafts can be differentiated toward bone and cartilage; (iii) LIFT allows printing of cell densities high enough for the promotion of chondrogenesis; (iv) with LIFT three-dimensional scaffold-free autologous tissue grafts can be fabricated keeping their predefined structure, and (v) predifferentiated MSCs survived the complete printing procedure and kept their functionality. We believe that our results will find important applications in stem cell biology and tissue engineering.
Introduction
From the technical point of view the production of stem cell niches and tissue constructs is very challenging with respect to their complexity. First, one needs a technology that allows the generation of defined 3D microstructures copying original tissue templates. By this means, there is a demand to develop natural substitutes instead of traditional 3D scaffolds, since they limit oxygen exchange within the tissue. 2 Second, the structure has to be formed out of a material that is not only compatible with the cells but also enables the exchange of nutrients and soluble factors. Further, material elasticity and forces have to be taken into account since these parameters are known to influence stem cell differentiation. 1 Third, the cells have to be arranged in 3D, enabling the formation of close cell–cell as well as cell–matrix contacts and interactions. Fourth, cell differentiation has to be guided and controlled within this complex 3D system. As differentiation is dependent on the initial cell density, the cell constructs have to be formed out of a defined, variable, and high cell amount.3,4
During the last decade, computer-aided deposition of biological materials has been investigated as a potential technique for engineering of tissue replacements. In general, all existing bioprinting approaches5,6 can be separated into two groups: orifice7–11 and orifice-free techniques. 12 Among the orifice-free techniques laser printing based on laser-induced forward transfer (LIFT) possesses the capability to print cell suspensions with a wide range of viscosities and high cell densities. LIFT has previously been proven for printing single to tens of cells simultaneously without any observable damage to pheno- and genotype.12–16 These properties and the current opinion3,4,11,12,17,18 that high cell densities lead to accelerated tissue self-assembly and in vitro maturation make laser printing an attractive technique for biofabrication.
On the basis of our previous result 15 this study combines the benefits of laser printing (LIFT) with mesenchymal stem cells (MSCs), which present a self-renewal and differentiation capacity into different tissue lineages. A natural hydrogel consisting of plasma and alginate served as the cell matrix material. After establishing the right laser processing parameters, we proved that laser printing did not negatively influence MSC behavior and differentiation into osteogenic and chondrogenic lineages. As a highlight of this work, we generated 3D constructs of nondifferentiated and predifferentiated MSCs embedded in the matrix material, which could be differentiated toward bone and cartilage tissue grafts. The differentiated 3D cell constructs kept their predefined shape even after several weeks in culture. By removal of the matrix material after 2 weeks in culture, autologous 3D tissue grafts were obtained. These results open possibilities for the realization of complex multi-cellular tissue grafts that could be transplanted in vivo. Moreover, laser-engineered 3D autologous stem-cell constructs can serve as an in vitro model of stem cell niches.
Materials and Methods
Material preparation
Materials
Chemicals were obtained from Sigma-Aldrich (Deisenhofen, Germany), unless otherwise noted. Cell culture media and supplements were from Biochrom (Berlin, Germany). Cell culture flasks and plates were from TPP-Techno Plastic Products AG (Trasadingen, Switzerland). Other plastic wares were from Corning (New York). Primer sequences are given in Supplemental Materials and Methods (available online at
Preparation of the hydrogel
For all conducted printing experiments a hydrogel consisting of alginate and ethylenediaminetetraacetic acid (EDTA) blood plasma obtained from the same cell donor was used. For cell suspensions 4 wt% alginate was dissolved in 0.15 M NaCl solution and sterilized by filtration with a 0.8 μm (pore size) filter. This solution was mixed 1:1 (v/v) with filtered blood plasma, resulting in 2 wt% alginate. The hydrogel for the collector slide was adjusted in the same way but with less alginate concentration (1 wt% alginate).
Preparation of cell-hydrogel layer on the donor slide
The cultivated MSCs were trypsinized and resuspended in a certain volume of cell medium. Afterward, the cell concentration was determined with a cell counter (Beckman Coulter™). The cells were centrifuged at 300 g for 5 min and the supernatant was removed. The cell pellet was resuspended in 45 μL of 2 wt% alginate. The obtained cell suspension was pipetted on the gold-coated donor slide. This square glass plate with a size of 26 mm and a thickness of 1 mm was coated with a 55–60-nm-thick gold layer using plasma-enhanced sputter deposition. The cell suspension was dispersed on the gold surface with a blade coater to form a homogenous layer of ~65 μm thickness. Then, the cell-coated donor slide was fixed in a metal frame and mounted upside-down in the setup.
Preparation of hydrogel layer on the collector slide
The collector slide, a square glass plate with a size of 25.4 and 1 mm thickness, was sterilized in an ethanol bath, cleaned with isopropanol, and then irradiated with UV-C for 5 h. Afterward, the collector slide was coated with 60 μL of 1 wt% alginate hydrogel and subsequently wetted with a 0.1 M CaCl2 solution, resulting in a layer thickness of ~90 μm. After gelation for several minutes, the coated collector slide was fixed in a metal frame and mounted in the setup facing the cell-coated donor slide in close proximity (500 μm).
