
Abstract
Complex microenvironmental stimuli influence neural cell properties. To study this, we developed a three-dimensional (3-D) neural culture system, composed of different populations including neurons, astrocytes, and neural stem cells (NSCs). In particular, these last-mentioned cells represent a source potentially exploitable to test drugs, to study neurodevelopment and cell-therapies for neuroregenerations. On seeding on matrigel in a medium supplemented with serum and mitogens, cells obtained from human fetal brain tissue formed 3-D self-organizing neural architectures. Immunocytochemical analysis demonstrated the presence of undifferentiated nestin+ and CD133+ cells, surrounded by β-tub-III+ and GFAP+ cells, suggesting the formation of niches containing potential human NSCs (hNSCs). The presence of hNSCs was confirmed by both neurosphere assay and RT-PCR, and their multipotentiality was demonstrated by both immunofluorescent staining and RT-PCR. Flow cytometry analysis revealed that neurosphere forming cells originating from at least two different subsets expressing, respectively, CD133 and CD146 markers were endowed with different proliferative and differentiation potential. Our data implicate that the complexity of environment within niches and aggregates of heterogeneous neural cell subsets may represent an innovative platform for neurobiological and neurodevelopmental investigations and a reservoir for a rapid expansion of hNSCs.
Introduction
The finding of neural stem cell (NSC) in the human adult and fetal CNS2–6 has offered wider possibilities to understand cellular, molecular, and developmental mechanisms of neurodegenerative diseases. NSCs are clonogenic cells endowed with self-renewal and multipotential differentiative capability. NSCs can be isolated and extensively propagated in vitro as multicellular aggregates called neurospheres by using mitogenic factors in a chemically defined media.7–15
Neurosphere culture is a well-known system for large-scale production of neural cells for use in cell replacement therapies and to assay for and characterize NSCs. However, this culture method suffers from several disadvantages, such as its apparent sensitivity to variations in the methodological procedure and neurosphere heterogeneous nature. In fact, variations in the culturing method used may affect both the proliferation capacity 16 and the positional cues that the cells are exposed to. 17 In addition, differences in both the composition of cell types and the cells' properties within each neurosphere can arise. Diverse constituents or concentrations of factors in the media, the method and frequency of passaging, whether the neurosphere is dissociated before differentiation, and the number of passages after isolation can affect cells growth.18–21
Proliferation, survival, and differentiation of stem cells is regulated by both environmental and cell-autonomous signals.22,23 In vivo, the stem cells “niche” is made up by the external signals that control stem cell fate.24,25 A stem cell niche is the native cellular microenvironment characterized by three primary features: cell secreted factors, cell–cell interaction, and the extracellular matrix (ECM). 23 Neurosphere cultures should grant some of these niche signals that may be important for NSC maintenance, proliferation, and survival. Nonetheless, due to the complex nature of multicellular aggregates, this system is not suitable for the study of signaling pathways involved in the neurodevelopment.
In several studies focused on studying corticogenesis, embryonic stem cells (ES cells) have emerged as a precious tool to study developmental biology, thus allowing the in vitro recreation of events of organogenesis in controlled and reproducible conditions. The resulting data demonstrate for the first time that a self-organizing cytoarchitecture in vitro can mimic a 3-D-brain-like structure, and they represent a powerful system to elucidate some of the fundamental mechanisms of cortical patterning.
Using only in vitro protocols to recapitulate some of the early key steps leading to forebrain and cortical development, Gaspard and colleagues have shown that ES cells can be differentiated into cortical progenitors and, subsequently, cortical pyramidal neurons, following similar pathways as in vivo. 26
Using a different approach, Eiraku and colleagues 27 cultured ES cells as bowls of cells capable of differentiation into cortical-like progenitors, and confirmed the sequential generation of distinct types of cortical neurons, and observed a cytoarchitecture, which remembers a nascent cortical primordium.
Recently, Conti has described a niche independent symmetrical self-renewal of adherent NSC derived from primary CNS tissue and ES cells. 28
The survival of network formation synaptogenesis and neurite outgrowth had been evaluated in vitro in planar culture29,30; however, the presence of three-dimensional (3-D) growth/interactions and matrix is crucial. The development of a 3-D adherent culture system to study such basic neurobiological phenomena represents a fundamental step to increase our knowledge.
Here, we described a 3-D adherent culture system of human fetal neural tissue in the presence of serum, which mimics the organization of neural tissue. This system is also a “reservoir” of human fetal NSCs (hNSCs) and allows the study of some neurodevelopmental diseases, as it is possible to derive hNSCs also from pathological fetuses.31–33
Materials and Methods
Human fetal brain cultures
Research on human tissue with informed consent was authorized by the Ethics Committee of Fondazione IRCCS Istituto Neurologico “Carlo Besta” and Fondazione IRCCS Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena (PoliMaRe).
Human fetal brain specimens were isolated from twenty 11–12-week-old human fetuses according to the ethical guidelines of the European Network for Transplantation. Briefly, the brain tissue was mechanically minced and then incubated with 0.625 wU/mL Liberase™ Blendzime II (Roche Diagnostic) for 1 h at 37°C under gentle shaking. The resulting cells suspension was plated at 2×105 cells/cm2 on Growth Factor Reduced Matrigel (GFR MTG) (BD Pharmingen) coated T25 culture flasks in the presence of NSC serum-free medium (HM) and supplemented with 10% fetal bovine serum (FBS) (Gibco). In particular, HM was composed of Dulbecco's modified Eagle's medium/F12 (Euroclone) optimized for the culture of NSCs, containing 10 μg/L basic fibroblast growth factor (Boehringer Mannheim) and 20 μg/L human recombinant epidermal growth factor, glutamine (2 mM), glucose (0.6%), sodium bicarbonate (3 mM), HEPES buffer (5 mM) (all from Gibco), transferrin (100 μg/mL), insulin (25 μg/mL), putrescine (60 μM), selenium chloride (30 nM), and progesterone (20 nM). This medium has been developed for the isolation and the expansion of human NSCs.13,34 After 4–6 weeks, cultures were trypsinized, and cells were seeded into a new GFR MTG coated flask at splitting ratio 1:2. Human fetal brain cells (hFBCs) could be maintained for up to 30 passages in vitro without showing any particular modification. Forty days in vitro (DIV) hFBCs were used as indicated for all experiments. Experiments were performed in triplicate.
