
Abstract
Introduction:
Acute liver failure has high mortality due to donor organ shortages. A bioartificial liver could “bridge the gap” to transplant or spontaneous recovery. Alginate encapsulation of HepG2 cells enables cell spheroid formation, thus providing sufficient functional biomass. Cryopreservation (CryoP) of these spheroids would allow an off-the-shelf capability for unpredictable emergency use. Cell death during CryoP often results from intracellular ice formation, after supercooling. An ice nucleating agent (INA), crystalline cholesterol, was trialled to reduce supercooling and subsequent cryoinjury.
Materials and Methods:
Spheroids were cooled in a controlled rate freezer in 12% dimethylsulfoxide/Celsior +/− INA, and sample temperatures were recorded throughout. Viability was assessed using fluorescent staining with image analysis, cell number by nuclei count, function using assays to detect liver-specific protein synthesis and secretion, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reduction, and broad-spectrum cytochrome P450 activity.
Results:
Spheroids cryopreserved without INA displayed latent cryoinjury in the first 6 h after thawing. INA reduced supercooling during CryoP and also latent cryoinjury. Cell numbers, viability, and function as measured over 72 h post-thaw were all improved when INA was present during CryoP.
Introduction
BALs comprise a cellular component, and the concept is that the large range of functions (metabolic, synthetic, and detoxification) performed by the liver may be replaced by hepatocytes contained within an extracorporeal circuit against which ALF patient plasma could be perfused. Primary human hepatocytes are considered the gold standard but are unavailable in sufficient numbers for BAL, as any available organs would be utilized for OLT. Nonhuman hepatocytes, such as porcine, carry a risk of zoonoses 8 but have been used in previous clinical trials, despite the persistent risk of porcine endogenous retrovirus transmission. 9 An alternative cell source is human-derived cell lines such as HepG2s, used here, or their subclone, C3A, which have been utilized in a clinical trial albeit with limited success. 10
Monolayer cultures of neither C3A nor HepG2 cells are sufficiently functional to treat ALF but can be available in sufficient number (∼7×1010: equivalent to 30% of a human liver), and the function of HepG2s is significantly upregulated by alginate encapsulation. 8–9 days after encapsulation, three dimensional (3D) encapsulated liver spheroids (ELS) with a tissue-like structure form, 11 which retain function in liver failure plasma, as would be required. 12 Additionally, 3D cultures mean greater cell densities are achieved, thus allowing for a more compact BAL and increasing ease of use within a clinical setting.
Since onset of ALF is unpredictable, an “off-the-shelf” BAL is desirable. In our current system, sufficient functional biomass is achieved after 8–9 days culture at significant cost; it would be disadvantageously expensive to continually culture ELS for the eventuality of ALF onset. By developing a cryopreservation (CryoP) protocol, performance-competent ELS could be stored at low temperatures available for immediate recovery. Recovery of cryopreserved ELS should be characterized to ensure that functional ELS can be provided in the crucial early time period after ALF diagnosis. Sufficient immediate recovery of ELS at early time points (i.e., to 48 h) is crucial to the success of any BAL and has not been previously demonstrated in other existing BALs.
Successful CryoP of cell suspensions (including HepG2s) is possible when appropriate cooling rates and cryoprotectants (CPAs) are used. The optimal cooling rate of each cell is unique to type and depends on several factors including the cells' permeability to water. 13 At rapid cooling rates, sufficient cell dehydration cannot occur, and intracellular ice formation (IIF) is likely.14,15 Conversely, at slow cooling rates, cells may be exposed to high solute concentrations that may disrupt the chemical equilibrium of cells. 16 However, CryoP of multi-cellular complexes such as tissues, organs, and ELS is more complex and recoveries are typically worse. 17 This may be due to an increased susceptibility of multi-cellular complexes to the effects of ice, both intra and intercellular. Multi-cellular complexes are more prone to IIF, as water transport characteristics differ from single cell suspensions and ice may propagate between cells. 18 Similarly, intercellular ice is problematic in multi-cellular complexes where function often depends on maintenance of tissue structure. For example, formation of intercellular ice crystals during CryoP of skin distorts structure, resulting in poor recovery. 19
IIF is likely to occur after supercooling (when a liquid cools to below its equilibrium freezing point), as this disrupts cooling control. 8 Although supercooling can be reduced by inducing nucleation close to the equilibrium freezing point of the mixture using “seeding.” 20 this approach does not lend itself to large volume samples, as required here. Alternatively, heterogeneous INA could be used, which facilitate the liquid-to-crystalline transition of water by “ordering” water molecules.21,22 INA have been previously identified, including silver iodide 23 and microbial proteins, 24 and both have been employed as INA during CryoP. Microbial INA were not considered suitable for BAL because of concerns over their suitability in products destined for human therapy. Cholesterol crystals (CholC) have also been identified as INA 21 but have not, to our knowledge, previously been utilized during CryoP of multi-cellular complexes such as ELS.
Our study, therefore, aimed at characterizing the chosen optimized ELS CryoP protocol in terms of ice nucleation and functional recoveries (to 72 h) and investigating whether application of CholC could improve post-thaw outcomes.
Materials and Methods
Chemicals
Unless otherwise stated, all chemicals were sourced from Sigma.
HepG2 monolayer cell culture
HepG2 cells from the ECACC were maintained in monolayer culture in modified MEM-alpha medium (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone), 50 U/mL penicillin, and 50 μg/mL streptomycin (Lonza) replenished every 2–3 days. Cells were counted using a hemocytometer, and viability was assessed using trypan blue dye exclusion.