Printing setup
Bioprinting setup based on LIFT
A detailed description of the laser printing setup based on LIFT (see Fig. 1A) has previously been published. 15 Briefly, two coplanar glass slides are used. The upper one, referred to as donor slide, is covered underneath with a laser-absorbing gold layer and, subsequently, a layer of cell containing material to be transferred. Laser pulses are focused through the glass slide onto the gold layer, which is evaporated locally at the focal point. A high gas pressure is generated that propels the subjacent cell containing material toward the lower glass slide, referred to as collector slide. All material transfer experiments in this study are carried out under normal air conditions.
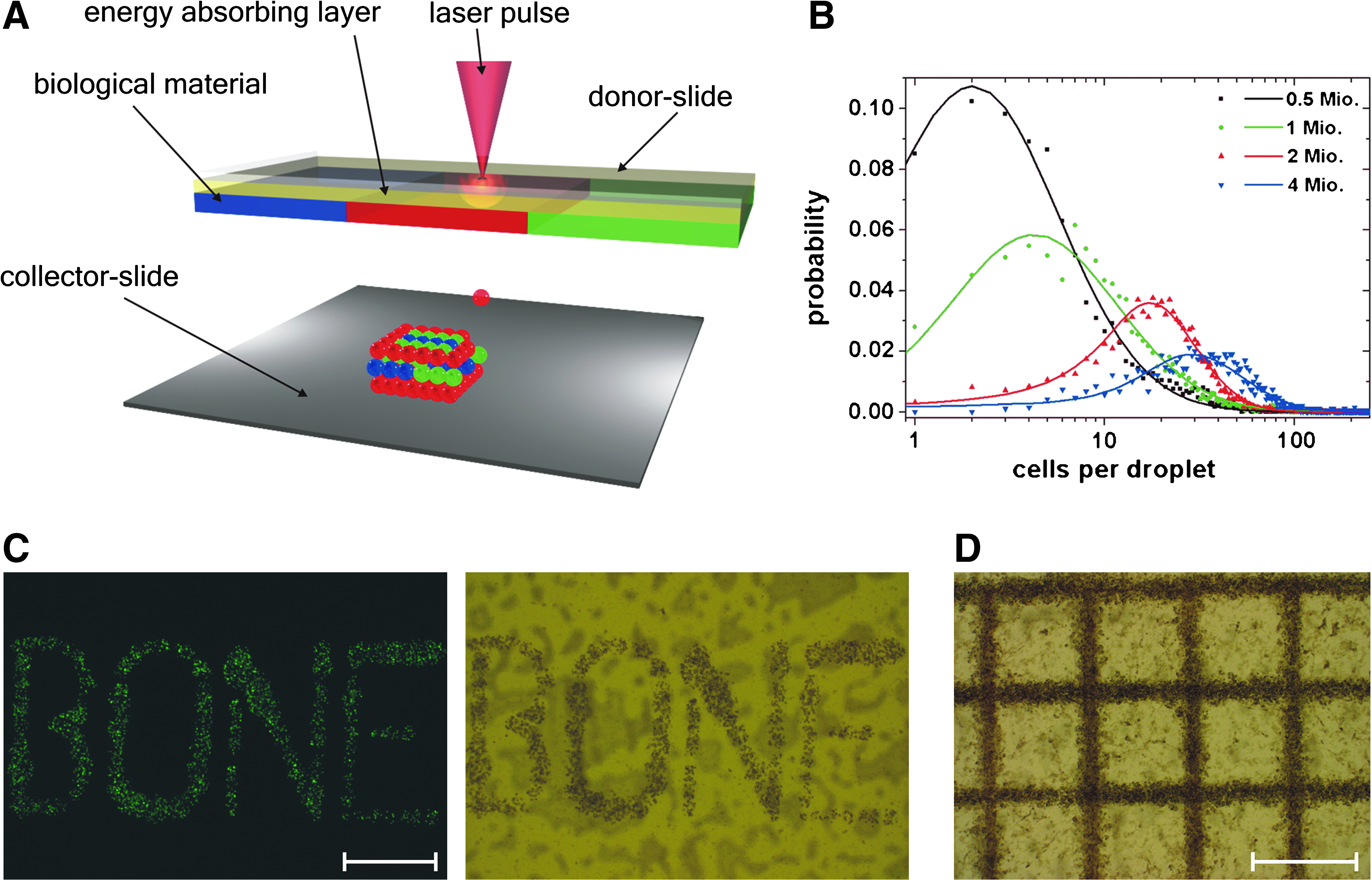
Laser printing setup, variation of cell density, and printed stem cell patterns.