Adhesion test
On enzymatic digestion, freshly isolated monocellular suspensions were resuspended in HM +10% FBS and seeded at different cell densities on uncoated or previously coated T25 culture flasks with different ECM proteins including Collagen Type I and IV (5 μg/cm2), plasma fibronectin (FN) (1 μg/cm2) (Sigma-Aldrich), and GFR MTG (0.2 mg/cm2). Flasks were then incubated for 6 and 24 h; and at the end of each incubation time, floating cells were aspirated, and the adherent hFBCs were detached with trypsin and counted using a hemocytometer.
Immunofluorescence
HFBCs seeded at optimal cell density on a GFR MTG coated chamber slide were cultured in HM +10% FBS for 40 days as described. The cultures were fixed and treated as previously reported. 35 Specific antibodies used were rabbit anti-human GFAP (1:500), NG2 (1:400) (Chemicon), Gal-C (1:500), von Willebrand factor (vWf) (1:80), CD31 (1:100), CD105 (1:100) (Sigma-Aldrich), mouse anti-human β-tub-III (1:100), Nestin (1:100) (Chemicon), and Flk-1/KDR (1:100) (Santa Cruz Biotechnology). Cultures were then washed twice with PBS and incubated with an appropriate secondary antibody: anti-rabbit IgG (1:1000), anti-mouse IgG (1:500) (Cy2 and Cy3; Jackson Immunoresearch) for 45 min at room temperature. After washing, cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/mL) diluted 1:15 for 20 min at room temperature in the dark. Cells were then rinsed twice with PBS, mounted with Fluorsave™ (Calbiochem). For 3-D reconstruction, a stack of ∼100 optical sections covered the entire thickness of the cellular layers of HFBCs. The distance between sections was 1 μm. 3-D digital images were made using a Leica TCS SP2 AOBS (Leica Microsystems) confocal lazer scanning microscope.
Flow cytometry analysis
HFBCs were characterized for endothelial markers expression by means of flow cytometry analysis (FACS). Briefly, 105 hFBCs were incubated with mouse antibodies to the following human antigens: CD133/2, CD34 (Miltenyi Biotec), CD31, CD105, Flk-1/KDR, vWf (all purchased from BD Pharmingen), and CD146 (CABRU). The controls were isotype-matched mouse IgG. The cytometry analyses were done with FACS calibur flow cytometer and Cell Quest software (BD Pharmingen).
Isolation, growth, and differentiation of hNSCs derived from hFBCs
After 40 DIV of culture, hFBCs were detached with collagenase type I (0.1% w/v) (Gibco) to obtain single-cell suspensions that were seeded into culture flasks in the presence of serum-free HM. These conditions promote the formation of neurospheres. Neurosphere cultures were routinely propagated and cryopreserved, thus maintaining multipotentiality. They were induced to differentiate into neural phenotypes in differentiating medium, that is, mitogen-free HM containing 2% FBS. 36
Immunomagnetic separation of hFBCs subpopulations
After 40 DIV of culture, hFBCs were detached with collagenase type I (0.1% w/v) (Gibco) to obtain a suspension of single cells. Around 107 hFBCs/mL were resuspended in PBS/0.1% BSA and then mixed with 25 μL (107 beads, ratio cell/beads 1:1) magnetic beads (Dynal, Biotech) coated with different antibodies: anti-CD146 (CABRU), anti-CD34, anti-CD105, and anti-Flk-1/KDR, according to the manufacturer's instructions (Pan mouse IgG Kit; Dynal). For CD133 immunoselection, we used Miltenyi Kit (Biotec). All the positive cell selections were cultured in serum-free HM to evaluate growth and neurospheres formation. This experiment was performed on 10 different hFBCs preparations and no difference, statistically significant, was observed between the samples.
Neurosphere forming assay
For neurosphere forming assay (NSA), hNSCs were dissociated at different passages; and single cells were plated in six-well plates at a density of 2×104 cells per well in serum-free HM containing 0.8% (wt/vol) methylcellulose.32,37 The cell clusters (diameter >2 mm) number was counted at 3 weeks after plating under a Nikon Eclipse TE300 inverted microscope equipped with a Zeiss Axiovision device camera.
Reverse transcriptase-polymerase chain reaction
Three separate RNA extractions were performed on hFBCs-, CD133+-, and CD146+-derived hNSCs, and separately processed. Total RNA from different hNSC subpopulations was isolated with TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Specific primers for human β-III-tubulin, 35 GFAP, 6 CD133, 32 SOX2, 32 SOX9, 38 and PAX6 39 were previously reported. Human GalC primers were as follows: sense 5′- GCTCATTATCCTGGAACCCATTC-3′ and anti-sense 5′- AGACAGTTGGCCATTTAGAGAAA-3′. GAPDH 36 was used as internal control. To guarantee accuracy, each reaction was repeated thrice.
Spectral karyotyping analysis
For spectral karyotyping (SKY) studies, five hFBCs at P1 and P30, and five hFBC-derived hNSCs lines at P10 were arrested at the metaphase by colcemid (Sigma-Aldrich), incubated in hypotonic buffer, and then fixed in a methanol/acetic acid mixture. SKY was performed by following standard protocols. 40
Proliferation assay
Nonselected, CD133+-, and CD146+-derived neurospheres were mechanically dissociated, counted, subsequently resuspended at the concentration of 2.5×105 cells/mL in serum-free HM, and incubated at 37°C. At every in vitro passage, cells were recovered and counted. The growth rate (GR) was calculated following the formula GR=(nk+1-nk)/nk, where the n=cell number and k=count number. Values represent a measure of the increment.
Differentiation assay
Nonselected, CD133+-, and CD146+-derived neurospheres were plated on GFR MTG-coated glass chamber slides (Nunc), in HM without mitogens plus 2% FBS. 6 After that, cells were fixed at 7 DIV; and the presence of neural markers was analyzed by means of immunostaining 35 : GFAP (rabbit, 1:500), NG2 (rabbit, 1:200), and β-III-tubulin (mouse, 1:100) (purchased from Chemicon). Three separate immunofluorescence analyses were performed on CD133- and CD146-positive hNSCs; positive cells were counted in a blind manner.