Alginate encapsulation
HepG2 monolayer cultures at 80% confluence (passage number 40–60) were trypsinized and resuspended in modified MEM-alpha medium at 1×106cells/mL. This cell suspension was diluted 1:1 with 2% alginate solution and filtered into a 50 mL syringe through a 50 μm filter to remove cell clumps. The syringe was attached to the syringe pump of an Inotech IER-20 encapsulator (Inotech), and cell-alginate mix was passed through a 200 um nozzle at 5 mL/min, 1295 Hz vibrational frequency to yield beads of diameter 475 μm +/−45 (n=50 +/− standard deviation [SD]). The droplets fell into a 0.204 M calcium chloride/0.15 M sodium chloride polymerization buffer and were stirred for 10 min. The beads were removed from polymerization buffer by filtration through a 200 μm mesh and washed using Dulbecco's modified Eagle's medium (Invitrogen) to remove any residual polymerization buffer.
Encapsulated liver spheroid culture
Beads were resuspended in culture medium at a ratio of 1:32 in 6-well plates, and medium was replenished every 2–3 days unless otherwise stated. At the time of CryoP, each alginate bead contained between 20 and 25 spheroids, each of which contained ∼20–25 cells.
Cryopreservation
250 μL beads were exposed to 1 mL CPA medium (12% w/v dimethyl sulfoxide in Celsior solution [SangStat]) +/−1.1 mg/mL CholC for 10 min at 4°C and transferred to 1.8 mL cryovials (Nunc) in 1.25 mL volumes. An R204 Cell Freezer controlled rate freezer (CRF) (Planer) was used to perform a predetermined protocol (0°C for 8 min, −2°C/min to −8°C, −35°C/min to −28°C, −2.5°C

Freezing program and temperature of samples during cooling. Freezing protocol (solid line) applied was: 0°C for 8 min, −2°C/min to −8°C, −35°C/min to −28°C, −2.5°C
Temperature measurements
Cooling profiles were recorded during CryoP using Type K thermocouples and logged using a TC-08 data logger (Pico Technology) in “dummy” cryovials containing 1.25 mL of CPA medium, n=3 separate runs per condition +/− range.
Determination of equilibrium freezing point
The equilibrium freezing point of the CPA media was determined using modulated differential scanning calorimetry (DSC) with a Q2000 calorimeter (TA Instruments). About 80 μL aliquots were sealed inside high-volume stainless steel pans, compared with an empty reference pan. Analysis was at 5°C/min with modulation of 1°C/min. Data were analyzed using Universal Analysis software (TA Instruments), n=3 separate runs +/− range.
Quantification of cell numbers recovered from beads
Beads were washed twice with Hank's balanced salt solution, dissolved using 16 mM EDTA/0.15 M NaCl, and centrifuged at 13,000 rcf for 5 min. Supernatant was removed, and pellets were resuspended in phosphate buffered saline. Cell concentrations were measured using the Nucleoview System (Sartorius Stedim).
Cell membrane viability assay
About 250 μL beads were washed with 1 mL PBS/Ca2+/Mg2+ (Lonza) and resuspended in 500 μL PBS/Ca2+/Mg2+. Beads were incubated with 10 μL of 1 mg/mL fluorescein diacetate (FDA/dimethyl sulfoxide) and 20 μL of 1 mg/mL propidium iodide (PI/water) for 90 s at room temperature. ELS were washed twice with PBS/Ca2+/Mg2+ and loaded onto a microscope slide. Images bisecting the center of the beads were captured using Lucia Imaging Software with a DX1200 camera at exposures of 64 ms (FDA) and 250 ms (PI), ×4 magnification. Each of the time membrane viabilities were assessed, five separate fields were captured. Quantification was performed using Lucia imaging software, which assessed the density of each stain within an individual field, and viability was calculated as described below:
Liver-specific protein synthesis and secretion
Albumin, alpha-1-fetoprotein (AFP), alpha-1-antitrypsin (AAT), and fibrinogen synthesis and secretion over 24 h were quantified from 5 mL conditioned medium samples by sandwich enzyme-linked-immunosorbent-assay (ELISA) using species-specific capture antibodies (Abcam) and horseradish peroxidase-linked detection antibodies (Abcam). Detection limits were 200 to 6.25 ng/mL. Optical density (492 nm) was measured (Anthos HTIII plate reader [Labtech]), and data were analyzed using Biolise software (Labtech). Results are normalized to viable cell number (determined from nuclei counts and quantitative image analysis).
Broad-spectrum CYP activity
Broad-spectrum cytochrome P450 (CYP) activity was assessed using 7-ethoxycoumarin de-ethylation (ECOD) assay. ELS were induced to increase CYP activity 26 using 20 μM indirubin 24 h before ECOD assay. ELS were incubated with 200 μM 7-ethoxycoumarin (substrate) under normal culture conditions, for 6 h before 7-hydroxycoumarin (product), was back-extracted using chloroform/0.01 M NaOH. Detection range was 50–0.8 μM 7-hydroxycoumarin. Fluorescence (370 nm excitation/450 nm emission) was measured (Fluoroskan Ascent fluorescence platereader) (Thermo Labsystems). Results are normalized to viable cell number (determined from nuclei counts and quantitative image analysis).
MTT assay
General oxidative metabolism in ELS was assessed by reduction of tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT). ELS were incubated with 0.75 mg/mL MTT under normal culture conditions for 3 h. ELS were removed from alginate as described earlier, supernatant was discarded, and crystals were dissolved by shaking for 30 min with 4 mM hydrochloric acidified-isopropanol. ELS were centrifuged once more, and optical density of supernatant (570 nm) was measured as described earlier. Results were normalized to total protein and assessed using bicinchoninic acid assay. 27
Experimental plan
Unfrozen control ELS were prepared by identical treatment to cryopreserved samples (i.e., exposure to CPA media on ice, transfer to cryovials, two washes with media, and return to culture) but without exposure to sub-zero temperatures. Previous experiments have determined that these processes alone do not affect ELS viability. All assays were performed at equivalent time points for unfrozen and cryopreserved ELS.