The applied laser is an Nd:YAG-laser (DIVA II; Thales Laser, Orsay, France) with 1064 nm wavelength, a pulse duration of 10 ns (FWHM), and 20 Hz repetition rate. Laser pulses are focused with a 60 mm achromatic lens, producing an ablation spot size of 40 μm in diameter. Adapted to the cell/hydrogel layer thickness of about 65 μm, the laser fluence is set to 2.15 J cm2 and measured continuously during the transfer process.
The carrier for the donor slide allows quick replacement of donor slides coated with different cell-containing materials. The collector slide is assembled on a movable stage to provide controlled positioning with respect to the donor slide. The gap between the donor and collector slides is set to 500 μm with an accuracy of 5 μm, which is monitored by a camera.
Isolation, culturing, differentiation, and cell handling after printing
Isolation and culturing of MSCs
Porcine bone marrow-derived MSCs were isolated from the femur and tibia bones of domestic pigs (6- to 12-month-old), obtained from a local abattoir. The bone marrow was flushed out the bones by a 10 mL syringe and placed in 40 mL complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% fetal bovine serum, 100 U mL−1 of penicillin, and 100 μg mL−1 streptomycin (complete DMEM), and centrifuged at 400 g for 10 min to pellet the cells and remove the fat layer. Cell pellets were resuspended in 10 mL of red blood cells lysing solution reagent (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA, pH 7.4) to remove erythrocytes, 40 mL of complete DMEM was added, and the cells were pelleted at 400 g for 10 min. The MSCs containing pellets were resuspended in complete DMEM, and isolated cells were seeded at a density of 5 × 104 nucleated cells per cm2 into 75 cm2 culture flasks. The cells were cultured in complete DMEM at 37°C in a humidified atmosphere containing 95% air and 5% CO2. MSCs were purified by plastic adherence; nonadherent cells were removed by medium exchange after 2 days. Medium was changed twice weekly. After 10–14 days in primary culture, cells became confluent and were detached with 0.05% trypsin/0.02% EDTA for 5 min at 37°C, and subsequently replated at 5 × 103 cells per cm2 for continued passaging. MSCs from each passage were kept in liquid nitrogen for long-term storage. For the laser printing experiments, MSCs at passage 3–8 were used.
Osteogenic differentiation
About 7.5 × 103 cells per cm2 were seeded on 24-well plates and cultured for 3 weeks in DMEM supplemented with 15% fetal bovine serum, 0.1 μM dexamethasone, 10 mM β-glycerophosphate, 1% ITS + Premix (BD Biosciences, Heidelberg, Germany), and 50 μM ascorbate-2-phosphate. Medium changes were performed twice weekly. To quantify osteogenic differentiation, alkaline phosphatase (ALP) activity and calcium mineralization were measured at different times.
Chondrogenic differentiation
About 2 × 105 cells were pelleted by centrifugation to the bottom of a 15-mL conical polypropylene tube. The pelleted micromass was cultured for 3 weeks in DMEM supplemented with 0.1 μM dexamethasone, 1 mM sodium pyruvate, 0.17 mM ascorbate-2-phosphate, 0.35 mM proline, 10 ng mL−1 transforming growth factor-beta 3 (RELIATech, Braunschweig, Germany), and 1% ITS + Premix. Medium changes were performed twice weekly, and chondrogenesis was assessed by Alcian blue staining and by immunohistochemical analysis for aggrecan and collagen type II.
Isolation of chondrocytes
Articular cartilage samples from the metatarsal joints 6- to 12-month-old pigs were cut into small pieces, followed by enzymatic digestion with 2.5 mg mL−1 collagenase A in complete DMEM for 18 h at 37°C. The resulting suspension was filtered through a 100 μm nylon mesh filter and centrifuged at 1000 g for 10 min. The cell pellet was washed twice in complete DMEM and the isolated chondrocytes were seeded at a density of 8.5 × 104 nucleated cells per cm into 175 cm2 culture flasks. The cells were cultured in complete DMEM at 37°C in a humidified atmosphere containing 95% air and 5% CO2. First-passage chondrocytes were used for the laser printing experiments.
Cell handling after printing
For all reported quantitative analysis, the cells were treated in the following way: An adequate amount of cells was printed. Directly after the laser printing process, the printed cells were rinsed off the collector slide with additional medium into a tube, counted, and distributed to the corresponding well plates for further analysis. The cells that remained on the donor slide underwent the same procedure—except the LIFT transfer—and were collected as control cells.
For all reported analysis of the 3D grid pattern, the cells were handled as followed: Directly after printing of the MSC grid pattern, the pattern on the collector slide was kept in a 0.1M CaCl2 solution for 3 min. Afterward, the collector slide with the jellied MSC pattern was placed in a Petri dish filled with additional medium and kept in an incubator for further analysis.
Additional and supplemental methods
Estimation of the grid volume
Frozen sections (7 μm) from 30 independent grid patterns, generated under the same conditions (laser fluence of 2.15 J cm2, hydrogel thickness (donor slide) of 65 μm, and 2 × 106 initial cell density), were measured. The recorded grid height was 300 ± 40 μm (at a line width of 150 ± 35 μm), which results in an averaged grid volume of 4.1 × 109 μm3.