Calcium uptake/glutamate sensitivity
In the presence of glutamate, neuronal cells are able to take up calcium. Given the essential role of calcium in neuronal function, a calcium assay could be useful to identify neurons. First, we treated hFBCs in HM +10% FBS for 6 days with forskolin 10 μM (Sigma-Aldrich) and isobutylmethylxanothine (IBMX) 0.1 mM (Sigma-Aldrich). The calcium assay was performed as previously described.41,42 Briefly, hFBCs were rinsed with sterile Hanks solution (Mascia Brunelli). DAPI (1 μg/mL) and 2 mM Oregon Green 488 Bapta-1 (Invitrogen) were diluted in Hanks solution, 1:15 and 1:200 respectively, and added to the cells after the removal of the Hanks solution. The cells remained in darkness for 1 h at room temperature. Then, Oregon Green 488 Bapta-1 (Invitrogen) and DAPI were removed, and 60 mM glutamic acid in Hanks Solution was added. Images were photographed on the Nikon Inverted Eclipse TE300. Fields of neuron were taken every 10 min, from 0 min to 1 h.
Monoamines and glutamate assay
Culture medium (4 mL) was removed, and 400 μL of 1M HClO4 were added (final HClO4 concentration, 0.1M) and stored at 4°C in 15 mL tubes. After 15 min, tubes were spun for 5 min at 5000g, and the supernatant was frozen at −20°C until analysis.
Cells were (2×106 cells) rinsed twice with ice-cold PBS (Gibco) and homogenized in 1.5 mL Eppendorf tubes by sonication for 10 s in 500 μL of ice-cold 0.1 M HClO4. After 15 min, homogenates were centrifuged as just described, and supernatant was collected in 0.5 mL Eppendorf tubes and frozen at −20°C until analysis.
20 μL of supernatant obtained from cells homogenate or culture medium were injected into the high-performance liquid chromatography (HPLC) apparatus equipped with a reverse-phase column and an electrochemical detector, and monoamines were determined as previously described.43,44 Dopamine (DA), noradrenaline (NA), and serotonin (5-HT) were analyzed.
Electron microscopy
For electron microscopy, hFBCs (105 cells/mL) cultured in 24 mm Transwell® with 0.4 μm Pore Polyester Membrane Insert (Corning Incorporated) were fixed after 40 DIV in 3% glutaraldehyde 0.1 M Na Cacodylate, pH 7.4, postfixed in 2% osmium tetroxide in s-collidine buffer, pH 7.1, and processed for embedding in EPON-12 (Ted Pella). Thin sections were stained with uranyl acetate and lead citrate and analyzed with a Jeol transmission electron microscope.
Statistical analysis
Data are presented as mean±SD. Statistical analysis was performed using Wilcoxon rank sum test (p<0.05).
Results
HFBCs to ECM interactions