Statistics
Statistical analyses were performed by one-way ANOVA and posthoc student's t-test using Excel software. For viability, nuclei counts, albumin synthesis, and secretion experiments, four independent experiments were performed; and data are expressed as a fraction of the unfrozen control, shown as n=4 experiments +/−SD. For all other assays, a single, independent experiment was performed, and data are shown as a mean of five samples +/−SD.
Results
Throughout the results section, cell number, viability, and function are assessed between 0 and 72 h postwarming. For a BAL, recovery of cryopreserved ELS should be rapid and for these reasons, the earlier time points (to 48 h postwarming) may be considered most relevant.
Cooling profiles and nucleation during CryoP
CPA media temperatures within “dummy” cryovials were recorded during CryoP. For ease of interpretation, Figure 1 displays results from a typical cooling run; and temperature profiles of only two vials (one containing standard-, and one containing INA-modified-CPA-medium) and CRF chamber are shown. Both standard- and INA-modified-CPA-media temperature lagged slightly behind the chamber temperature. Nucleation in both vials is indicated by release of latent heat of crystallization and occurred at −22.1 +/−2.4°C in standard-CPA-medium and −8.5 +/−1.46°C in INA-modified-CPA-medium. The true equilibrium freezing point of CPA-medium was −4.5 +/−0.23°C as determined by DSC. Supercooling was calculated as the difference between mean nucleation and equilibrium freezing point temperatures and was, on average, 17.6°C for standard-CPA-medium, 4°C for INA-modified-CPA-medium.
Measurement of ELS membrane viability after CryoP
Membrane viability of ELS cryopreserved using standard-CPA-medium was assessed between 0 and 6 h (Fig. 2) and then at 24, 48, and 72 h after thawing for ELS cryopreserved in both standard and INA-modified-CPA-medium (Fig. 3). PI staining, indicating nonviable cells, increased gradually over the first 6 h in ELS cryopreserved in standard medium; membrane integrity was quantified using image analysis and fell from ∼80% immediately post-thaw to ∼50% after 6 h.
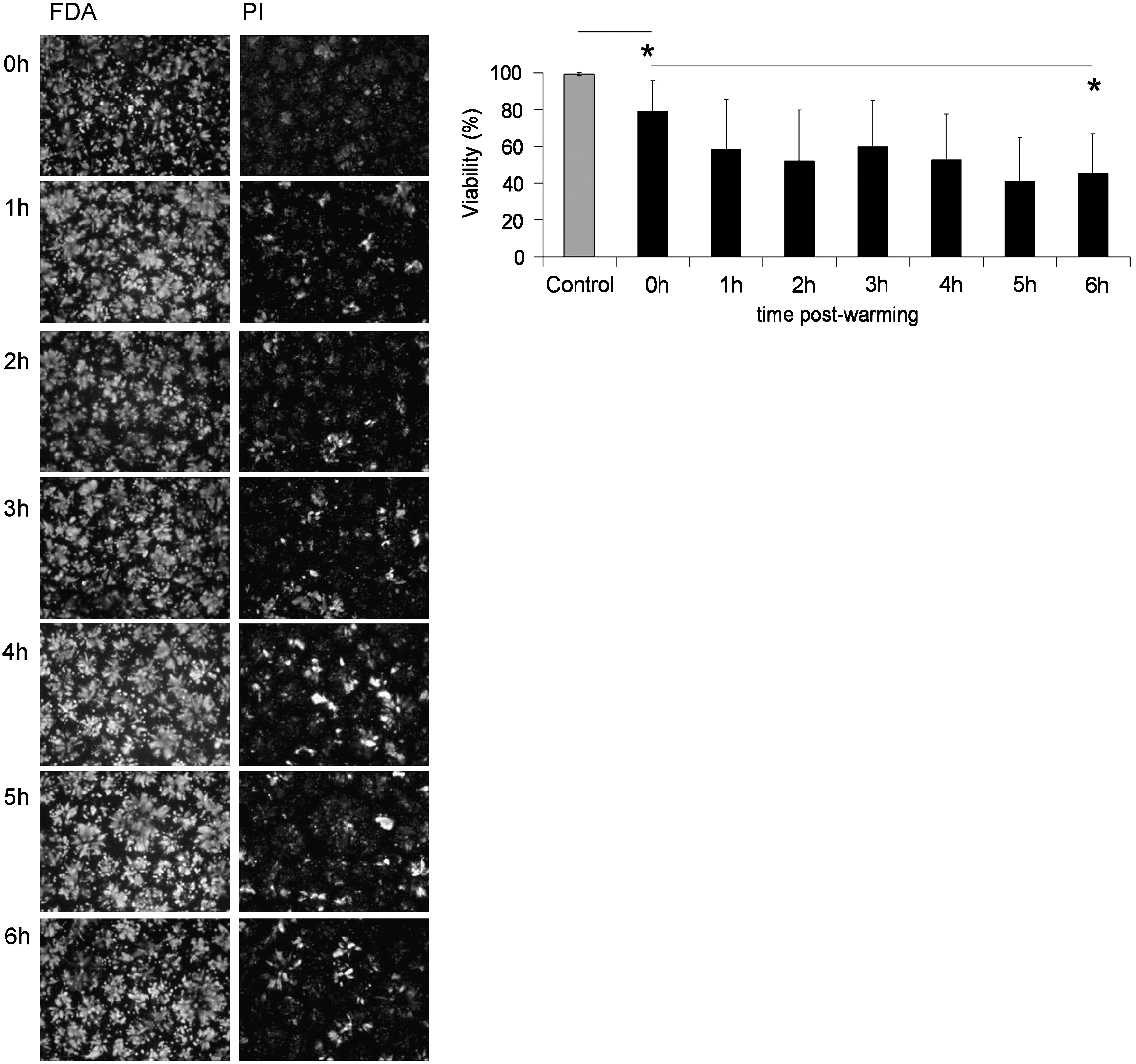
Viability in the first 6 h after CryoP. Images show FDA and PI-vital dye staining of ELS cryopreserved in standard-CPA-media to 6 h after CryoP. Viability was quantified as a percentage using image analysis software. Original magnification×4. Exposure times: 64 ms (FDA) and 250 ms (PI). Scale bar=1 mm. *p<0.05. CryoP, cryopreservation; FDA, fluorescein diacetate; PI, propidium iodide, ELS, encapsulated liver spheroids.