Supplemental materials and methods
Details of staining, proliferation, DNA, reverse-transcription-polymerase chain reaction (RT-PCR), and quantitative absorption analysis are summarized in the Supplemental Materials and Methods.
Statistical analysis
Statistical analyses of all data for comparison were carried out using Student's unpaired two-sample t-test. p-Values of time series data are given as the minimum of the p-values obtained at different points in time. Values are reported as means ± standard error of the mean if not otherwise noted.
Results
Parameter study for laser bioprinting with LIFT
We investigated probability distributions of the number of cells in printed hydrogel droplets depending on the initial cell density at a constant laser fluence of 2.15 J cm2 and fixed layer thickness (65 μm) of the cell containing hydrogel at the donor slide (Fig. 1B). To evaluate the amount of cells per printed droplet, Calcein AM stained MSCs were suspended in the hydrogel. Hydrogel droplets were printed in a square array of 31 × 31 droplets with a 600 μm spacing between them. Afterward, the number of cells in each droplet was counted; the obtained results are shown in Figure 1B. As one can see, by changing the initial cell density on the donor slide, different numbers of cells per droplet can be printed. Further, by changing the laser fluence it is also possible to vary the droplet size and the number of transferred cells; for example, an initial cell density of 2 × 106 cells results at a laser fluence of 0.93 J cm2 in 3 ± 2 cells per droplet (mean ± SD [standard deviation]) and 1.2 J cm2 in 10 ± 7 cells per droplet (mean ± SD; n = 600 cell containing droplets per laser fluence were evaluated). Thus, using the donor slide with the same initial cell density, different cell amounts per single shot can be transferred (Fig. 1C, D). The grid pattern shown in Figure 1D, with a total size of 6 × 6 mm, was used as a standard structure in all laser printing experiments reported below. Using the data shown in Figure 1B (for 2 × 106 cells) as well as the averaged grid volume, the printed cell density in the grid pattern can be estimated as 46,000 ± 23,000 cells μL−1 (mean ± SD). Such high cell densities allow accelerated in vitro tissue maturation. 18
Investigation of cell impairments after laser printing
To determine whether no cell impairments are induced by the laser printing process via cell acceleration and impact, as well as possible cell damage due to laser radiation, the proliferation ability of the printed cells over 5 days has been investigated. The obtained results for printed and nonprinted (control) MSCs are shown in Figure 2A, and no difference in the proliferation of printed and control cells could be observed (p ≥ 0.16).
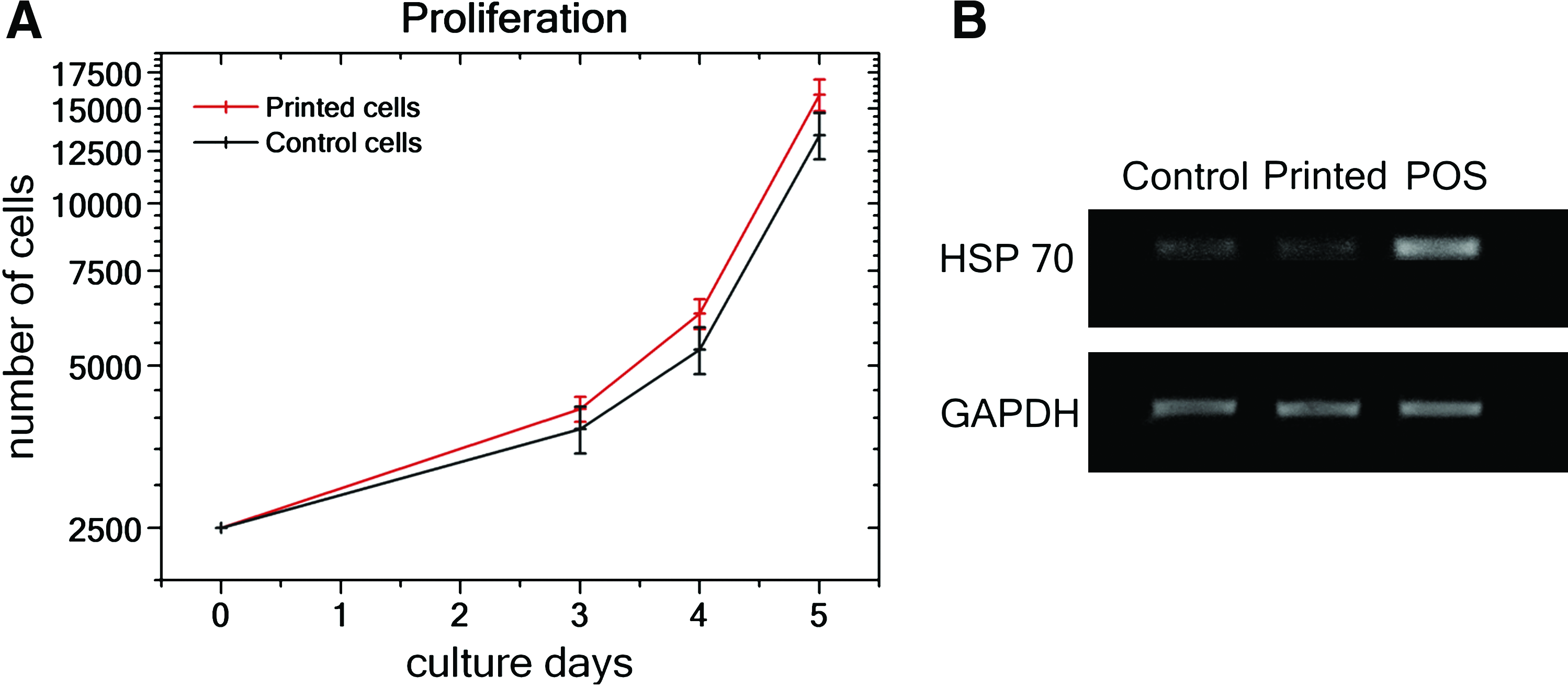
Statistical assessment of MSCs' proliferation ability and their heat-shock gene expression after laser printing.