At first, we established the best substrata to support hFBCs adhesion and proliferation. Thus, culture flasks were coated with ECM proteins, including collagen type I and IV, FN, and GFR MTG. The GFR MTG is well suited for studies that require a more-defined and characterized reconstituted basement membrane than Matrigel.
As shown in Figure 1A, hFBCs rapidly attached (within 6 h) on GFR MTG and, to a lesser extent, to FN. In contrast, neither collagen I and IV, nor culture plastic was effective even after longer incubation (24 h). Next, we assayed the optimal growth culture conditions and found that hFBCs proliferation was dependent on the initial number of cells seeded (best cell density seeding ranged between 1×105 and 2.5×105 cells/cm2) (Fig. 1B). The addition of 10% FBS promoted the formation of focal 3-D overgrowth zones. These zones expanded and connected to each other to form a complex 3-D cellular network (Fig. 1C). Four-five weeks later, these 3-D self-organizing neural architectures appeared well organized and covered the entire surface of the underlying cell monolayer (Fig. 1D). At optimal cell density seeding and culture conditions, generally 3 weeks were necessary to obtain this distinctive structural formation. Under our culture conditions, hFBCs were not cell-cell contact growth inhibited but could continuously proliferate. The capacity of hFBCs to organize 3-D self-neural architectures was not a transient process but could be propagated in vitro by passing cells into new GFR MTG–coated culture flasks. In addition, using SKY analysis, we were able to determine that hFBCs at P1 and P30 have a normal human karyotype (data not shown). HFBCs, therefore, represent genetically stable cell lines.
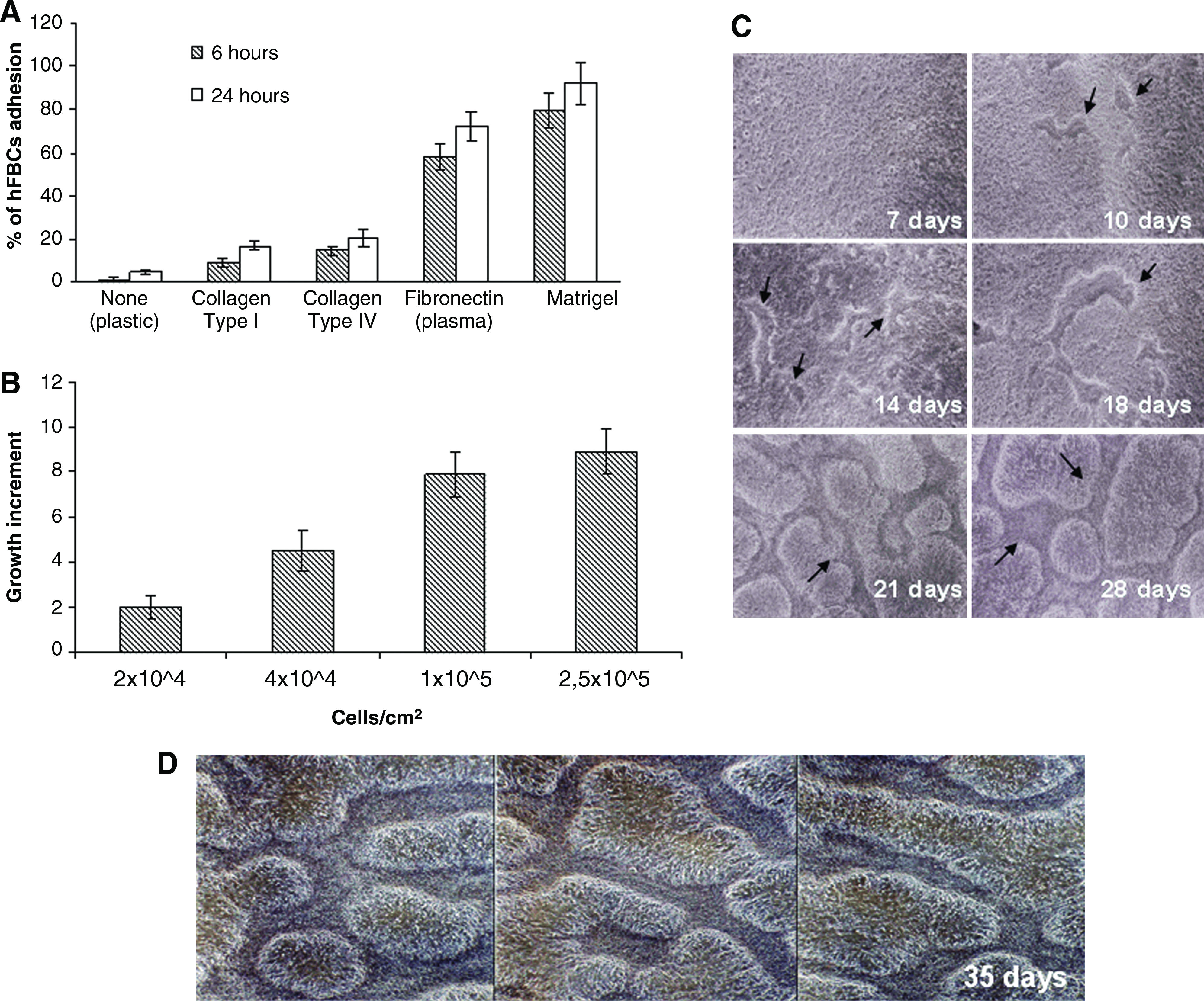
hFBCs produce 3-D self-organizing neural architectures under specific culture conditions.
3-D Self-organizing neural architectures
We dissected the 3-D structures and analyzed the cytoarchitecture, using confocal microscopy; pictures were taken as Z-stack with slices of 1 μm. The entire complex cellular organization is shown in Figure 2A: the pattern of β-tub-III-expression revealed a complex network of neurons, whereas spread GFAP-labeled astrocytes were mainly located in the inner layers of the structure. Only rare cells were immunoreactive for NG2, an immature oligodendrocyte marker and Gal-C, a mature oligodendrocyte marker, respectively (Fig. 2B, C).
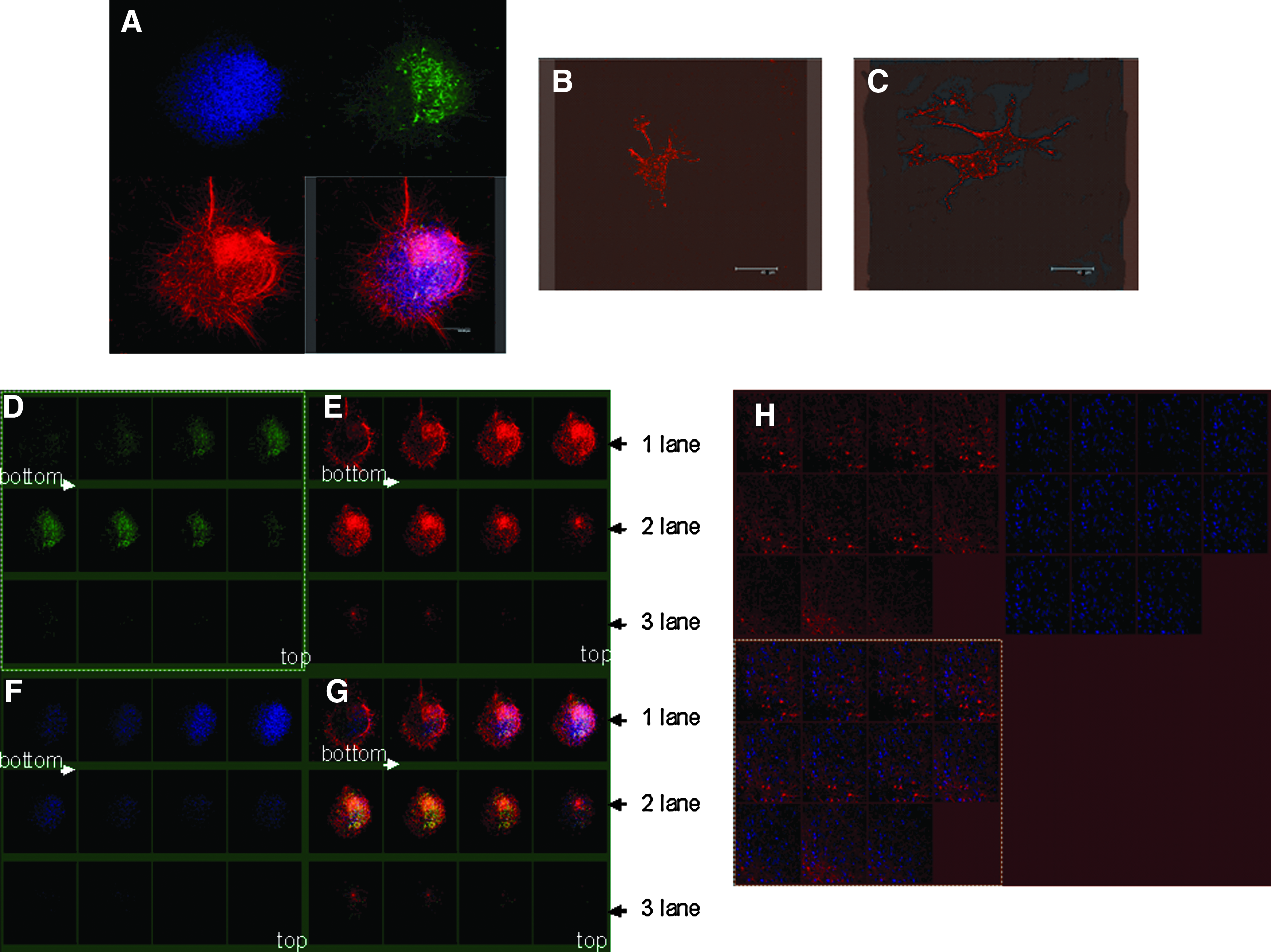
Cellular organization of hFBCs culture. Several areas (n=30) of 3-D self-organizing neural architectures were examined by confocal analysis to analyze the cellular distribution and organization.