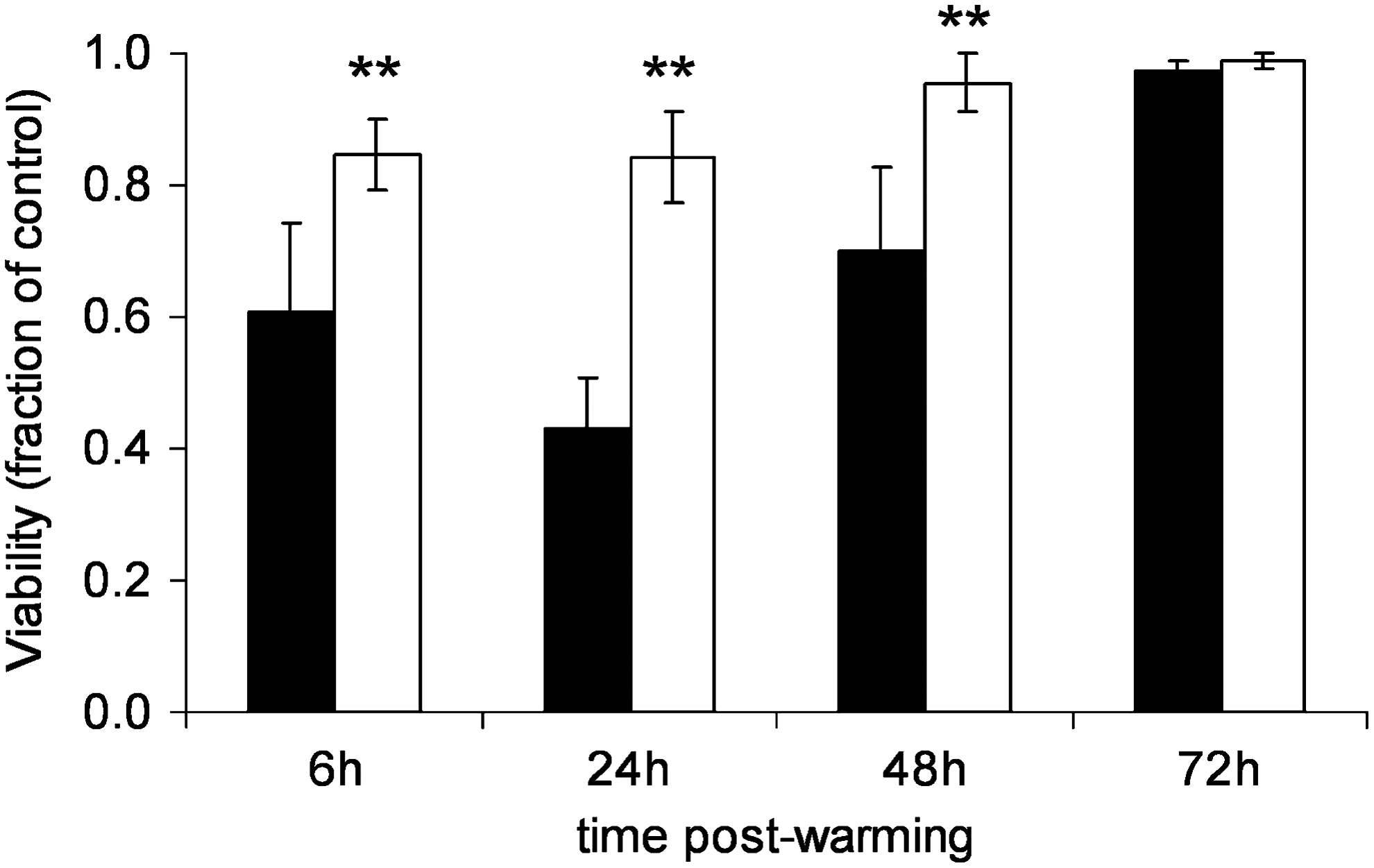
Effect of CholC on viability after CryoP. Viability of ELS cryopreserved either in standard-CPA-media (black bars) or in INA-modified-CPA-media (white bars) was quantified as a percentage at times indicated and at equivalent time points for unfrozen ELS. Results from cryopreserved ELS are expressed as a fraction of unfrozen ELS at an equivalent time point. Values are means for five samples from four independent experiments +/− SD. **p<0.01. CholC, Cholesterol crystals; SD, standard deviation.
At 24 to 72 h postwarming, unfrozen control ELS cultured displayed high viability (∼95%). However, ELS cryopreserved with standard-CPA-medium showed decreased membrane viability characterized by high levels of PI staining at both 6 and 24 h postwarming: ∼60% and ∼40% viable, respectively. By 48 h, PI staining was reduced relative to FDA staining, which increased quantified viability to ∼70%, that improved further by 72 h: ∼ 95%. Conversely, ELS cryopreserved with INA-modified-CPA-medium showed less PI staining; and viability was better maintained, at ∼85% that of control, at both 6 and 24 h. Again, viability improved over time; and by 48 h postwarming, viability was ∼95%.