However, laser printing may still damage the cells with respect to protein denaturation by laser heating, stress, and DNA damage effects during cell acceleration and impact. By the method of RT-PCR, gene expression of the heat-shock protein (HSP-70) by laser printed and control cells were compared. The same, negligibly small level of HSP-70 gene expression (Fig. 2B) for both printed and nonprinted control cells has been recorded in contrast to the positive control, where HSP expression was induced by keeping MSCs at 45°C for 15 min. Further, the genotoxicity was investigated with the comet assay method where the tailmoment characterizes the amount of damaged DNA. It was shown that tailmoments of the laser-printed cells (1.18 ± 0.13) are similar (p = 0.45) to their nonprinted control cells (1.34 ± 0.15).
Examination of differentiation potential into multiple lineages after laser printing
To assess possible alterations in the differentiation potential of stem cells, laser-printed and nonprinted control MSCs were characterized by their ability to differentiate into multiple lineages, such as osteogenic and chondrogenic.
The osteogenic differentiation of printed and control MSCs was evaluated by measuring the ALP activity and the amount of accumulated calcium over several weeks. Bone-specific ALP activity detected after 3 days in culture, expressed as p-nitrophenol nmol min−1 dish−1, is shown in Figure 3A. No significant difference between printed and nonprinted control cells (p ≥ 0.36) was observed. The subsequent calcium accumulation was evident after 1 week (Fig. 3B), increased over time, and showed no difference between printed and nonprinted control cells (p ≥ 0.13). Figure 3A and B demonstrates bone-specific curve progression 19 proving the osteogenic potential of printed MSCs. These findings were confirmed by the RT-PCR method (Fig. 3C, D). In contrast to unstimulated stem cells (MSC), similar gene expression levels of osteocalcin (OC) and ALP from printed and nonprinted control MSCs after 7 and 21 days in osteogenic culture were observed (Fig. 3C). OC is used as a biochemical marker for the bone formation process. Higher OC levels correspond to higher bone mineral density (calcium). The decline in ALP activity between 14 and 21 days correlates with the increased calcium accumulation.
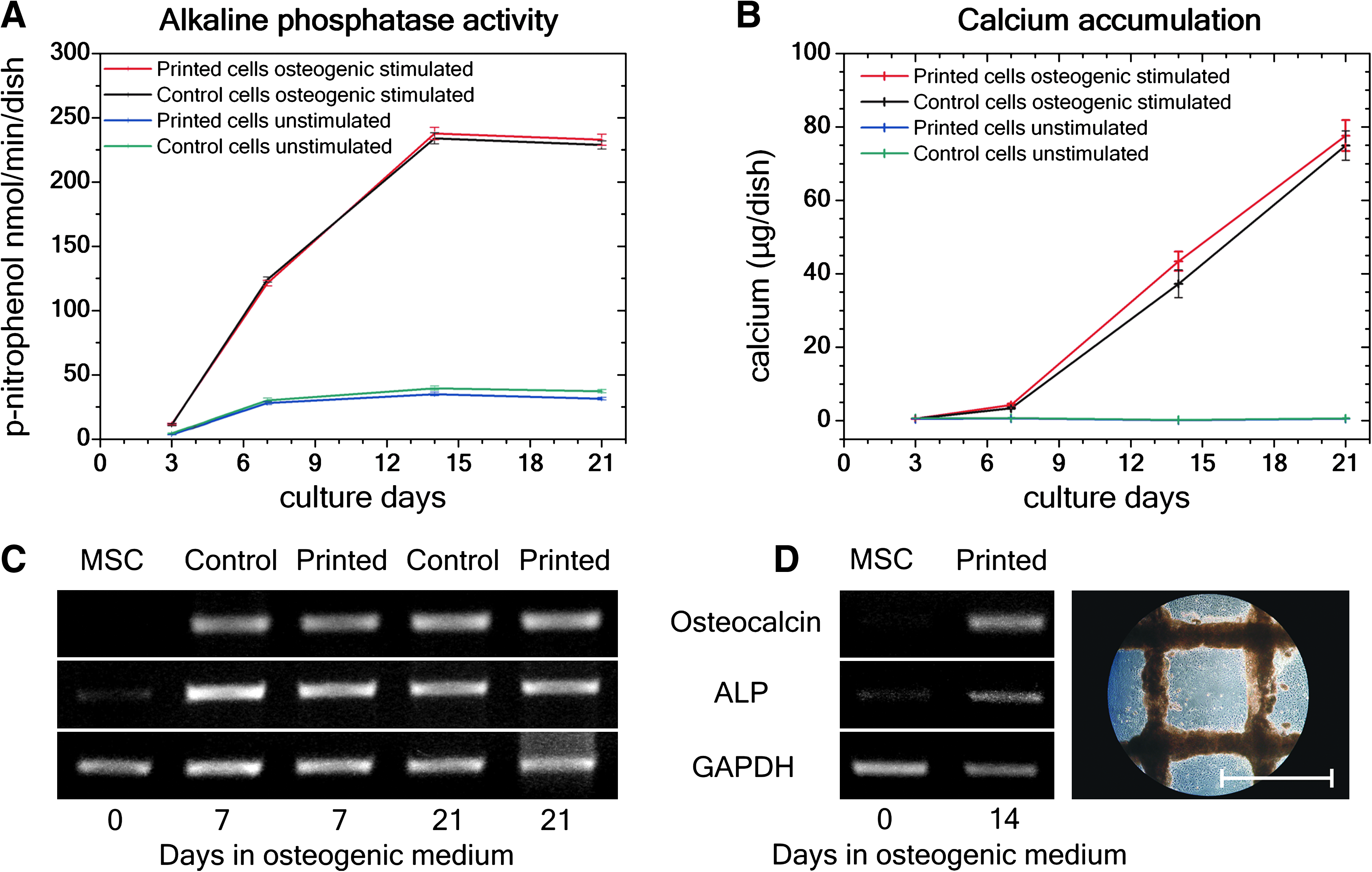
Osteogenic differentiation potential of printed and nonprinted control MSCs.