3-D self-organizing neural architectural areas appeared to be formed by several layers of cells (Fig. 2): in particular, β-tub-III+ cells seemed to be organized as a complex net underlying a stratum of GFAP+ cells (Fig. 2G). A first layer anchored to GFR MTG (Fig. 2D–G lane 1) and was enriched with β-tub-III+ cells, and a minority of GFAP+ cells; a second layer was composed of GFAP+ cells and β-tub-III+ cells (Fig. 2–G lane 2) and a third layer, with fewer cells, was composed of only β-tub-III+ cells (Fig. 2D–G lane 3). Several Nestin+ cells were distributed throughout the different layers with no specific localization (Fig. 2H). It is worth noting that the total number of Nestin+ cells was not reduced by FBS addition to HM.
Neurochemical characteristics of hFBCs
Neuronal cells are able to take up calcium in the presence of glutamate. 45 HFBCs' ability to take up calcium in the presence of glutamate was tested by examining the fluorescence of a visible light–excitable intracellular calcium indicator, which fluoresces when it binds calcium.
To increase cyclic AMP, we added IBMX, which is a phosphodiesterase inhibitor, to the culture. 46 It has been demonstrated that an increase in intracellular cAMP promotes neuronal signaling mechanisms.
HFBCs did not show any fluorescence at 0 min after the addition of glutamate (Fig. 3). We observed a rapid increase of fluorescence between 0 and 10 min (Fig. 3), and the fluorescence remained constant up until 60 min. The cell body of hFBCs was mainly fluorescent.
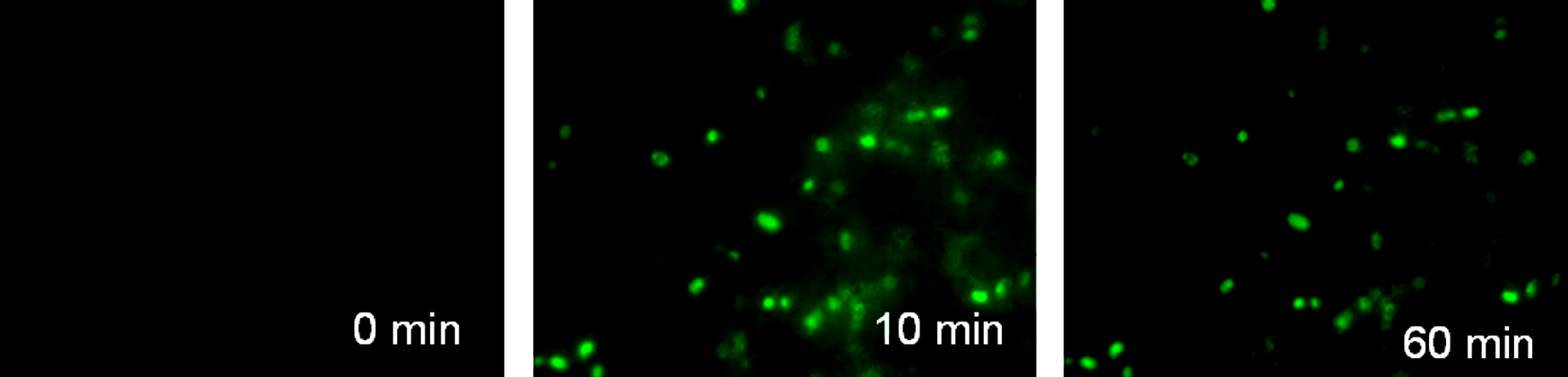
Glutamate sensitivity/calcium uptake of hFBCs. HFBCs treated with Oregon Green 488 Bapta-1. Glutamate was added to the culture, and pictures were taken at the following time intervals: 0 min, 10 min, 60 min (magnification 200×). Color images available online at
Moreover, we evaluated the concentrations of monoamines in hFBCs cell homogenate and supernatant. Cellular content of NA was 34 fmol/sample. DA and 5-HT were not detectable in cells. Monoamines were present in the supernatant of cells. NA was 6.3 fmol/sample, DA was 4 fmol/sample, and 5-HT was 2.9 fmol/sample.
Electron microscopy of 3-D self-organizing neural architectures
Ultrastructural studies of 40 DIV hFBCs demonstrated multilayered aggregates of neuronal cells embedded within intricate networks of axonal processes (Fig. 4). Nuclei were oval shaped and contained distinct nucleoli within an abundant euchromatin matrix. Cytoplasmic organelles primarily consisted of free ribosomes, mitochondria, Golgi complex, and a few cisternae of rough endoplasmic reticulum. The axonal process (Fig. 4A) contained a prominent microtubular cytoskeleton originating at the axonal hillock, near the cell body. Axonal microtubules (Fig. 4B) were arranged in parallel arrays and formed tracks, along which elongated mitochondria and round transport vesicles were frequently identified. Most distal axonal terminals formed bulbous enlargements (Fig. 4C) containing mitochondria and transport vesicles. Occasional synaptic type junctions between vesicle-containing processes and cell bodies were observed (Fig. 4D). Interspersed between neuronal cells were stromal-type cells. The nuclei and cell bodies of these cells showed no distinct ultrastructural features. Stromal cells were, however, recognizable by their cytoplasmic process, which characteristically contained abundant intermediate filaments and no microtubules (Fig. 4E, F).