Measurement of ELS viable cell numbers after CryoP
ELS were harvested using EDTA, cell nuclei were counted, and viable cell numbers were calculated (Fig. 4). At all time points, viable cell numbers of ELS were significantly higher when INA-modified-CPA-medium was used cf. to ELS cryopreserved using standard-CPA-medium, with values of ∼0.65-fold of control at 24, 48, and 72 h. Using standard-CPA-medium, viable cell numbers were ∼0.3-, 0.35-, and 0.45-fold of control at 24, 48, and 72 h, respectively.

Effect of CholC on viable cell numbers after CryoP. Viable cell numbers of ELS cryopreserved either in standard-CPA-media (black bars) or in INA-modified-CPA-media (white bars) were quantified, at 24 h intervals to 72 h postwarming and at equivalent time points for unfrozen ELS, as 106 viable nuclei per mL alginate. Results from cryopreserved ELS are expressed as a fraction of unfrozen ELS at equivalent time points. Values are means for five samples from four independent experiments +/−SD. **p<0.01.
ELS protein synthetic function after CryoP
Synthetic function of ELS after CryoP was assessed using ELISAs to quantify albumin, AFP, fibrinogen, and AAT and expressed as total production (Fig. 5B i–iv) and product per viable cell (Fig. 5A i–iv). Up to 24 h, per cell protein release was higher in ELS cryopreserved using standard-CPA-medium than INA-modified-CPA-medium. Thereafter, per cell albumin and fibrinogen release was comparable between both cryopreserved groups and the control. Similarly, per cell AFP and AAT production of ELS cryopreserved with INA-modified-CPA-medium returned to levels comparable to those of the control by 72 h. However, AFP and AAT release of ELS cryopreserved using standard-CPA-medium fell over time to ∼0.25- and 0.4-fold of control, respectively.
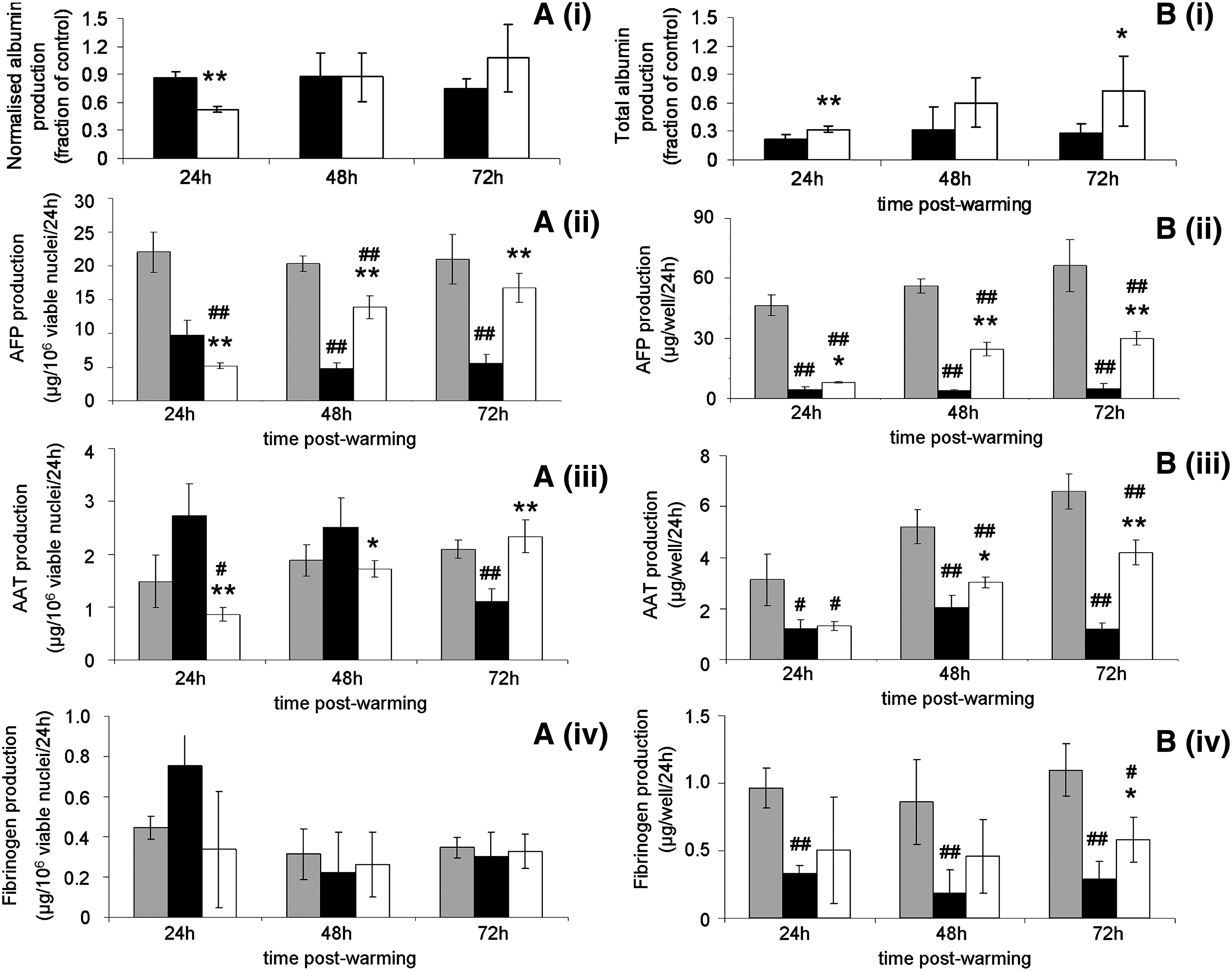
Effect of CholC on synthetic function after CryoP. Albumin
Total albumin production of cryopreserved ELS using standard-CPA-medium was ∼0.3-fold of control between 24 and 72 h; but using INA-modified-CPA-medium, total albumin production of cryopreserved ELS was improved from recovered to ∼0.9-fold control by 72 h. Similarly, with standard-CPA-medium, AFP secretion was ∼0.1-fold of control between 24 and 72 h but was improved by 48 h and maintained to 72 h using INA-modified-CPA-medium to 0.5-fold of control. Total fibrinogen and AAT secretion were similar for both cryopreserved cohorts at 24 h at ∼0.4-fold of control. Thereafter, however, total secretion of both fibrinogen and AAT was improved using INA-modified-CPA-medium to ∼0.6-fold of control, approximately double cf. ELS cryopreserved using standard-CPA-medium. Total synthesis and secretion of all four proteins was improved at all time points using INA-modified CPA-medium as compared with standard-CPA-medium.