To promote chondrogenic differentiation for qualitative and quantitative analyses, pelleted micromasses from printed and nonprinted control MSCs were formed and cultured under chondrogenic conditions over several weeks. Comparable presences of type II collagen and aggrecan were detected by immunofluorescence staining as well as Alcian blue staining (indicator of sulfated glycosaminoglycan [sGAG]-rich extracellular matrix) from printed and nonprinted control cells after chondrogenic culture conditions for 21 days (Fig. 4A). Also the sGAG assay (Fig. 4B) demonstrated the presence of sGAG after 21 days in culture and no differences between printed and nonprinted control cells (p ≥ 0.92). After 21 days, similar gene expression levels of type II collagen and aggrecan were identified by printed and nonprinted control cells, in contrast to unstimulated stem cells (MSC). Moreover, printed cells possessed gene expression intensities comparable to that of primary chondrocytes (Fig. 4C, D).
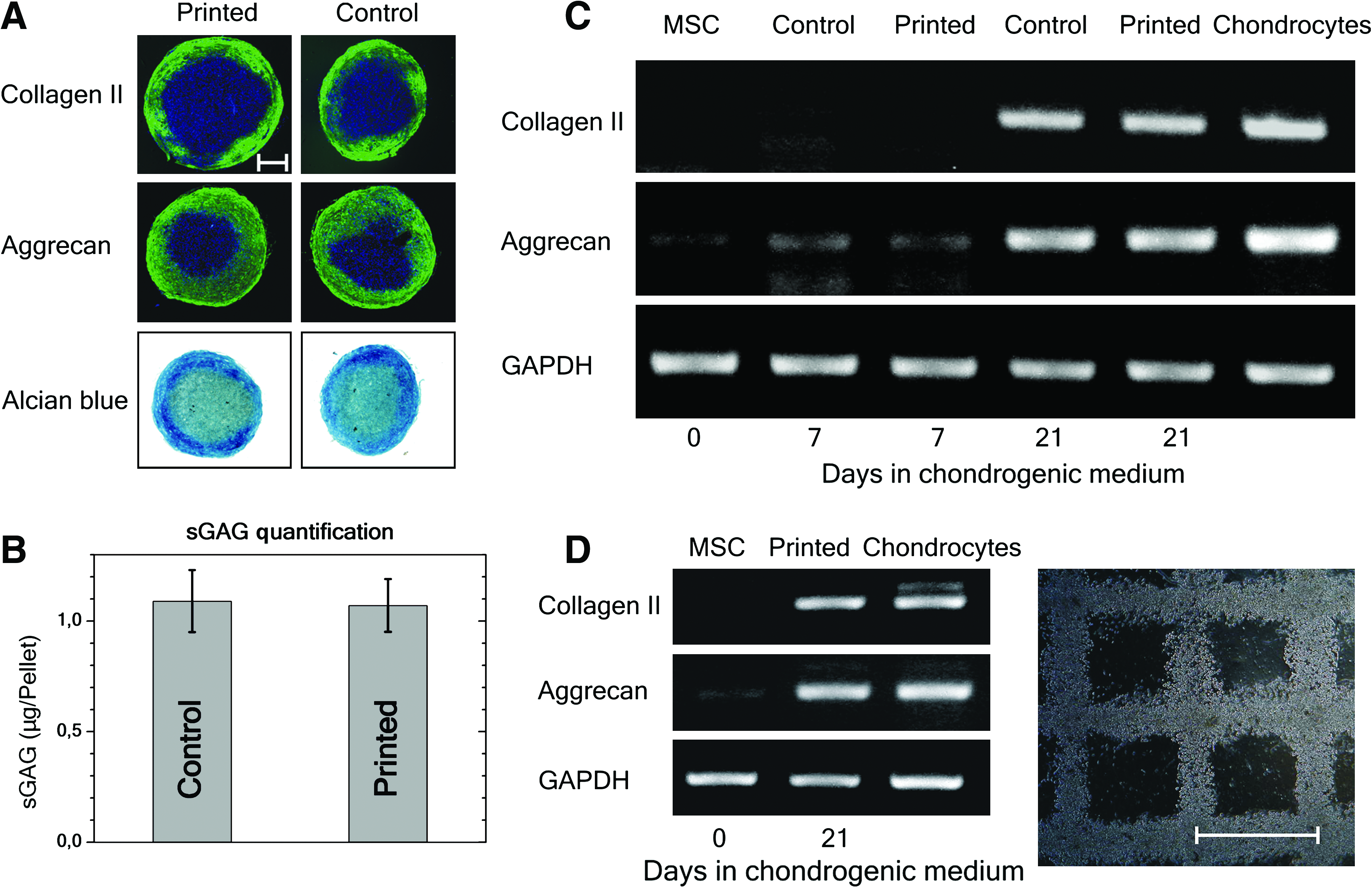
Chondrogenic differentiation potential of printed and nonprinted control MSCs.
Evaluation of the printed 3D patterns
After establishing the right processing parameters for printing MSCs and analyzing possible effects on MSCs induced by laser printing, a 3D grid pattern embedded in alginate hydrogel was generated (Fig. 5A). First, the differentiation potential into osteogenic and chondrogenic lineages was investigated by cultivating the 3D grid of MSCs under certain culture conditions. Similarly to the results mentioned above, the differentiation continued. After 2 weeks under osteogenic conditions, OC and ALP gene expression levels were measured (Fig. 3D). Interestingly, the accumulation of calcium phosphates correlated with the printed cell density. An increased accumulation of calcium phosphates, induced by higher cell densities at intersection points of horizontal and vertical lines, is shown in Figure 5B for osteogenic differentiation of printed MSCs. Moreover, the gene expression levels of type II collagen and aggrecan were documented from MSCs cultivated for 21 days under chondrogenic conditions in the 3D grid pattern (Fig. 4D). Thereby, the control MSCs, which were printed in the 3D grid pattern and kept under normal culture conditions, showed an increase of aggrecan expression, indicating the chondrogenic differentiation direction (see Supplemental Fig. S1, available online at

Printed grafts after several weeks under chondrogenic and osteogenic culture conditions.