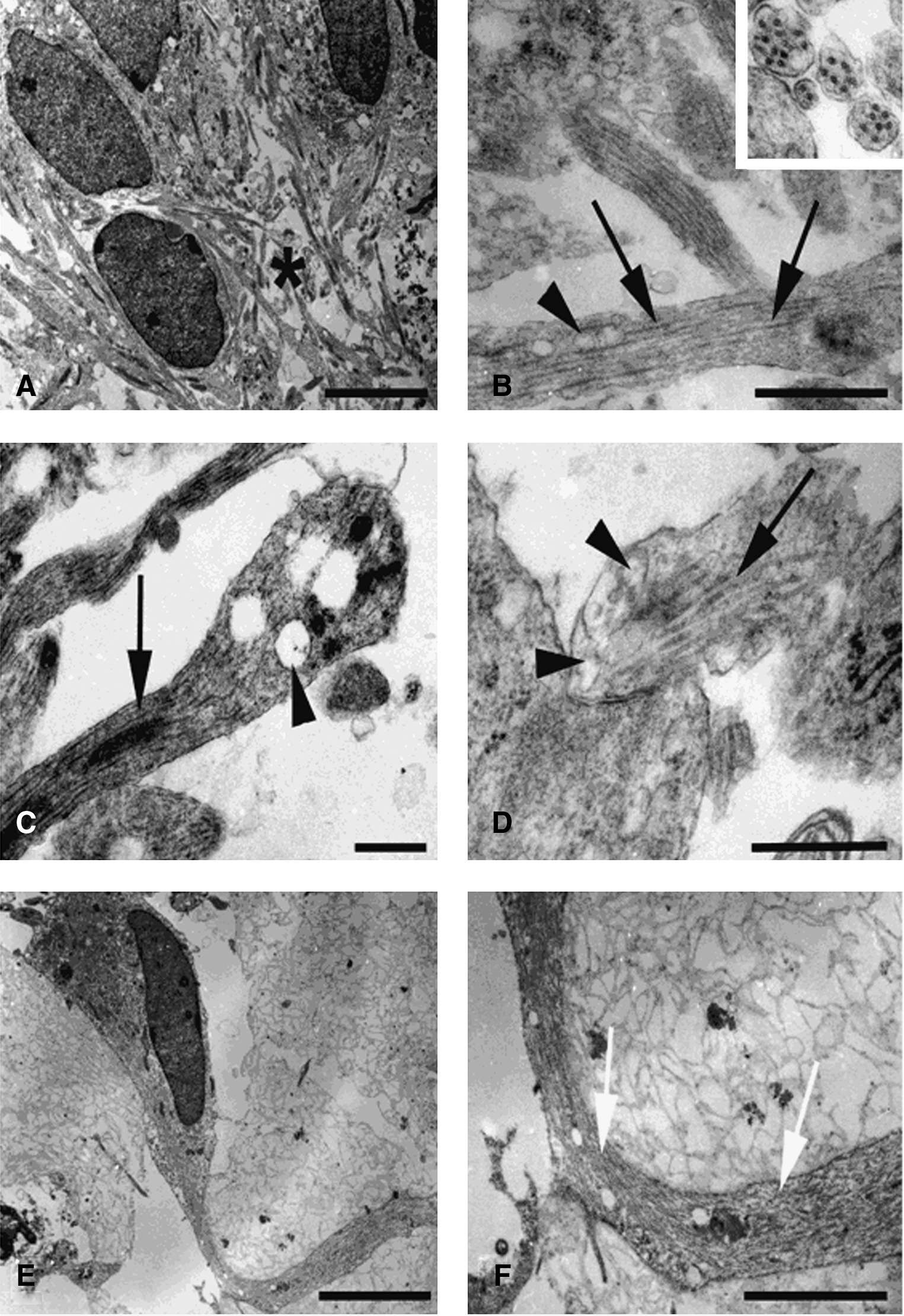
Axons, synapsis, and filaments. Electron micrographs of 40 days in vitro hFBCs showing neuronal
HNSCs derived from hFBCs isolation, growth, and differentiation
The absence of cell-cell contact growth inhibition suggested the presence of immature neural progenitor cells.
After cell dissociation, a part of the hFBCs was cultured in a serum-free medium containing mitogens (HM), which are known to support the hNSCs proliferation. After five DIV, the serum-free medium supported selective survival and growth of cells that aggregated in spheres (data not shown). These primary spheres were collected and re-plated in fresh medium. The cultures were then passed by mechanical cell dissociation every seven DIV.
We found that the proliferative potential of hNSCs derived from adherent cultures (hFBC-derived hNSCs) was higher than hNSCs isolated in standard conditions (Fig. 5A). 34
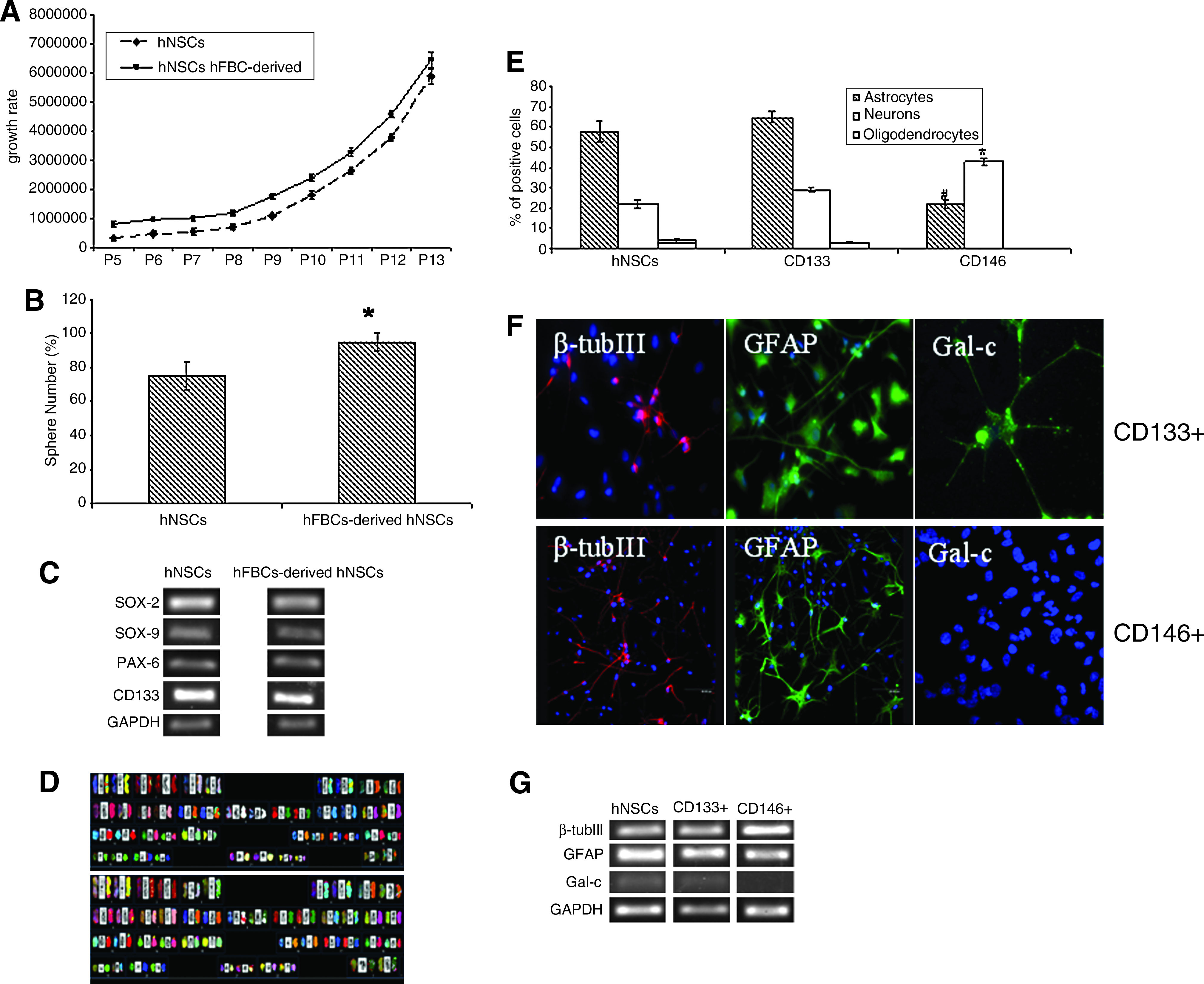
Proliferation and characterization of hFBC-derived hNSCs and characterization of CD133+- and CD146+-derived hNSC subpopulations.