ELS detoxification function after CryoP
Broad-spectrum CYP activity of ELS after CryoP was assessed using ethoxycoumarin conversion assay and expressed as product per viable cell (Fig. 6A) and total product (Fig. 6B). Per viable cell activity was similar for both cryopreserved groups at 24 h at ∼0.35-fold that of the control. By 48 h, per cell activity of ELS cryopreserved in standard-CPA-medium had improved to 0.5-fold that of the control; whereas for ELS cryopreserved using INA-modified-CPA-medium, values were significantly higher at 0.8-fold that of the control. By 72 h, both cryopreserved groups displayed similar per cell activity at 0.8-fold that of the control. Total CYP activity was improved at times to 72 h post-thaw using INA-modified-CPA-medium at approximately twice that of ELS cryopreserved using standard-CPA-medium.

Effect of CholC on cytochrome P450 activity after CryoP. Broad-spectrum CYP activity was quantified using ECOD assay at 24 h intervals to 72 h postwarming and at equivalent time points for unfrozen ELS. Results for unfrozen ELS (gray bars) and ELS cryopreserved in either standard-CPA-media (black bars) or INA-modified-CPA-media (white bars) are displayed either as total activity
ELS oxidative metabolism after CryoP
Metabolic function of ELS after CryoP was assessed using MTT assay and expressed as product per mg protein (Fig. 7A) and total product (Fig. 7B). ELS cryopreserved using standard-CPA-medium showed decreased MTT reduction per mg protein at both 24 h cf. the control at 0.2-fold, respectively. ELS cryopreserved using INA-modified-CPA-medium showed improved MTT reduction per mg protein at 24 h at 0.35-fold that of the control. By 72 h, both cryopreserved groups displayed similar MTT reduction per mg protein to the control. The total MTT product was improved at all three time points when INA-modified-CPA-medium was used by ∼30% over that of ELS cryopreserved with standard-CPA-medium.
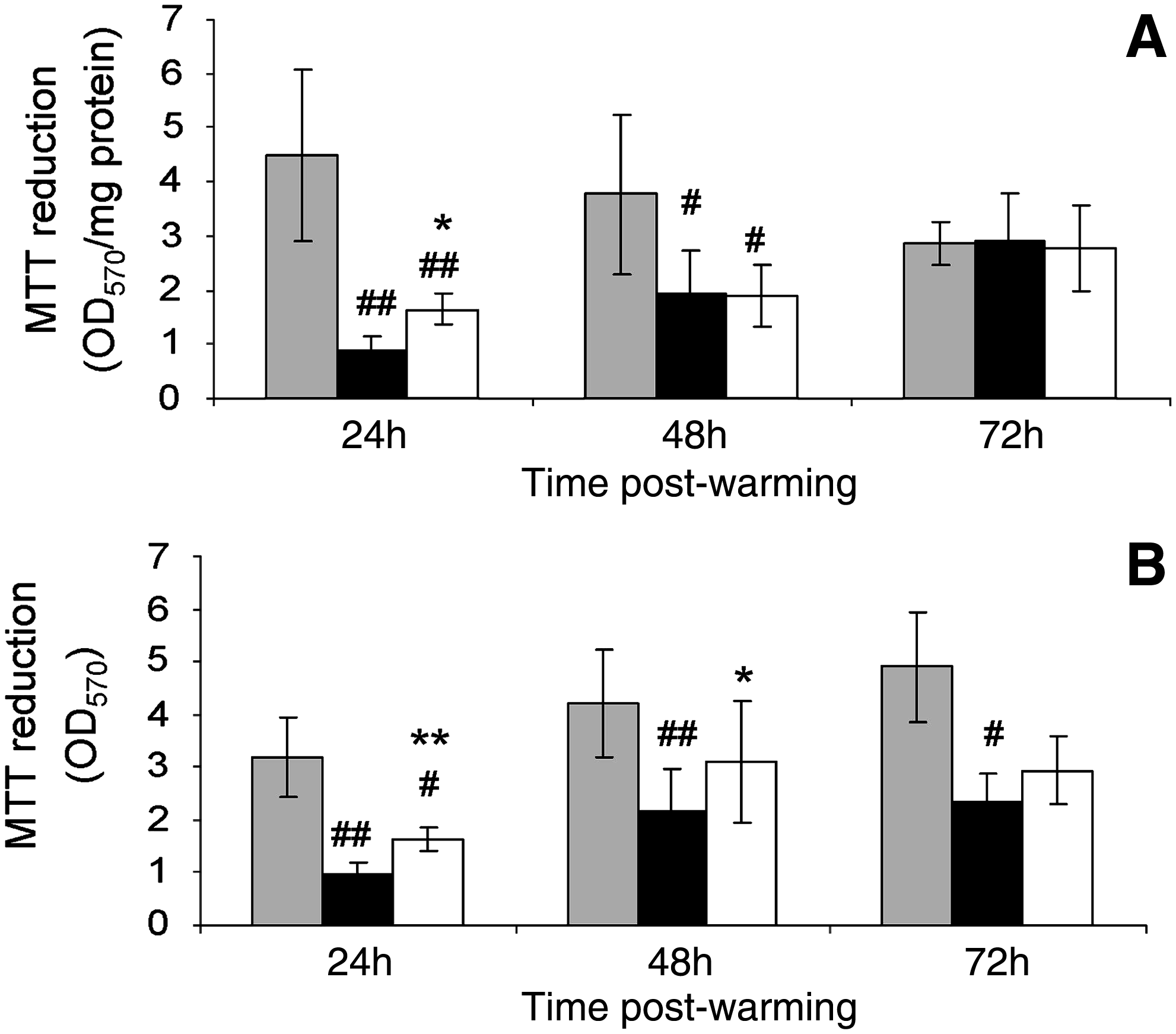
Effect of CholC on metabolic activity after CryoP. Metabolic activity was assessed using MTT assay at 24 h intervals to 72 h postwarming and at equivalent time points for unfrozen ELS. Results for unfrozen ELS (gray bars) and ELS cryopreserved in either standard-CPA-media (black bars) or INA-modified-CPA-media (white bars) are displayed either as total activity
Discussion
For any BAL to be clinically relevant, it should be available “off-the-shelf,” which could be achieved using CryoP. CryoP of parenchymal liver cells has often been attempted, but large discrepancies in reported success exist.28,29 To some extent, this will depend on the assay used. Typically, simple membrane integrity assays overestimate success compared with more complex functional assays. An alternative explanation for these discrepancies is the time after CryoP at which success was gauged. For BAL, in particular, where ELS function should remain optimal throughout the early post-thaw period (in the region of 24–48 h), time-related characterization of ELS after CryoP is important.