Second, the viability of the printed 3D grafts was evaluated by Live/Dead®-assay (Invitrogen) (Fig. 5A). We observed a smaller quantity of dead cells and higher cell proliferation rates by keeping the printed grafts in normal culture medium for about 24 h before applying lineage-specific culture conditions. To increase the number of dead cells and investigate their influence on the printed graft, MSCs were differentiated before printing in osteogenic lineage (leading to partial cell apoptosis during calcification
20
). After 7 days the differentiation was stopped and the predifferentiated MSCs were printed in a grid pattern. Then, the osteogenic differentiation was continued. In this case, the live/dead-staining (see Supplemental Fig. S2, available online at
Discussion
Tissue functions are dependent on the arrangement, connections, and interactions of their composed cells and matrix elements in two and three dimensions. To get more insights into tissue behavior, reconstruction, and regeneration, an in vitro model is necessary that mimics the natural architecture of the tissue. This ideal model should be also conferrable to stem cells, since stem cell fate is regulated by an appropriate 3D environment called niche. 21 Among all approaches to create a defined 3D construct of cells, computer-controlled laser printing techniques as an orifice-free procedure was described to have additional benefits. 12 We previously demonstrated that a precise arrangement of different cell types in 2D pattern is possible. Further, we found that the stem cells were not negatively affected by the laser printing procedure. 15
In this study, we focused on the biofabrication of 3D autologous tissue grafts consisting of MSCs. MSCs are found in many adult tissues and represent an attractive cell pool due to their self-renewing ability, high proliferation capacity, and mesodermal differentiation potential. 22 Since MSCs are expected to regenerate many tissues like bone and cartilage, 23 these cells are very attractive for tissue engineering. Before producing 3D stem-cell constructs, we explored the required laser processing parameters and whether the laser printing procedure has any effect on the impairment and on MSC differentiation into multiple lineages. These factors are the major requirements for all bioprinting techniques and are essential for the successful fabrication of functioning 3D stem cell constructs.