Next, to determine the frequency of neurosphere formation, we initially plated single cells in HM containing methylcellulose to guarantee that each colony was derived from a single cell and, consequently, clonal in origin. We found that hFBC-derived hNSCs form neurospheres at a higher frequency than hNSCs (Fig. 5B).
The originating neurospheres expressed the same panel of neural precursor and stem cell markers such as Sox2, Sox9, PAX6, and CD133 compared with control hNSCs (Fig. 5C). Further, they were able to grow and differentiate into neuronal cell lineages similar to neurospheres produced using a standard method (data not shown).
On removal of the mitogens, after the addition of 2% FBS and plating on a coverslip precoated with MTG, the hFBC-derived hNSCs differentiated into neural phenotypes (i.e., astrocytes, neurons, and oligodendrocytes) (data not shown), a property of hNSCs.
In addition, proliferation rates, NSA, and differentiation potential of hFBC-derived hNSCs were evaluated at both early and late passages (passage 10 and passage 30, respectively). The growth and differentiation capability of hFBC-derived hNSCs were not significantly different from controls at either passage (data no shown).
SKY of five hFBC-derived hNSCs lines confirmed the normal, nonaltered karyotype of cells after 10 passages (Fig. 5D).
Isolation of CD133+ or CD146+ hNSCs from the culture by immunomagnetic selection
In a previous study, we demonstrated that hNSCs express several vascular markers. 36 Thus, we first evaluated by FACS the presence of cells expressing vascular markers in freshly isolated and in cultured hFBCs (after 40 DIV). Freshly isolated hFBCs contained up to 30%±2% of cells expressing CD34 followed by CD31, vWf, CD146, and Flk-1/KDR, which all ranged between 10%±3%. CD105 and, particularly, CD133 were expressed in very low percentages (3.5%±0.2% and 1.2%±0.13% respectively). In contrast, cultured hFBCs showed a significant reduction of all endothelial markers with the exception for CD105 and CD133. Interesting, the lack of vWf and CD31 expression indicated the possible loss of mature endothelial cells (ECs) during hFBCs expansion. CD146 and Flk-1/KDR were greatly reduced but still detectable (2.6%±0.24% and 0.5%±0.05%, respectively).
To isolate hFBC-derived hNSCs, we applied a procedure based on immunomagnetic beads separation. Successful separation of different hFBCs phenotypes was obtained using CD34, Flk-1/KDR, CD105, CD146, and CD133. However, it was interesting to observe that only CD133 and CD146 subsets were able to grow and generate “neurosphere forming” cells. We characterized these two sub-populations of “neurosphere forming” hFBCs. We next assessed their differentiation spectrum. As shown in Figure 5E, both subpopulations efficiently differentiated into β-tubulin-III (neurons) and GFAP (astrocytes) positive cells; whereas Gal-C positive cells were detectable only in the CD133+ subpopulation (Fig. 5E, F). This multipotentiality of hFBC-derived hNSCs confirms their nature of tripotent NSCs. We observed no significant difference between the pattern of differentiation of hNSCs and the one of CD133+ hNSCs (Fig. 5E) as demonstrated also by RT-PCR (Fig. 5G).
The number of neurospheres originated from CD146, although initially higher than from CD133, declined rapidly, and a remarkable reduction of neurosphere forming cells was observed around passages 4 to 5. In contrast, both CD133 and nonimmunoselected-derived neurospheres continued to grow and to give rise to new neurospheres for >30 passages (data not shown).
Discussion
In this study, we have developed a 3-D adherent culture model composed of astrocytes, neurons, and stromal cells within a factor-reduced matrix as an in vitro model of neural tissue. We have also demonstrated that hFBCs may provide an alternative source of multipotent, self-renewing karyotypically stable hNSCs.
CNS precursors derived from human embryonic, fetal, and adult cadaveric tissues have been shown to proliferate when isolated and cultured as two-dimensional adherent cultures.28,47 Traditional culture systems cannot reproduce the complex microenvironment formed, in vivo, by the cells' connections to each other and to surrounding matrix. In vivo, the fate of the stem cell is controlled by external signals that jointly constitute the stem cell “niche”. 25 This niche maintains a balance of quiescence, cell commitment, and self-renewal in resident stem cells. Homeostatic cell turnover of cells and cell proliferation after a stimulation, such as injury, within neurogenic niches are regulated by cell-to-cell contact, cell-to-ECM interactions, and growth factor signaling.