Due to the conformation of ELS within alginate, attachment was not a relevant assay, so dual staining to assess membrane viability (using FDA and PI) was employed here. FDA stains the cytoplasm of metabolically active cells, whereas PI stains nuclei of cells with permeable plasma membranes. Using objective image analysis, viability can be quantified immediately after CryoP and subsequently on repeated occasions, post-thaw, to provide an important, rapid assessment to supply early information concerning the quality of the biomass in the BAL.
Here, dual-staining of ELS was performed hourly to 6 h after CryoP (Fig. 2). We detected a progressive decline in viability postwarming after CryoP using standard-CPA-medium, indicating latent cryo-injury. This phenomenon, previously called CryoP-induced delayed onset cell death (CIDOCD), has been recognized in other cell types 30 and been attributed to both apoptosis and necrosis 31 developing over time postwarming, emphasizing that immediate assessment of ELS viability after CryoP is not necessarily representative or reliable of BAL function.
CryoP of single cell suspensions, including hepatocytes, has been studied for many years for use in various applications including BALs,29,32–40 although cell injury is still evident, with function notably affected.29,41–43 This is often attributed to IIF that occurs during cooling when cells have had insufficient time to dehydrate before nucleation/ice formation and “freezable” water persists intracellularly.13,14,44 The risk of IIF is compounded in a 3D system such as ELS for two reasons. First, during CryoP of multi-cellular complexes, such as ELS, transport of water and solutes differs from that of individual cells. It has been shown in similar spheroid systems that cells centrally located within spheroids require more time to dehydrate by the required degree than those peripherally located, because water sequentially moves across cell layers in response to the osmotic gradient.45,46 Second, we know that cells within ELS are connected via gap junctions, 47 and it has been shown experimentally in other interconnecting cell types that IIF may propagate via these channels.18,48 This means that once ice forms in an individual cell within a spheroid, rapid propagation of potentially lethal IIF through the entire cell mass is possible.
Strategies to avoid IIF in the first instance include using a slow cooling rate, as here. Ice formation outside ELS excludes solutes, increasing the osmolarity of the unfrozen solution surrounding the cells. In response, ELS dehydrate by exosmosis as the mobile cell water content leaves ELS. This means that there is less water contained within ELS to potentially undergo IIF. 16 However, if supercooling occurs, IIF may occur and propagate throughout the ELS. This is because ice nucleation is an exothermic reaction and when this latent heat of crystallization is released, this effectively delays the sample from cooling while the CRF chamber temperature continues to decrease with the applied cooling protocol. The greater the supercooling, the greater is the difference between sample and chamber temperature and ELS are exposed to a faster cooling rate than planned, meaning that ELS are less likely to be able to dehydrate sufficiently on a kinetic basis, resulting in an increased likelihood of IIF.28,13
This has been previously observed during CryoP of single cell suspensions of hepatocytes25,49,13 and other cell types. 18 Thus, the additional strategy of including a rapid cooling step, as part of a multi-step cooling protocol as used here, is commonly used with the aim of triggering ice nucleation. This may reduce supercooling to some level and allow good control of cooling rates in the samples during conventional, linear slow cooling. 25 However, these strategies were not sufficient to avoid injury in ELS here, and better control of ice nucleation using INA was essential to achieve high functional recoveries post-thaw.
Homogenous ice nucleation occurs at low temperatures, ∼−40°C so nucleation at any temperature above this (as here) is considered heterogeneous 16 and may be facilitated using INA. INA possess favorable surface interfaces for ordering of water molecules, and as the number of “ordered” water molecules increases at the INA surface, the more likely nucleation is to occur nearer the equilibrium freezing point (i.e., less likely to supercool) and, thus, cell death as a result of IIF is less likely. 50
Steroids, such as CholC, have been previously identified as INAs, 21 but this is, to our knowledge, the first example of its use for CryoP of liver cells or spheroids. CholC were included in the CryoP medium at a concentration of 1.1 mg/mL. In preliminary experiments, this concentration was shown to be sufficient for consistent ice nucleation (see Fig. 1), and higher concentrations did not increase efficacy, presumably because a sufficient number of surface “seeding planes” were present to favor ice nucleation. Additionally, if CholC were used at higher concentrations, this may present an unnecessary metabolic challenge to ELS.