Another critical parameter important for tissue formation and stem cell differentiation is the initial cell density.3,4,18 By changing the laser pulse energy, we were able to control the printed cell density in a precise and reproducible manner. It is well known that the promotion of chondrogenesis 24 requires high cell densities, which now can easily be achieved by laser printing.
Further, we analyzed whether the laser printing procedure induces cell impairments. We demonstrated that the laser printing process does not cause negative effects on MSC proliferation. Moreover, no changes in the immunophenotype such as stress protein (HSP-70) expression or DNA strand breaks were induced. Comparable results have already been described by us for other cell types. 15 After laser printing the differentiation potential of MSCs into multiple lineages was investigated. In these experiments no spontaneous differentiation of MSCs that may occur due to mechanical forces during the printing process was observed. Several experiments were performed to determine osteogenic and chondrogenic characteristics. The expression levels of OC and ALP activity and the observed calcium accumulation proved that the printed stem cells differentiated toward bone. The chondrogenic differentiation was confirmed by mRNA expression of type II collagen and aggrecan as well as the quantification of sGAG, which can be found in a large extent in the extracellular matrix of cartilage. In a sum, we demonstrated that laser printing does not induce any cell impairment or negatively affect MSC differentiation. In this manner, we were able to exclude all possible side effects that may have been caused by the laser printing process.
The big challenge in our work was the biofabrication of functioning 3D MSC grafts. As shown in Figure 5A, a grid pattern out of MSCs forming close cell-to-cell contacts could be generated by laser printing. It needs to be mentioned that any desired and controllable 3D arrangement of the cells is possible. Embedded in the matrix material, the grafts kept their predefined structure. The printed grid patterns were successfully differentiated over several weeks into bone and cartilage tissue grafts. This result indicates that the exchange of nutrients and soluble factors within the 3D graft was high enough to promote cellular behavior. However, further experiments are needed to explore in detail oxygen exchange, and the like, which was described to play a critical parameter in 3D tissue substitutes. 2 Further, the possibility of computer-controlled assembly of high density cell grafts offers many advantages for chondrogenic tissue engineering compared to current procedures like centrifugal 25 or molding techniques, 18 which possess limited shape-forming abilities.
Cartilage is an attractive tissue for bioprinting approaches, since it is an avascular and aneural tissue, and the development of new clinical therapies is needed due to limited self renewal possibilities from the adult cartilage. Moreover, cartilage is characterized by an organized zonal cell-matrix distribution and density, which can be duplicated by a layer-by-layer approach. Similar to the presented grid pattern (height of 300 ± 40 μm), different grid section with adjustable cell densities and/or phases of differentiation can be printed on top of each other with a CaCl2 layer printed in between to induce gelification. The final height of the structure mainly depends on the mechanical properties of the applied hydrogel, which have to be adjusted for each cell type. However, more fundamental studies are needed to figure out how 3D stem cell constructs of various sizes interact with different cell densities and cell–cell as well as cell–zone interactions in a jellied hydrogel environment.
We also demonstrated that an increase in the initial cell density improved MSC differentiation. This coincides with the results already observed by other groups.3,4 Interestingly, laser-printed MSCs, which were not cultivated under chondrogenic conditions, also presented an increase in aggrecan expression level in 3D arrangement. Even though no expression of collagen type II, which is another proof for chondrocytes, was observed, this finding indicates that MSCs in 3D grafts already change toward the chondrogenic direction. Further studies are needed to clarify this topic, as some studies report that cell behavior in 3D differs significantly from 2D.2,26 The correlation between 3D environment and differentiation direction is of huge interest in stem cell biology with respect to stem cell niches. For that research purposes, the laser printing technique seems to be a very promising instrument.
Moreover, we found that predifferentiated MSCs survived the complete printing procedure, kept their ability to proliferate, and continued their differentiation after laser printing into osteogenic lineage. This result opens new possibilities for future investigations of 3D stem cell constructs with variable differentiation levels and controlled cell densities. In this manner, stem cell interactions and influences could be documented. Another advantage of our method is the demonstrated ability to form scaffold-free 3D grafts, which is an alternative approach to scaffold-based 3D tissue engineering.
Footnotes
Acknowledgments
This work was supported by funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for the Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy) and Sonderforschungsbereich Transregio 37 (Mikro- und Nanosysteme in der Medizin).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.