Our system, based on 3-D adherent human fetal brain cultures, could provide the conditions and the signals typical of neurogenic niches, such as survival, maintenance, and proliferation, which could be relevant for NSC for long-term studies.
We established karyotypically stable culture composed of the whole mixture of cells derived after enzymatic dissociation of human fetal brain specimens. The presence of hNSCs/neural precursors was monitored by in situ analysis of nestin positive cells; by the expression at mRNA level of SOX2, SOX9, PAX6, and CD133; and by the NSA. Highly proliferative hFBCs cultures were obtained mostly on MTG and, to a minor extend, on FN as substrata, thus indicating an important role of β1-integrin in brain maturation and striatal development.48,49 The addition of serum to the medium significantly increased the proliferation of hFBCs and shortened the time for monolayer formation, thereby preserving the genetic integrity of hFBCs at early and late passages. These conditions together with an optimal cell density seeding were necessary to obtain the formation of 3-D self-organizing neural architectural structures. The spontaneous rearrangement of dissociated hFBCs into organized 3-D neural architectures was not a transient property but could be repeated by passing the cells into new tissue culture flasks. The analysis of the cellular composition demonstrated the presence of both mature and immature neural cells. In particular, neurons seemed to form a basal layer on which mature astrocytes were anchored, whereas very few immature oligodendrocytes and several immature nestin positive cells were randomly distributed among the layers. The presence of hNSCs was confirmed by neurosphere assay and RT-PCR, and their multipotentiality was demonstrated by immunofluorescent staining. It is worth noting that, although serum has been previously used to induce terminal differentiation of hNSCs, 34 in our conditions, the presence of serum did not influence the presence of nestin-positive cells and neural precursor and stem cell markers, essentials for hNSC proliferation, multipotency, and neurogenesis.34,38,50–52 This seems to indicate that the proximity of neighboring mature neural cells may counteract the effect of the serum to induce hNSCs terminal differentiation. The contact between the NSC and adjacent supporting cells is supposed to be important, as illustrated in the drosophila germ cell niches.53,54
Glutamate, the main excitatory neurotransmitter in the brain of mammalians, increases the concentration of Ca2+ in neurons via activation of N-methyl-D-aspartate (NMDA) and non-NMDA receptors.55,56 Some physiological processes depend on [Ca2+] changes induced by glutamate, that is, long-term potentiation. 57 The formation of neural connections in much of the developing nervous system often involves Ca2+ entry through the NMDA glutamate receptor. 58 Voltage-dependent calcium entry at the synapse plays a major role in initiating neurotransmitter release and in regulating synaptic function. In this study, a Ca2+-sensitive fluorescent dye has been used to monitor glutamate-induced [Ca2+] changes in hFBCs. An increased intracellular fluorescence indicated by a visible light–excitable intracellular calcium indicator revealed calcium uptake induced by glutamate in HFBCs cultures. Additionally, through electron microscopy, we observed axonal terminals containing mitochondria and transport vesicles, synaptic-type junctions between vesicle-containing processes, and cell bodies.
Neurotransmitters seem to play a important role in adult hNSC niches.59–61 In this study, we show that hFBCs cultures contained monoamines. The capability of the hFBCs in vitro to synthesize and release neurotrasmitters was measured by reverse-phase HPLC. These data indicate that hFBCs cultures possess neuron-like functions under these experimental conditions.
Ii and colleagues demonstrate the presence of simultaneous neurogenesis and vasculogenesis from common stem cells, which may play a role in regenerative organogenesis. 62 It is worth noting in our study that we isolated specific subpopulations of hNSCs/progenitors from hFBCs, according to the expression of CD13351 and CD146. 63 However, the CD146+-derived subpopulation were different from CD133+-derived hNSCs: (a) CD146+ cells differentiated into astrocytes and neurons but not into oligodendrocytes, (b) CD146+ showed a lower self-renewal potential. The inability to give rise to oligodendrocytes by CD146+ subpopulation suggested that neurospheres originated from CD146-positive cells could be constituted by more mature neural progenitors with a reduced capacity of differentiation into different neural lineages. Otherwise, the processes that regulate primary oligodendrocyte progenitor differentiation and proliferation seem to be conserved in CD133+ subpopulation. We, therefore, conclude that CD146 recognizes a hFBC-derived hNSCs subset residing in fetal brain tissue. Besides this important finding, our data suggest that fetal brain can contain a heterogeneous pool of progenitors. Further, the diversity of hFBC-derived hNSCs subsets in the human fetal brain may, in part, explain the unique complexity of the human cerebral cortex. The presence of several subsets of neural stem/progenitor cells in vivo has been previously reported.64,65 Our data demonstrate that CD146-expressing hNSCs are a pool of progenitors which proliferate slowly residing in the human fetal brain. In any case, we have to consider that cells cultured in vitro acquire characteristics or phenotypes that could be different from in vivo condition.
In conclusion, our system for hNSCs in vitro expansion demonstrated at least three important advantages: (1) our method of culture permits the survival of both hNSCs and mature brain cell counterparts and, thus, leads to a deeper investigation of the mechanism and of the role of neighboring mature neural cells on hNSCs' differentiation and proliferation; (2) a great number of hNSCs could be obtained in a short period of incubation; and (3) our culture conditions lead hFBCs to spontaneously organize complex 3-D structures in which different neurogenic cell subsets can concomitantly survive.
These hNSCs can proliferate for extended periods of time 6 and, therefore, offer an alternative tool to investigate the neurodevelopment even in pathological conditions.
Although our focus has been on setting-up a 3-D model as an alternative and complex source of hNSCs, it is also noteworthy that our 3-D adherent culture system may be useful to study healthy and pathological, neurobiological, and neuro-developmental phenomena by more precisely simulating in vivo interactions with the precision and control of in vitro systems.
Footnotes
Acknowledgments
The authors thank Dr. Andrea Smith for the English review of the article. We thank Deborah Jones for her excellent technical assistance with the electron microscopy studies. This work was supported by Fondazione IRCCS Istituto Neurologico “Carlo Besta” (LR8); the Italian Ministry of Health (RF2008.22); National Heart, Lung, and Blood Institute (HL52585); and Merit Review Grant, Department of Veterans Affairs Medical Research Service.
Disclosure Statement
No competing financial interests exist.