Demonstrably, by utilizing CholC as an INA, ELS membrane viability was improved (Fig. 3). Viability measured at 6 h postwarming was reduced to around 60% that of the control using standard-CPA-medium but was improved to 85% with INA-modified-CPA-medium. By 24 h postwarming, membrane viability had fallen to 40% using standard-CPA-medium; but by using INA, this CIDOCD was limited and viability was maintained at ∼85%. By 48 h postwarming, viabilities improved in both groups, although ELS cryopreserved with INA-modified-CPA-medium still maintained a significant viability advantage of ∼20% compared with those cryopreserved using standard-CPA-medium. Overall viability may be improved by either an increased proportion of live cells or decrease of dead cells within the total population. The former will occur as HepG2s continue to proliferate within alginate beads. The latter may also occur as lethally damaged PI-positive cells degenerate and no longer exist for staining. Similarly, nonlethally-damaged PI-positive cells may regain membrane integrity after initiation of repair processes in response to cryo-injury, although this is less likely. That this was a true advantage in viable cell numbers was confirmed using cell counts (Fig. 4). At both 24 and 48 h post-thaw, viable cell numbers were almost double when INA-modified-CPA-medium was used. Given that success of BAL will depend on sufficient functional cell mass in the bioreactor, this is a significant finding.
Maintenance of ELS function after CryoP is equally important. Liver functions are wide ranging, including synthetic, detoxification, and metabolic functions. Here, we assessed synthetic function from synthesis and secretion of four liver-specific proteins (Fig. 5). ELS are capable of albumin synthesis at levels comparable to that of primary human hepatocytes. 11 Albumin and other liver-specific proteins may be simply measured by ELISA without the addition of further components to culture medium.
Liver-specific protein synthesis and secretion per viable cell at 72 h in ELS cryopreserved in INA-modified-CPA-media was comparable with those of the unfrozen controls; but at 24 and 48 h, was generally lower. For ELS cryopreserved in standard-CPA-media, per viable cell synthesis and secretion was higher than ELS cryopreserved in INA-modified-CPA-media for albumin and AFP at 24 h, and higher even than the unfrozen control for fibrinogen and AAT at 24 h. This is an interesting finding and likely results from unregulated release of protein synthesized before CryoP from dying cells through permeable plasma membranes, resulting from IIF. This is more apparent for fibrinogen and AAT, as these proteins are synthesized in far lower quantities than albumin or AFP; and so, the proportion of presynthesized protein release from dying cells is larger compared with de novo synthesis from surviving cells. Despite this, total synthesis and secretion of ELS cryopreserved in INA-modified-CPA-medium was improved over those cryopreserved in standard-CPA-medium at all time points for all four proteins. This resulted from many more cells surviving CryoP. Again, given that for the BAL, total liver-specific protein secretion is important, this finding is key.
We assessed detoxification function using ECOD assay to assess broad-spectrum CYP activity (Fig. 6). It is well known that HepG2 cells possess only limited CYP activity due to decreased mRNA levels. 26 However, CYP activity may be upregulated in HepG2s by both chemical induction using 20 μM indirubin 26 and 3D culture as ELS. 11 Both approaches were utilized here, and CYP activity was able to be reliably quantified.
Per viable cell CYP activity was decreased in both cryopreserved groups by at least 50%. We could find no previous information concerning the effect of CryoP on CYP function in HepG2 cells, either as suspensions or as 3D cultures. However, this value is comparable to similarly cryopreserved suspensions of rat hepatocytes. 51 Again, after 24 h, a recovery period occurred over which per cell detoxification function improved. ELS viability is reduced by ∼60% by CryoP using standard-CPA-medium, which may influence the inducibility in these ELS, which could account for the longer recovery period compared with ELS cryopreserved in INA-modified-CPA-medium. However, this is not observed in suspensions of primary human hepatocytes where inducibility is maintained despite (an admittedly smaller) 20% loss in viability. 52 More likely is that IIF in ELS cryopreserved in standard-CPA-medium disrupted ELS structure and since the structural arrangement of ELS contributes to upregulation of CYP activity, this is another possible cause of reduced CYP activity after CryoP. Overall total detoxification of ELS is significantly improved using INA-modified-CPA-medium compared with standard-CPA-medium due to greater ELS survival and improved per viable cell function at both 24 and 48 h.
We assessed general metabolism using the MTT assay (Fig. 7). Again, metabolic function was reduced in both cryopreserved cohorts at 24 h, particularly in ELS cryopreserved in standard-CPA-medium; and once again, a recovery period was observed thereafter. Similarly, metabolism was increased using INA-modified-CPA-medium. The effect of CholC was not as evident in this assay, which perhaps reflects the decreased sensitivity and nonspecificity of this assay with regard to ELS.
In summary, CryoP of ELS is possible with recoveries of high cell viability and function, but only if CIDOCD is avoided. Optimized-CryoP schemes, in which an INA such as CholC are used to minimize likelihood of IIF, may be essential for success during scale-up of the CryoP protocol to volumes needed for clinical liver support. However, cell injury is still evident even with INA. Our preliminary results indicate caspase activation in ELS after CryoP even with INA, and this will be the focus of future work. Finally, the approach of including INA during CryoP may prove useful for multi-cellular constructs in other tissue-regeneration applications.
Footnotes
Acknowledgments
We thank the Liver Group Charity and the Garfield Weston Foundation for funding this study, the Wellcome Trust for fluorescent microscope facilities (Wellcome grant number 066327), Dr Shiyu Yang (Division of Surgery and Interventional Science, UCL Medical School) for use of fluorescence platereader, and also Kiran Malik, Chinwe Duru, and Dr. Paul Matejtschuk (NIBSC, a Center of the Health Protection Agency) for use of DSC equipment.
Disclosure Statement
No competing financial interests exist.